Metal Oxides Applied to Thermochemical Water-Splitting for Hydrogen Production Using Concentrated Solar Energy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
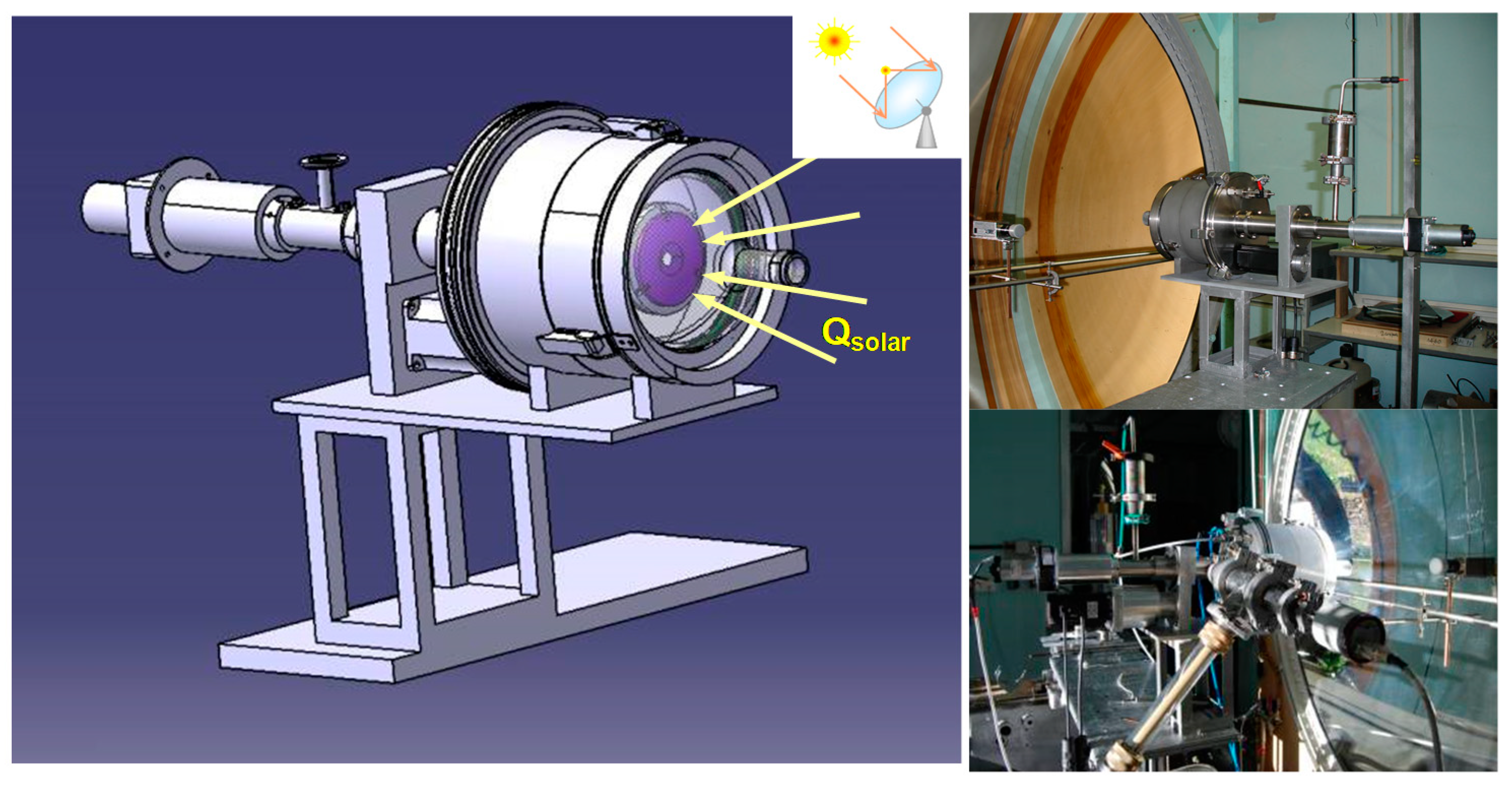{kind=link}
{kind=link}
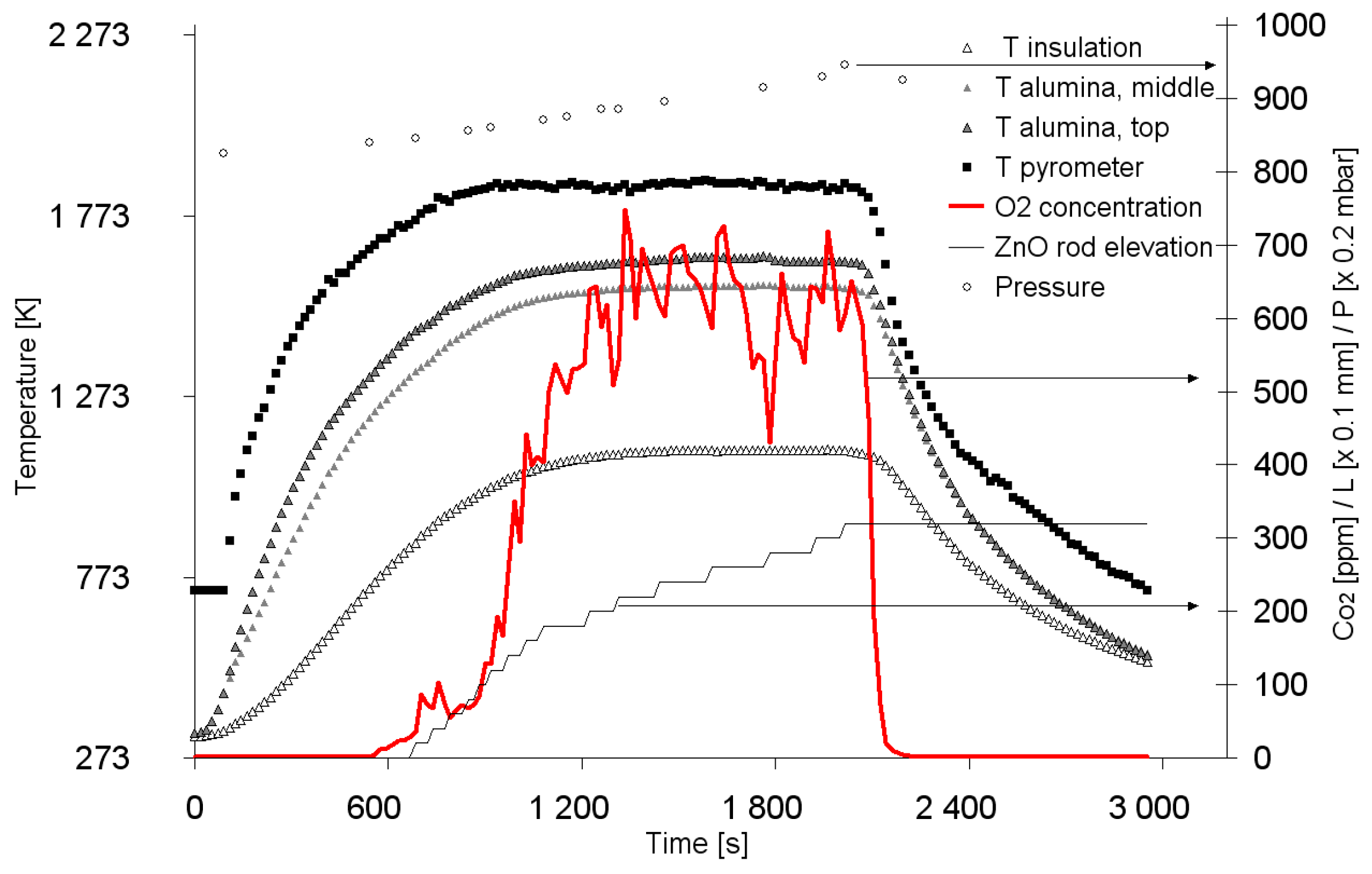{kind=link}
{kind=link}
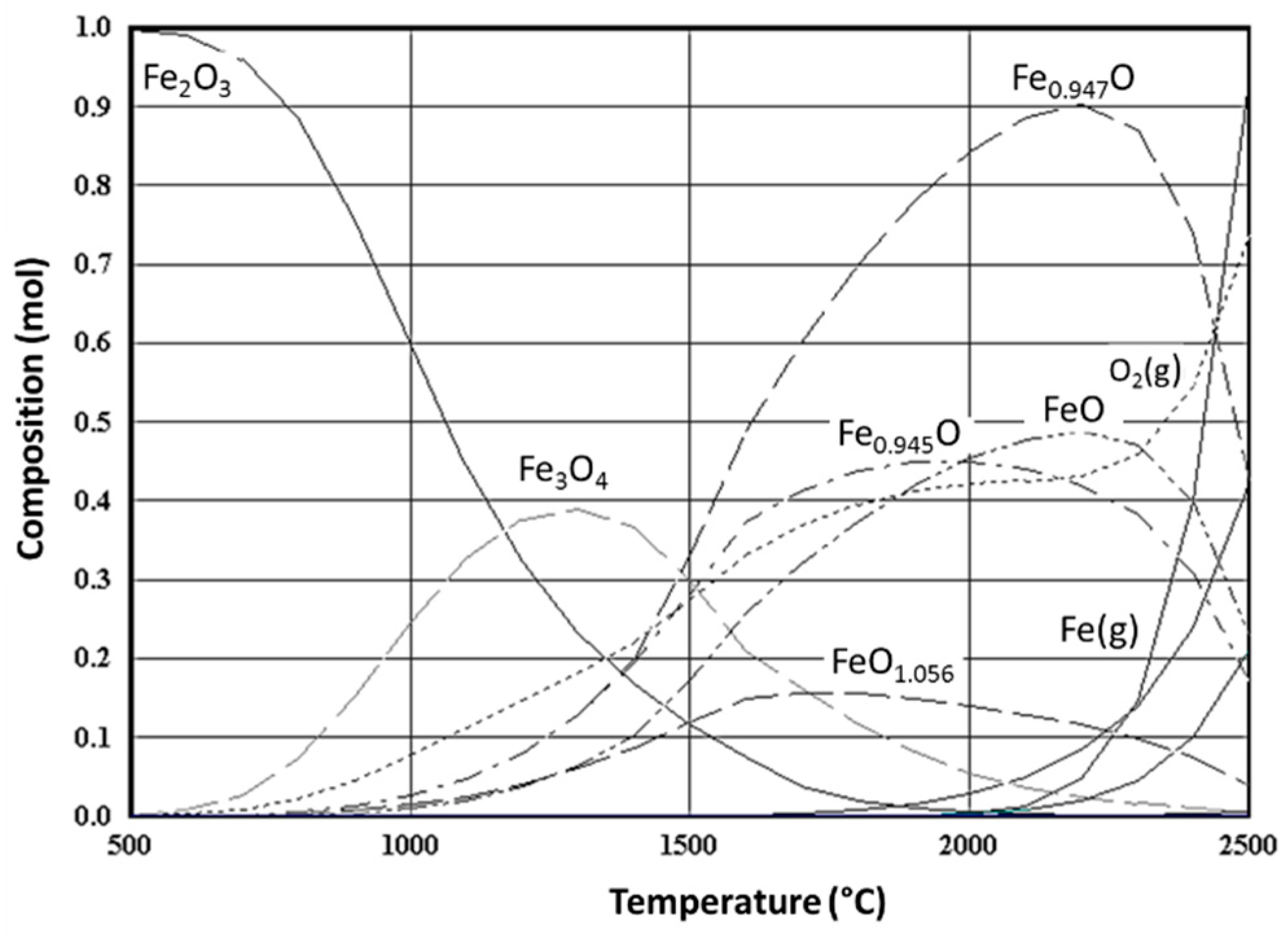{kind=link}
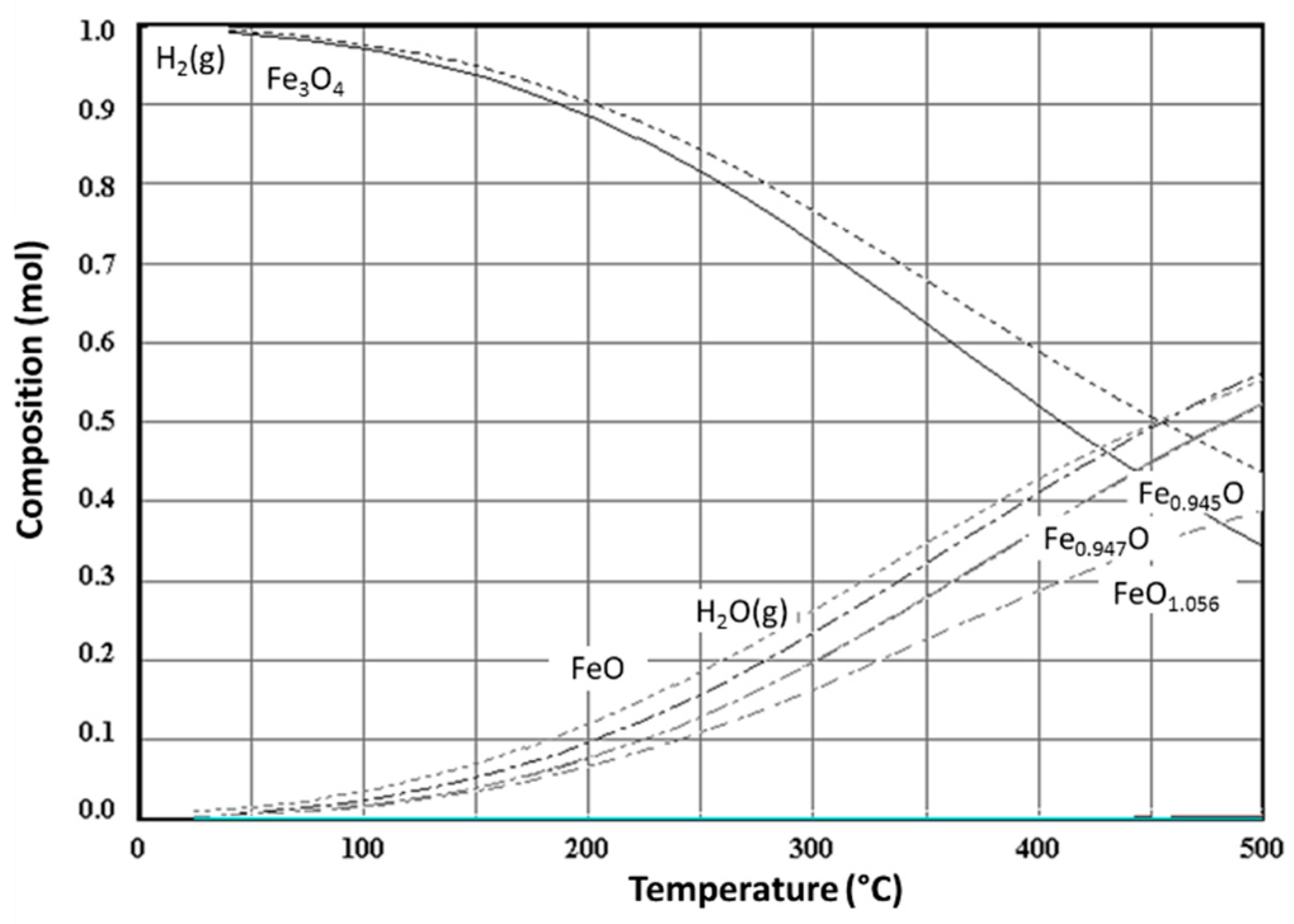{kind=link}
{kind=link}
{kind=link}
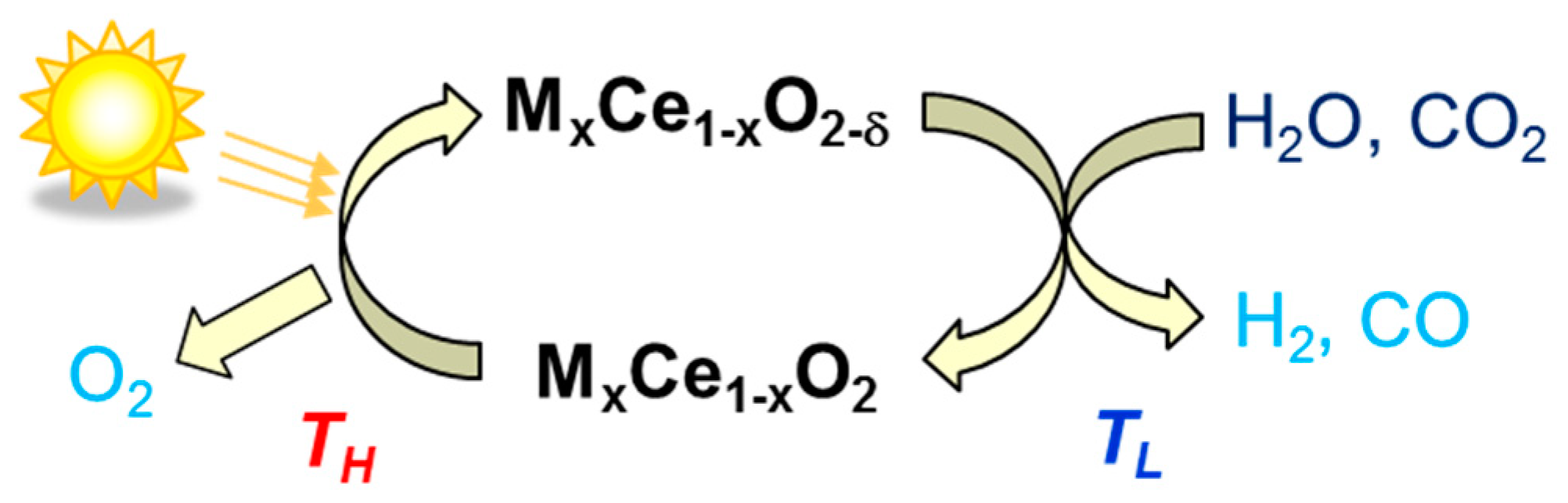{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
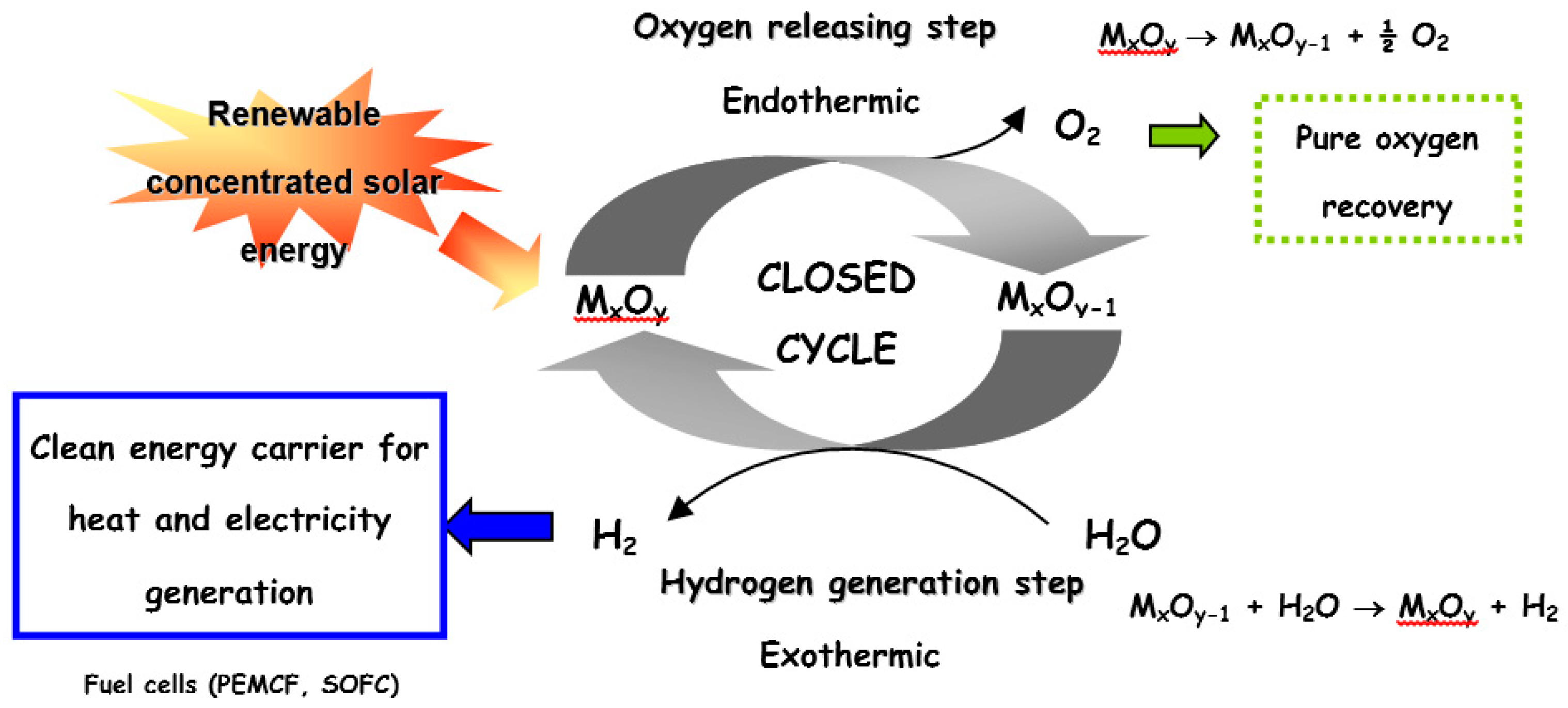
:1. Introduction
2. Volatile Metal Oxide Cycles
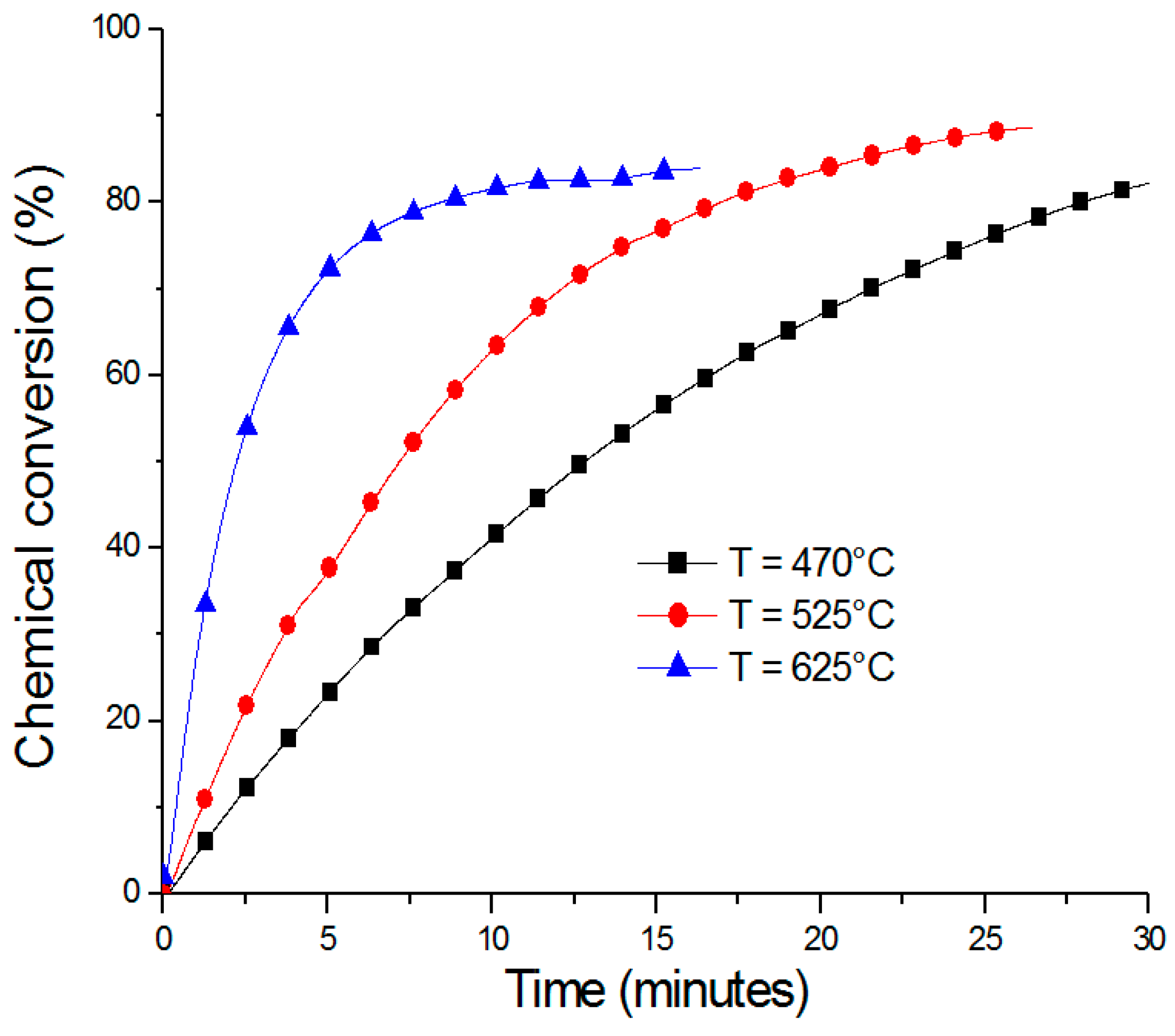
2.1. ZnO/Zn Cycle
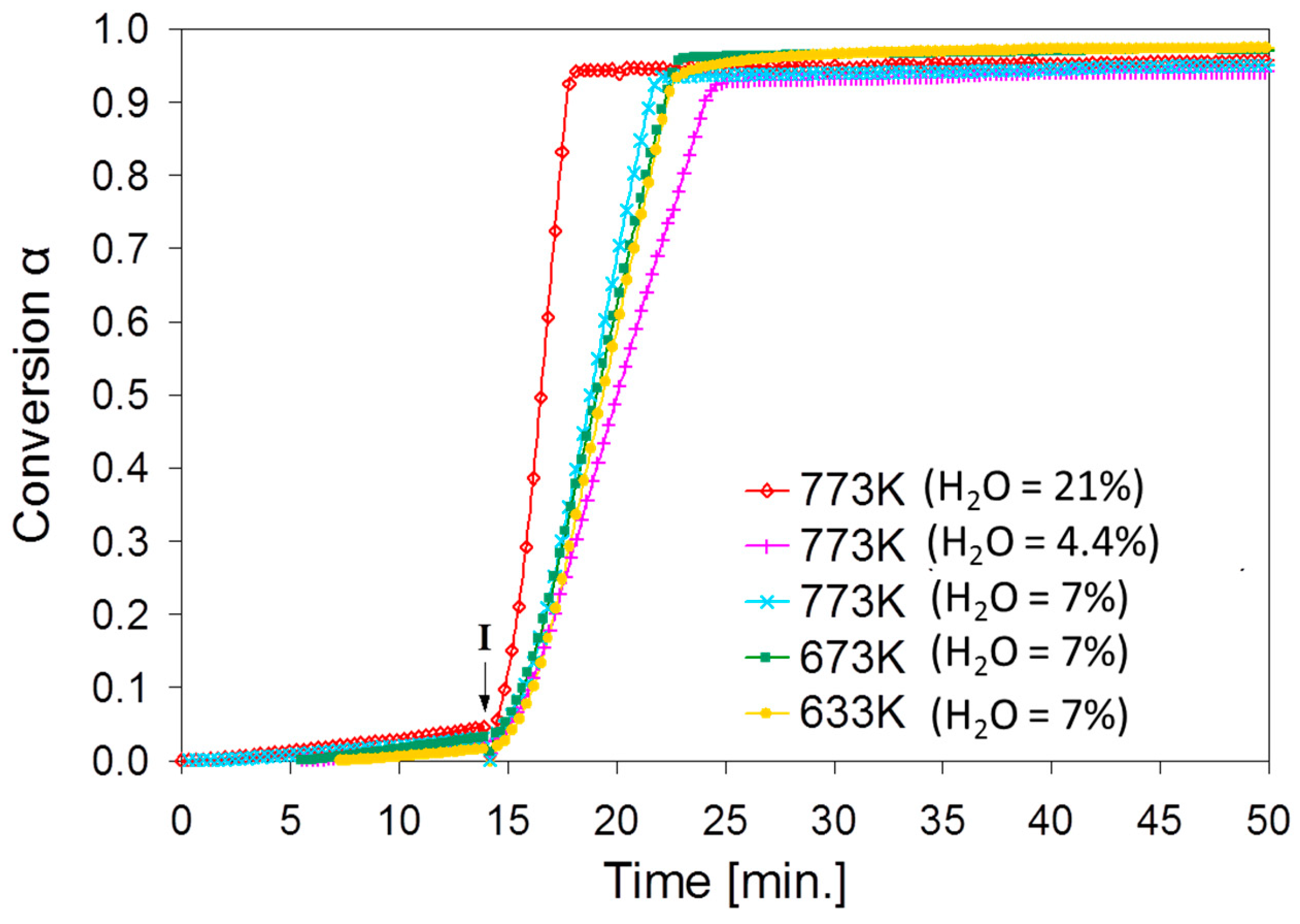
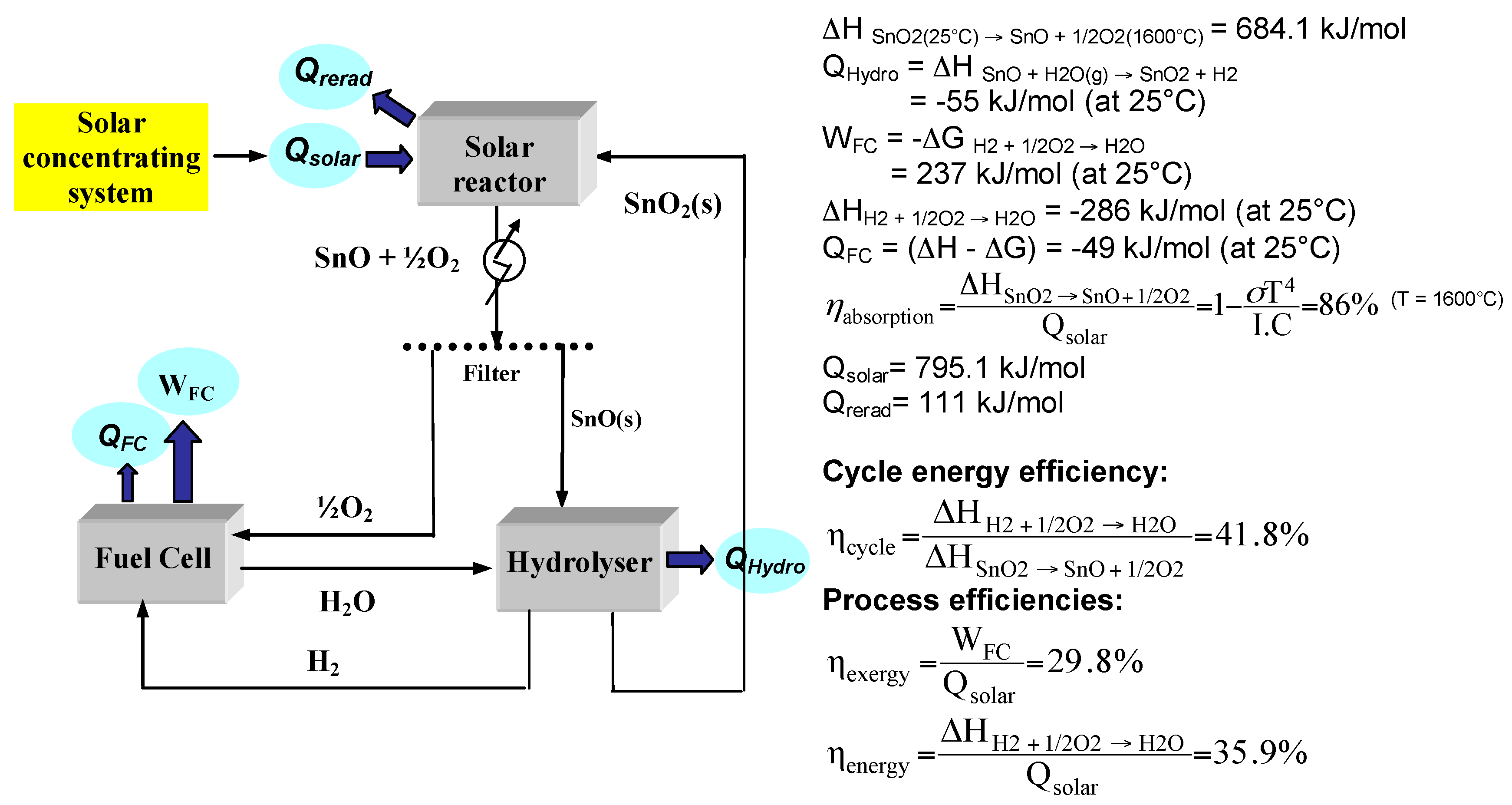
2.2. SnO2/SnO Cycle
3. Non-Volatile Metal Oxide Cycles
4. Conclusions
Funding
Conflicts of Interest
References
- Yadav, D.; Banerjee, R. A review of solar thermochemical processes. Renew. Sustain. Energy Rev. 2016, 54, 497–532. [Google Scholar] [CrossRef]
- Yilmaz, F.; Tolga Balta, M.; Selbaş, R. A review of solar based hydrogen production methods. Renew. Sustain. Energy Rev. 2016, 56, 171–178. [Google Scholar] [CrossRef]
- Villafán-Vidales, H.I.; Arancibia-Bulnes, C.A.; Riveros-Rosas, D.; Romero-Paredes, H.; Estrada, C.A. An overview of the solar thermochemical processes for hydrogen and syngas production: Reactors, and facilities. Renew. Sustain. Energy Rev. 2017, 75, 894–908. [Google Scholar] [CrossRef]
- Agrafiotis, C.; Roeb, M.; Sattler, C. A review on solar thermal syngas production via redox pair-based water/carbon dioxide splitting thermochemical cycles. Renew. Sustain. Energy Rev. 2015, 42, 254–285. [Google Scholar] [CrossRef]
- Beghi, G.E. A decade of research on thermochemical hydrogen at the Joint Research Centre, Ispra. Int. J. Hydrogen Energy 1986, 11, 761–771. [Google Scholar] [CrossRef]
- Funk, J.E. Thermochemical hydrogen production: Past and present. Int. J. Hydrogen Energy 2001, 16, 185–190. [Google Scholar] [CrossRef]
- Yalcin, S. A review of nuclear hydrogen production. Int. J. Hydrogen Energy 1989, 14, 551–561. [Google Scholar] [CrossRef]
- Bamberger, C.E.; Richardson, D.M. Hydrogen production from water by thermochemical cycles. Cryogenics 1976, 16, 197–208. [Google Scholar] [CrossRef]
- Bamberger, C.E. Hydrogen production from water by thermochemical cycles; A 1977 update. Cryogenics 1978, 18, 170–183. [Google Scholar] [CrossRef]
- Brown, L.C.; Besenbruch, G.E.; Lentsch, R.D.; Schultz, K.R.; Funk, J.F.; Pickard, P.S.; Marshall, A.C.; Showalter, S.K. High Efficiency Generation of Hydrogen Fuels Using Nuclear Power; GA-A24285; General Atomics: San Diego, CA, USA, December 2003. [Google Scholar]
- Kameyama, H.; Yoshida, K. Reactor design for the UT-3 thermochemical hydrogen production process. Int. J. Hydrogen Energy 1981, 6, 567–575. [Google Scholar] [CrossRef]
- Kameyama, H.; Tomino, Y.; Sato, T.; Amir, R.; Orihara, A.; Aihara, M.; Yoshida, K. Process simulation of ‘MASCOT’ plant using the UT-3 thermochemical cycle for hydrogen production. Int. J. Hydrogen Energy 1989, 14, 323–330. [Google Scholar] [CrossRef]
- O’Keefe, D.; Allen, C.; Besenbruch, G.; Brown, L.; Norman, J.; Sharp, R.; McCorkle, K. Preliminary results from bench-scale testing of a sulfur-iodine thermochemical water-splitting cycle. Int. J. Hydrogen Energy 1982, 7, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Oztürk, I.T.; Hammache, A.; Bilgen, E. An improved process for H2SO4 decomposition step of the sulfur-iodine cycle. Energy Convers. Mgmt. 1995, 36, 11–21. [Google Scholar] [CrossRef]
- Kubo, S.; Nakajima, H.; Kasahara, S.; Higashi, S.; Masaki, T.; Abe, H.; Onuki, K. A demonstration study on a close-cycle hydrogen production by the thermochemical water-splitting iodine-sulfur process. Nucl. Eng. Design 2004, 233, 347–354. [Google Scholar] [CrossRef]
- Kubo, S.; Kasahara, S.; Okuda, H.; Terada, A.; Tanaka, N.; Inaba, Y.; Ohashi, H.; Inagaki, Y.; Onuki, K.; Hino, R. A pilot test plan of the water-splitting iodine-sulfur process. Nucl. Eng. Design 2004, 233, 355–362. [Google Scholar] [CrossRef]
- Noglik, A.; Roeb, M.; Rzepczyk, T.; Hinkley, J.; Sattler, C.; Pitz-Paal, R. Solar Thermochemical Generation of Hydrogen: Development of a Receiver Reactor for the Decomposition of Sulfuric Acid. J. Sol. Energy Eng. 2009, 131, 011003. [Google Scholar] [CrossRef]
- Kolb, G.J.; Diver, R.B.; Siegel, N. Central-Station Solar Hydrogen Power Plant. J. Sol. Energy Eng. 2007, 129, 179–183. [Google Scholar] [CrossRef]
- Bilgen, C.; Broggi, A.; Bilgen, E. The solar Cristina process for hydrogen production. Sol. Energy 1986, 36, 267–280. [Google Scholar] [CrossRef]
- Sakurai, M.; Bilgen, E.; Tsutsumi, A.; Yoshida, K. Adiabatic UT-3 thermochemical process for hydrogen production. Int. J. Hydrogen Energy 1996, 21, 865–870. [Google Scholar] [CrossRef]
- Sakurai, M.; Miyake, N.; Tsutsumi, A.; Yoshida, K. Analysis of a reaction mechanism in the UT-3 thermochemical hydrogen production cycle. Int. J. Hydrogen Energy 1996, 21, 871–875. [Google Scholar] [CrossRef]
- Sakurai, M.; Bilgen, E.; Tsutsumi, A.; Yoshida, K. Solar UT-3 thermochemical cycle for hydrogen production. Sol. Energy 1996, 57, 51–58. [Google Scholar] [CrossRef]
- Graf, D.; Monnerie, N.; Roeb, M.; Schmitz, M.; Sattler, C. Economic comparison of solar hydrogen generation by means of thermochemical cycles and electrolysis. Int. J. Hydrogen Energy 2008, 33, 4511–4519. [Google Scholar] [CrossRef]
- Pregger, T.; Graf, D.; Krewitt, W.; Sattler, C.; Roeb, M.; Möller, S. Prospects of solar thermal hydrogen production processes. Int. J. Hydrogen Energy 2009, 34, 4256–4267. [Google Scholar] [CrossRef]
- Bilgen, E.; Bilgen, C. Solar hydrogen production using two-step thermochemical cycles. Int. J. Hydrogen Energy 1982, 7, 637–644. [Google Scholar] [CrossRef]
- Steinfeld, A.; Kuhn, P.; Reller, A.; Palumbo, R.; Murray, J.; Tamaura, Y. Solar-processed metals as clean energy carriers and water-splitters. Int. J. Hydrogen Energy 1998, 23, 767–774. [Google Scholar] [CrossRef]
- Abanades, S. Hydrogen production technologies from solar thermal energy. Green 2011, 1, 209–220. [Google Scholar] [CrossRef]
- Abanades, S.; Charvin, P.; Flamant, G.; Neveu, P. Screening of water-splitting thermochemical cycles potentially attractive for hydrogen production by concentrated solar energy. Energy 2006, 31, 2805–2822. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Lemort, F.; Flamant, G. Analysis of solar processes for hydrogen production from water-splitting thermochemical cycles. Energy Convers. Manag. 2008, 49, 1547–1556. [Google Scholar] [CrossRef]
- Romero, M.; Buck, R.; Pacheco, J.E. An update on solar central receiver systems, projects, and technologies. J. Sol. Energy Eng. 2002, 124, 98–108. [Google Scholar] [CrossRef]
- Dokiya, M.; Kotera, Y. Hybrid cycle with electrolysis using Cu-Cl system. Int. J. Hydrogen Energy 1976, 1, 117–127. [Google Scholar] [CrossRef]
- Wentorf, R.H.; Hanneman, R.E. Thermochemical hydrogen generation. Science 1974, 185, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ducarroir, M.; Tmar, M.; Bernard, C. Possibilités de stockage de l’énergie solaire à partir de sulfates. Revue Phys. Appl. 1980, 15, 513–528. [Google Scholar] [CrossRef]
- Kameyama, H.; Yoshida, K.; Kunii, D. A method for screening possible thermochemical decomposition processes for water using ΔG0 -T diagrams. Chem. Eng. J. 1976, 11, 223–229. [Google Scholar] [CrossRef]
- Lundberg, M. Model calculations on some feasible two-step water splitting processes. Int. J. Hydrogen Energy 1993, 18, 369–376. [Google Scholar] [CrossRef]
- Knoche, K.F.; Cremer, H.; Steinborn, G.; Schneider, W. Feasibility studies of chemical reactions for thermochemical water splitting cycles of the iron-chlorine, iron-sulfur and manganese-sulfur families. Int. J. Hydrogen Energy 1977, 2, 269–289. [Google Scholar] [CrossRef]
- Knoche, K.F.; Schuster, P. Thermochemical production of hydrogen by a vanadium/chlorine cycle. Part 1: An energy and exergy analysis of the process. Int. J. Hydrogen Energy 1984, 9, 457–472. [Google Scholar] [CrossRef]
- Knoche, K.F.; Cremer, H.; Steinborn, G. A thermochemical process for hydrogen production. Int. J. Hydrogen Energy 1976, 1, 23–32. [Google Scholar] [CrossRef]
- Ambriz, J.J.; Sibieude, F.; Ducarroir, M. Preparation of cadmium by thermal dissociation of CdO using solar energy. Int. J. Hydrogen Energy 1982, 7, 143–153. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar hydrogen production via a two-step water-splitting thermochemical cycle based on Zn/ZnO redox reactions. Int. J. Hydrogen Energy 2002, 27, 611–619. [Google Scholar] [CrossRef]
- Palumbo, R.; Ledé, J.; Boutin, O.; Elorza Ricart, E.; Steinfeld, A.; Muller, S.; Weidenkaff, A.; Fletcher, E.A.; Bielicki, J. The production of Zn from ZnO in a high-temperature solar decomposition quench process. I—The Scientific framework for the process. Chem. Eng. Sci. 1998, 53, 2503–2517. [Google Scholar] [CrossRef]
- Weidenkaff, A.; Steinfeld, A.; Wokaun, A.; Auer, P.O.; Eichler, B.; Reller, A. Direct solar thermal dissociation of zinc oxide: Condensation and crystallization of zinc in the presence of oxygen. Sol. Energy 1999, 65, 59–69. [Google Scholar] [CrossRef]
- Weidenkaff, A.; Reller, A.W.; Wokaun, A.; Steinfeld, A. Thermogravimetric analysis of the ZnO/Zn water splitting cycle. Thermochimica Acta 2000, 359, 69–75. [Google Scholar] [CrossRef]
- Möller, S.; Palumbo, R. Solar thermal decomposition kinetics of ZnO in the temperature range 1950–2400 K. Chem. Eng. Sci. 2001, 56, 4505–4515. [Google Scholar] [CrossRef]
- Möller, S.; Palumbo, R. The development of a solar chemical reactor for the direct thermal dissociation of zinc oxide. ASME J. Sol. Energy Eng. 2001, 123, 83–90. [Google Scholar] [CrossRef]
- Abanades, S.; Charvin, P.; Flamant, G. Design and simulation of a solar chemical reactor for the thermal reduction of metal oxides: Case study of zinc oxide dissociation. Chem. Eng. Sci. 2007, 62, 6323–6333. [Google Scholar] [CrossRef]
- Chambon, M.; Abanades, S.; Flamant, G. Design of a lab-scale rotary cavity-type solar reactor for continuous thermal reduction of volatile oxides under reduced pressure. ASME J. Sol. Energy Eng. 2010, 132, 021006. [Google Scholar] [CrossRef]
- Chambon, M.; Abanades, S.; Flamant, G. Thermal dissociation of compressed ZnO and SnO2 powders in a moving front solar thermochemical reactor. AIChE J. 2011, 57, 2264–2273. [Google Scholar] [CrossRef]
- Chambon, M.; Abanades, S.; Flamant, G. Solar Thermal Reduction of ZnO and SnO2: Characterization of the Recombination Reaction with O2. Chem. Eng. Sci. 2010, 65, 3671–3680. [Google Scholar] [CrossRef]
- Levêque, G.; Abanades, S. Kinetic analysis of high-temperature solid–gas reactions by an inverse method applied to ZnO and SnO2 solar thermal dissociation. Chem. Eng. J. 2013, 217, 139–149. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Neveu, P.; Lemort, F.; Flamant, G. Dynamic modeling of a volumetric solar reactor for volatile metal oxide reduction. Chem. Eng. Res. Design 2008, 86, 1216–1222. [Google Scholar] [CrossRef]
- Schunk, L.O.; Steinfeld, A. Kinetics of the thermal dissociation of ZnO exposed to concentrated solar irradiation using a solar-driven thermogravimeter in the 1800–2100 K range. AIChE J. 2009, 55, 1497–1504. [Google Scholar] [CrossRef]
- Perkins, C.; Lichty, P.; Weimer, A.W. Determination of aerosol kinetics of thermal ZnO dissociation by thermogravimetry. Chem. Eng. Sci. 2007, 62, 5952–5962. [Google Scholar] [CrossRef]
- Perkins, C.; Lichty, P.R.; Weimer, A.W. Thermal ZnO dissociation in a rapid aerosol reactor as part of a solar hydrogen production cycle. Int. J. Hydrogen Energy 2008, 33, 499–510. [Google Scholar] [CrossRef]
- Melchior, T.; Perkins, C.; Weimer, A.W.; Steinfeld, A. A cavity-receiver containing a tubular absorber for high-temperature thermochemical processing using concentrated solar energy. Int. J. Therm. Sci. 2008, 47, 1496–1503. [Google Scholar] [CrossRef]
- Haussener, S.; Hirsch, D.; Perkins, C.; Weimer, A.; Lewandowski, A.; Steinfeld, A. Modeling of a Multitube High-Temperature Solar Thermochemical Reactor for Hydrogen Production. J. Sol. Energy Eng. 2009, 131, 024503. [Google Scholar] [CrossRef]
- Haueter, P.; Moeller, S.; Palumbo, R.; Steinfeld, A. The production of zinc by thermal dissociation of zinc oxide-Solar chemical reactor design. Sol. Energy 1999, 67, 161–167. [Google Scholar] [CrossRef]
- Muller, R.; Haeberling, P.; Palumbo, R. Further advances toward the development of a direct heating solar thermal chemical reactor for the thermal dissociation of ZnO(s). Sol. Energy 2006, 80, 500–511. [Google Scholar] [CrossRef]
- Schunk, L.; Haeberling, P.; Wepf, S.; Wuillemin, D.; Meier, A.; Steinfeld, A. A Solar Receiver-Reactor for the Thermal Dissociation of Zinc Oxide. ASME J. Sol. Energy Eng. 2008, 130, 021009. [Google Scholar] [CrossRef]
- Koepf, E.; Villasmil, W.; Meier, A. Pilot-scale solar reactor operation and characterization for fuel production via the Zn/ZnO thermochemical cycle. Appl. Energy 2016, 165, 1004–1023. [Google Scholar] [CrossRef]
- Müller, R.; Steinfeld, A. H2O-splitting thermochemical cycle based on ZnO/Zn-redox: Quenching the effluents from the ZnO dissociation. Chem. Eng. Sci. 2008, 63, 217–227. [Google Scholar] [CrossRef]
- Keunecke, M.; Meier, A.; Palumbo, R. Solar thermal decomposition of zinc oxide: An initial investigation of the recombination reaction in the temperature range 1100–1250 K. Chem. Eng. Sci. 2004, 59, 2695–2704. [Google Scholar] [CrossRef]
- Berman, A.; Epstein, M. The kinetics of hydrogen production in the oxidation of liquid zinc with water vapor. Int. J. Hydrogen Energy 2000, 25, 957–967. [Google Scholar] [CrossRef]
- Wegner, K.; Ly, H.C.; Weiss, R.J.; Pratsinis, S.E.; Steinfeld, A. In situ formation and hydrolysis of Zn nanoparticles for H2 production by the 2-step ZnO/Zn water-splitting thermochemical cycle. Int. J. Hydrogen Energy 2006, 31, 55–61. [Google Scholar] [CrossRef]
- Weiss, R.J.; Ly, H.C.; Wegner, K.; Pratsinis, S.E.; Steinfeld, A. H2 production by Zn hydrolysis in a hot-wall aerosol reactor. AIChE J. 2005, 51, 1966–1970. [Google Scholar] [CrossRef]
- Ernst, F.O.; Tricoli, A.; Pratsinis, S.E.; Steinfeld, A. Co-synthesis of H2 and ZnO by in-situ Zn aerosol formation and hydrolysis. AIChE J. 2006, 52, 3297–3303. [Google Scholar] [CrossRef]
- Melchior, T.; Piatkowski, N.; Steinfeld, A. H2 production by steam-quenching of Zn vapor in a hot-wall aerosol flow reactor. Chem. Eng. Sci. 2009, 64, 1095–1101. [Google Scholar] [CrossRef]
- Abu Hamed, T.; Davidson, J.H.; Stolzenburg, M. Hydrolysis of Evaporated Zn in a Hot Wall Flow Reactor. J. Sol. Energy Eng. 2008, 130, 041010. [Google Scholar] [CrossRef]
- Lindemer, M.D.; Advani, S.G.; Prasad, A.K. Experimental investigation of heterogeneous hydrolysis with Zn vapor under a temperature gradient. Int. J. Hydrogen Energy 2017, 42, 7847–7856. [Google Scholar] [CrossRef]
- Weibel, D.; Jovanovic, Z.R.; Gálvez, E.; Steinfeld, A. Mechanism of Zn Particle Oxidation by H2O and CO2 in the Presence of ZnO. Chem. Mater. 2014, 26, 6486–6495. [Google Scholar] [CrossRef]
- Ernst, F.O.; Steinfeld, A.; Pratsinis, S.E. Hydrolysis rate of submicron Zn particles for solar H2 synthesis. Int. J. Hydrogen Energy 2009, 34, 1166–1175. [Google Scholar] [CrossRef]
- Funke, H.H.; Diaz, H.; Liang, X.; Carney, C.S.; Weimer, A.W.; Li, P. Hydrogen generation by hydrolysis of zinc powder aerosol. Int. J. Hydrogen Energy 2008, 33, 1127–1134. [Google Scholar] [CrossRef]
- Vishnevetsky, I.; Epstein, M. Production of hydrogen from solar zinc in steam atmosphere. Int. J. Hydrogen Energy 2007, 32, 2791–2802. [Google Scholar] [CrossRef]
- Abanades, S. Thermogravimetry analysis of CO2 and H2O reduction from solar nanosized Zn powder for thermochemical fuel production. Ind. Eng. Chem. Res. 2012, 51, 741–750. [Google Scholar] [CrossRef]
- Villafán-Vidales, H.I.; Abanades, S.; Montiel-González, M.; Romero-Paredes, H.; Arancibia-Bulnes, C.A.; Estrada, C.A. Transient heat transfer simulation of a 1 kWth moving front solar thermochemical reactor for thermal dissociation of compressed ZnO. Chem. Eng. Res. Design 2015, 93, 174–184. [Google Scholar] [CrossRef]
- Levêque, G.; Abanades, S. Investigation of thermal and carbothermal reduction of volatile oxides (ZnO, SnO2, GeO2, and MgO) via solar-driven vacuum thermogravimetry for thermochemical production of solar fuels. Thermochimica Acta 2015, 605, 86–94. [Google Scholar] [CrossRef]
- Abanades, S.; Charvin, P.; Lemont, F.; Flamant, G. Novel two-step SnO2/SnO water-splitting cycle for solar thermochemical production of hydrogen. Int. J. Hydrogen Energy 2008, 33, 6021–6030. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Lemont, F.; Flamant, G. Experimental study of SnO2/SnO/Sn thermochemical systems for solar production of hydrogen. AIChE J. 2008, 54, 2759–2767. [Google Scholar] [CrossRef]
- Chambon, M.; Abanades, S.; Flamant, G. Kinetic investigation of hydrogen generation from hydrolysis of SnO and Zn solar nanopowders. Int. J. Hydrogen Energy 2009, 34, 5326–5336. [Google Scholar] [CrossRef]
- Abanades, S. CO2 and H2O reduction by solar thermochemical looping using SnO2/SnO redox reactions: Thermogravimetric analysis. Int. J. Hydrogen Energy 2012, 37, 8223–8231. [Google Scholar] [CrossRef]
- Levêque, G.; Abanades, S.; Jumas, J.-C.; Olivier-Fourcade, J. Characterization of two-step tin-based redox system for thermochemical fuel production from solar-driven CO2 and H2O splitting cycle. Ind. Eng. Chem. Res. 2014, 53, 5668–5677. [Google Scholar] [CrossRef]
- Levêque, G.; Abanades, S. Thermodynamic and kinetic study of the carbothermal reduction of SnO2 for solar thermochemical fuel generation. Energy Fuels 2014, 28, 1396–1405. [Google Scholar] [CrossRef]
- Sturzenegger, M.; Nuesch, P. Efficiency analysis for a manganese-oxide-based thermochemical cycle. Energy 1999, 24, 959–970. [Google Scholar] [CrossRef]
- Abanades, S.; Flamant, G. Thermochemical hydrogen production from a two-step solar-driven water-splitting cycle based on cerium oxides. Sol. Energy 2006, 80, 1611–1623. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Bêche, E.; Lemont, F.; Flamant, G. Hydrogen production from mixed cerium oxides via three-step water-splitting cycles. Solid State Ionics 2009, 180, 1003–1010. [Google Scholar] [CrossRef]
- Bamberger, C.E.; Nichols, H. Basic chemistry of a new cycle, based on reactions of Ce(III) titanate, for splitting water. Int. J. Hydrogen Energy 1979, 4, 513–516. [Google Scholar] [CrossRef]
- Nakamura, T. Hydrogen production from water utilizing solar heat at high temperatures. Sol. Energy 1977, 19, 467–475. [Google Scholar] [CrossRef]
- Sibieude, F.; Ducarroir, M.; Tofighi, A.; Ambriz, J. High temperature experiments with a solar furnace. The decomposition of Fe3O4, Mn3O4, CdO. Int. J. Hydrogen Energy 1982, 7, 79–88. [Google Scholar] [CrossRef]
- Tofighi, A.; Sibieude, F.; Ducarroir, M.; Benezech, G. Thermal decomposition of magnetite in air at the focus of a solar furnace (Décomposition thermique à l’air de la magnétite au foyer d’un four solaire). Rev. Int. Htes Temp. Réfract. 1978, 15, 7–14. [Google Scholar]
- Tofighi, A.; Sibieude, F. Dissociation of magnetite in a solar furnace for hydrogen production. Tentative production evaluation of a 1000 kW concentrator from small scale (2 kW) experimental results. Int. J. Hydrogen Energy 1984, 9, 293–296. [Google Scholar] [CrossRef]
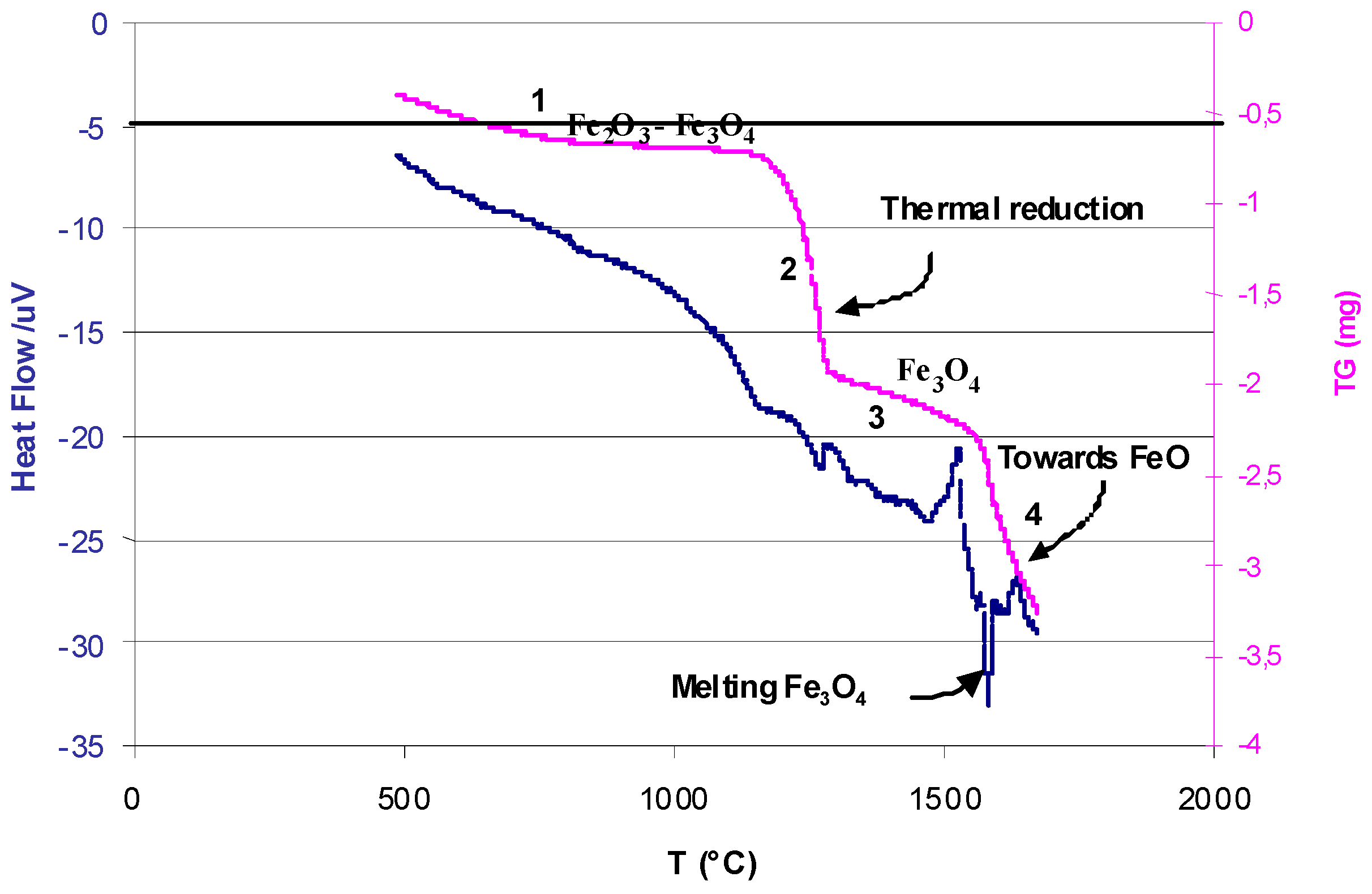
- Steinfeld, A.; Sanders, S.; Palumbo, R. Design aspects of solar thermochemical engineering—A case study: Two-step water-splitting cycle using the Fe3O4/FeO redox system. Sol. Energy 1999, 65, 43–53. [Google Scholar] [CrossRef]
- Weidenkaff, A.; Nüesch, P.; Wokaun, A.; Reller, A. Mechanistic studies of the water-splitting reaction for producing solar hydrogen. Solid State Ionics 1997, 101–103, 915–922. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Flamant, G.; Lemort, F. Two-step water-splitting thermochemical cycle based on iron oxide redox pair for solar hydrogen production. Energy 2007, 32, 1124–1133. [Google Scholar] [CrossRef]
- Charvin, P.; Abanades, S.; Lemort, F.; Flamant, G. Hydrogen production by three-step solar thermochemical cycles using hydroxides and metal oxide systems. Energy Fuels 2007, 21, 2919–2928. [Google Scholar] [CrossRef]
- Ehrensberger, K.; Frei, A.; Kuhn, P.; Oswald, H.; Hug, P. Comparative experimental investigations of the water splitting reaction with iron oxide Fe1-yO and iron manganese oxides (Fe1-xMnx)1-yO. Solid State Ionics 1995, 78, 151–160. [Google Scholar] [CrossRef]
- Tamaura, Y.; Steinfeld, A.; Kuhn, P.; Ehrensberger, K. Production of solar hydrogen by a novel, 2-step, water-splitting thermochemical cycle. Energy 1995, 20, 325–330. [Google Scholar] [CrossRef]
- Tamaura, Y.; Ueda, Y.; Matsunami, J.; Hasegawa, N.; Nesuka, M.; Sano, T.; Tsuji, M. Solar hydrogen production by using ferrites. Sol. Energy 1999, 65, 55–57. [Google Scholar] [CrossRef]
- Tamaura, Y.; Kojima, M.; Sano, Y.; Ueda, Y.; Hasegawa, N.; Tsuji, M. Thermodynamic evaluation of water splitting by a cation excessive (Ni, Mn) ferrite. Int. J. Hydrogen Energy 1998, 23, 1185–1191. [Google Scholar] [CrossRef]
- Inoue, M.; Hasegawa, N.; Uehara, R.; Gokon, N.; Kaneko, H.; Tamaura, Y. Solar hydrogen generation with H2O/ZnO/MnFe2O4 system. Sol. Energy 2004, 76, 309–315. [Google Scholar] [CrossRef]
- Kaneko, H.; Hosokawa, Y.; Kojima, N.; Gokon, N.; Hasegawa, N.; Kitamura, M.; Tamaura, Y. Studies on metal oxides suitable for enhancement of the O2-releasing step in water splitting by the MnFe2O4–Na2CO3 system. Energy 2001, 26, 919–929. [Google Scholar] [CrossRef]
- Kaneko, H.; Hosokawa, Y.; Gokon, N.; Kojima, N.; Hasegawa, N.; Kitamura, M.; Tamaura, Y. Enhancement of O2-releasing step with Fe2O3 in the water splitting by MnFe2O4–Na2CO3 system. J. Phys. Chem. Solids 2001, 62, 1341–1347. [Google Scholar] [CrossRef]
- Kaneko, H.; Ochiai, Y.; Shimizu, K.; Hosokawa, Y.; Gokon, N.; Tamaura, Y. Thermodynamic study based on the phase diagram of the Na2O–MnO–Fe2O3 system for H2 production in three-step water splitting with Na2CO3/MnFe2O4/Fe2O3. Sol. Energy 2002, 72, 377–383. [Google Scholar] [CrossRef]
- Kaneko, H.; Gokon, N.; Hasegawa, N.; Tamaura, Y. Solar thermochemical process for hydrogen production using ferrites. Energy 2005, 30, 2171–2178. [Google Scholar] [CrossRef]
- Tamaura, Y.; Kojima, N.; Hasegawa, N.; Inoue, M.; Uehara, R.; Gokon, N.; Kaneko, H. Stoichiometric studies of H2 generation reaction for H2O/Zn/Fe3O4 system. Int. J. Hydrogen Energy 2001, 26, 917–922. [Google Scholar] [CrossRef]
- Tamaura, Y.; Kaneko, H. Oxygen-releasing step of ZnFe2O4/(ZnO + Fe3O4)-system in air using concentrated solar energy for solar hydrogen production. Sol. Energy 2005, 78, 616–622. [Google Scholar] [CrossRef]
- Takahashi, Y.; Aoki, H.; Kaneko, H.; Hasegawa, N.; Suzuki, A.; Tamaura, Y. Oxygen-gas-releasing reaction of Zn ferrite by Xe lamp beam irradiation in air at 1800 K. Solid State Ionics 2004, 172, 89–91. [Google Scholar] [CrossRef]
- Aoki, H.; Kaneko, H.; Hasegawa, N.; Ishihara, H.; Suzuki, A.; Tamaura, Y. The ZnFe2O4/(ZnO+Fe3O4) system for H2 production using concentrated solar energy. Solid State Ionics 2004, 172, 113–116. [Google Scholar] [CrossRef]
- Tamaura, Y.; Uehara, R.; Hasegawa, N.; Kaneko, H.; Aoki, H. Study on solid-state chemistry of the ZnO/Fe3O4/H2O system for H2 production at 973–1073 K. Solid State Ionics 2004, 172, 121–124. [Google Scholar] [CrossRef]
- Kaneko, H.; Kojima, N.; Hasegawa, N.; Inoue, M.; Uehara, R.; Gokon, N.; Tamaura, Y.; Sano, T. Reaction mechanism of H2 generation for H2O/Zn/Fe3O4 system. Int. J. Hydrogen Energy 2002, 27, 1023–1028. [Google Scholar] [CrossRef]
- Kaneko, H.; Kodama, T.; Gokon, N.; Tamaura, Y.; Lovegrove, K.; Luzzi, A. Decomposition of Zn-ferrite for O2 generation by concentrated solar radiation. Sol. Energy 2004, 76, 317–322. [Google Scholar] [CrossRef]
- Ishihara, H.; Hasegawa, N.; Aoki, H.; Kaneko, H.; Suzuki, A.; Tamaura, Y. Two-step water splitting for H2 production with ZnII-MnII,III-FeIII spinel structure using concentrated solar heat. Solid States Ionics 2004, 172, 117–119. [Google Scholar] [CrossRef]
- Ishihara, H.; Kaneko, H.; Hasegawa, N.; Tamaura, Y. Two-step water-splitting at 1273–1623 K using yttria-stabilized zirconia-iron oxide solid solution via co-precipitation and solid-state reaction. Energy 2008, 33, 1788–1793. [Google Scholar] [CrossRef]
- Kaneko, H.; Yokoyama, T.; Fuse, A.; Ishihara, H.; Hasegawa, N.; Tamaura, Y. Synthesis of new ferrite, Al–Cu ferrite, and its oxygen deficiency for solar H2 generation from H2O. Int. J. Hydrogen Energy 2006, 31, 2256–2265. [Google Scholar] [CrossRef]
- Kodama, T.; Kondoh, Y.; Yamamoto, R.; Andou, H.; Satou, N. Thermochemical hydrogen production by a redox system of ZrO2-supported Co(II)-ferrite. Sol. Energy 2005, 78, 623–631. [Google Scholar] [CrossRef]
- Kodama, T.; Gokon, N.; Yamamoto, R. Thermochemical two-step water splitting by ZrO2-supported NixFe3−xO4 for solar hydrogen production. Sol. Energy 2008, 82, 73–79. [Google Scholar] [CrossRef]
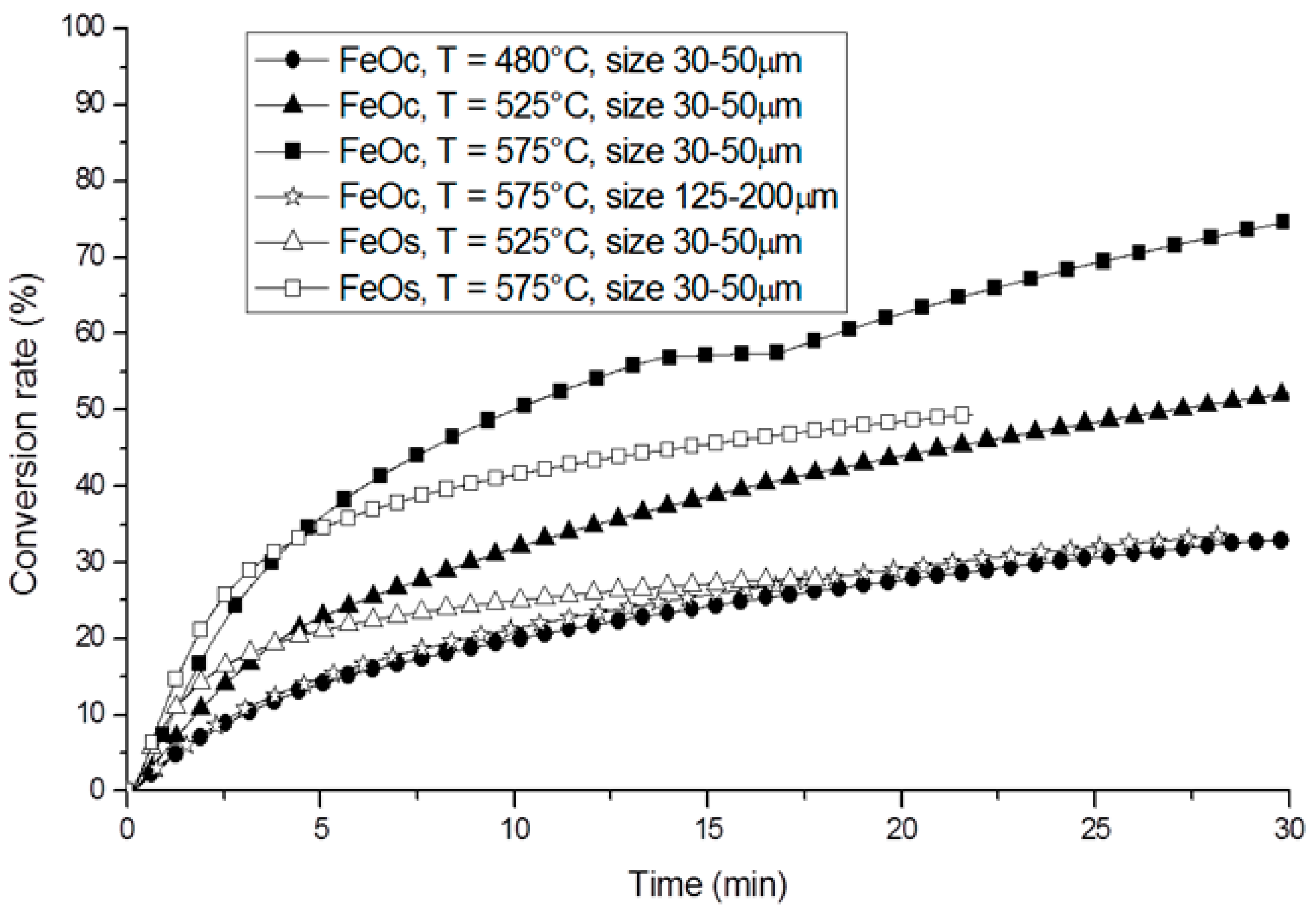
- Gokon, N.; Murayama, H.; Umeda, J.; Hatamachi, T.; Kodama, T. Monoclinic zirconia-supported Fe3O4 for the two-step water-splitting thermochemical cycle at high thermal reduction temperatures of 1400–1600 °C. Int. J. Hydrogen Energy 2009, 34, 1208–1217. [Google Scholar] [CrossRef]
- Gokon, N.; Murayama, H.; Nagasaki, A.; Kodama, T. Thermochemical two-step water splitting cycles by monoclinic ZrO2-supported NiFe2O4 and Fe3O4 powders and ceramic foam devices. Sol. Energy 2009, 83, 527–537. [Google Scholar] [CrossRef]
- Gokon, N.; Hasegawa, T.; Takahashi, S.; Kodama, T. Thermochemical two-step water-splitting for hydrogen production using Fe-YSZ particles and a ceramic foam device. Energy 2008, 33, 1407–1416. [Google Scholar] [CrossRef]
- Gokon, N.; Takahashi, S.; Yamamoto, H.; Kodama, T. Thermochemical two-step water-splitting reactor with internally circulating fluidized bed for thermal reduction of ferrite particles. Int. J. Hydrogen Energy 2008, 33, 2189–2199. [Google Scholar] [CrossRef]
- Kaneko, H.; Miura, T.; Fuse, A.; Ishihara, H.; Taku, S.; Fukuzumi, H.; Naganuma, Y.; Tamaura, Y. Rotary-Type Solar Reactor for Solar Hydrogen Production with Two-step Water Splitting Process. Energy Fuels 2007, 21, 2287–2293. [Google Scholar] [CrossRef]
- Han, S.B.; Kang, T.B.; Joo, O.S.; Jung, K.D. Water splitting for hydrogen production with ferrites. Sol. Energy 2007, 81, 623–628. [Google Scholar] [CrossRef]
- Roeb, M.; Sattler, C.; Klüser, R.; Monnerie, N.; de Oliveira, L.; Konstandopoulos, A.G.; Agrafiotis, C.; Zaspalis, V.T.; Nalbandian, L.; Steele, A.; et al. Solar Hydrogen Production by a Two-Step Cycle Based on Mixed Iron Oxides. J. Sol. Energy Eng. 2006, 128, 125–133. [Google Scholar] [CrossRef]
- Roeb, M.; Neises, M.; Säck, J.-P.; Rietbrock, P.; Monnerie, N.; Dersch, J.; Schmitz, M.; Sattler, C. Operational strategy of a two-step thermochemical process for solar hydrogen production. Int. J. Hydrogen Energy 2009, 34, 4537–4545. [Google Scholar] [CrossRef]
- Agrafiotis, C.; Roeb, M.; Konstandopoulos, A.G.; Nalbandian, L.; Zaspalis, V.T.; Sattler, C.; Stobbe, P.; Steele, A.M. Solar water splitting for hydrogen production with monolithic reactors. Sol. Energy 2005, 79, 409–421. [Google Scholar] [CrossRef]
- Agrafiotis, C.C.; Pagkoura, C.; Lorentzou, S.; Kostoglou, M.; Konstandopoulos, A.G. Hydrogen production in solar reactors. Catal. Today 2007, 127, 265–277. [Google Scholar] [CrossRef]
- Diver, R.B.; Miller, J.E.; Allendorf, M.D.; Siegel, N.P.; Hogan, R.E. Solar Thermochemical Water-Splitting Ferrite-Cycle Heat Engines. J. Sol. Energy Eng. 2008, 130, 041001. [Google Scholar] [CrossRef] [Green Version]
- Allendorf, M.D.; Diver, R.B.; Siegel, N.P.; Miller, J.E. Two-Step Water Splitting Using Mixed-Metal Ferrites: Thermodynamic Analysis and Characterization of Synthesized Materials. Energy Fuels 2008, 22, 4115–4124. [Google Scholar] [CrossRef]
- Fletcher, E.A. Solar thermal and solar quasi-electrolytic processing and separations: Zinc from zinc oxide as an example. Ind. Eng. Chem. Res. 1999, 38, 2275–2282. [Google Scholar] [CrossRef]
- Palumbo, R.D.; Fletcher, E.A. High-temperature solar electrothermal processing. Zinc from zinc-oxide at 1200–1675 K using a non-consumable anode. Energy 1988, 14, 319–332. [Google Scholar] [CrossRef]
- Souriau, D. Procédé et Dispositif pour L’utilisation D’énergie Thermique à Haute Température, en Particulier d’Origine Nucléaire. Device and Method for the Use of High-Temperature Heat Energy, in Particular of Nuclear Origin. Gaz de France Patent FR2135421, 22 December 1972. (In French). [Google Scholar]
- Vishnevetsky, I.; Epstein, M. Tin as a Possible Candidate for Solar Thermochemical Redox Process for Hydrogen Production. J. Sol. Energy Eng. 2009, 131, 021007. [Google Scholar] [CrossRef]
- Fletcher, E.A. Solar thermal processing: A review. J. Sol. Energy Eng. 2001, 123, 63–74. [Google Scholar] [CrossRef]
- Kodama, T. High-temperature solar chemistry for converting solar heat to chemical fuels. Prog. Energy Combust. Sci. 2003, 29, 567–597. [Google Scholar] [CrossRef]
- Segal, A.; Epstein, M. The optics of the solar tower reflector. Sol. Energy 2000, 69, 229–241. [Google Scholar] [CrossRef]
- Kaneko, H.; Miura, T.; Ishihara, H.; Taku, S.; Yokoyama, T.; Nakajima, H.; Tamaura, Y. Reactive ceramics of CeO2-MOx (M = Mn, Fe, Ni, Cu) for H2 generation by two-step water splitting using concentrated solar thermal energy. Energy 2007, 32, 656–663. [Google Scholar] [CrossRef]
- Abanades, S.; Legal, A.; Cordier, A.; Peraudeau, G.; Flamant, G.; Julbe, A. Investigation of reactive cerium-based oxides for H2 production by thermochemical 2-step water-splitting. J. Mater. Sci. 2010, 45, 4163–4173. [Google Scholar] [CrossRef]
- Miller, J.E.; Allendorf, M.D.; Diver, R.B.; Evans, L.R.; Siegel, N.P.; Stuecker, J.N. Metal oxide composites and structures for ultra-high temperature solar thermochemical cycles. J. Mater. Sci. 2008, 43, 4714–4728. [Google Scholar] [CrossRef]
- Chueh, W.; Haile, S.M. A thermochemical study of ceria: Exploiting an old material for new modes of energy conversion and CO2 mitigation. Phil. Trans. R. Soc. A 2010, 368, 3269–3294. [Google Scholar] [CrossRef]
- Chueh, W.C.; Haile, S.M. Ceria as a Thermochemical Reaction Medium for Selectively Generating Syngas or Methane from H2O and CO2. ChemSusChem 2009, 12, 735–739. [Google Scholar] [CrossRef]
- Kaneko, H.; Tamaura, Y. Reactivity and XAFS study on (1−x)CeO2–xNiO (x = 0.025–0.3) system in the two-step water-splitting reaction for solar H2 production. J. Phys. Chem. Solids 2009, 70, 1008–1014. [Google Scholar] [CrossRef]
- Kaneko, H.; Ishihara, H.; Taku, S.; Naganuma, Y.; Hasegawa, N.; Tamaura, Y. Cerium ion redox system in CeO2–xFe2O3 solid solution at high temperatures (1,273–1,673 K) in the two-step water-splitting reaction for solar H2 generation. J. Mater. Sci. 2008, 43, 3153–3161. [Google Scholar] [CrossRef]
- Meng, Q.-L.; Lee, C.-I.; Ishihara, T.; Kaneko, H.; Tamaura, Y. Reactivity of CeO2 based ceramics for solar hydrogen production via a two-step water-splitting cycle with concentrated energy. Int. J. Hydrogen Energy 2011, 36, 13435–13441. [Google Scholar] [CrossRef]
- Kaneko, H.; Taku, S.; Tamaura, Y. Reduction reactivity of CeO2-ZrO2 oxide under high O2 partial pressure in two-step water-splitting process. Sol. Energy 2011, 85, 2321–2330. [Google Scholar] [CrossRef]
- Lee, C.; Meng, Q.; Kaneko, H.; Tamaura, Y. Solar hydrogen productivity of ceria-scandia solid solution using two-step water-splitting cycle. J. Sol. Energy Eng. 2013, 135, 11002–11009. [Google Scholar] [CrossRef]
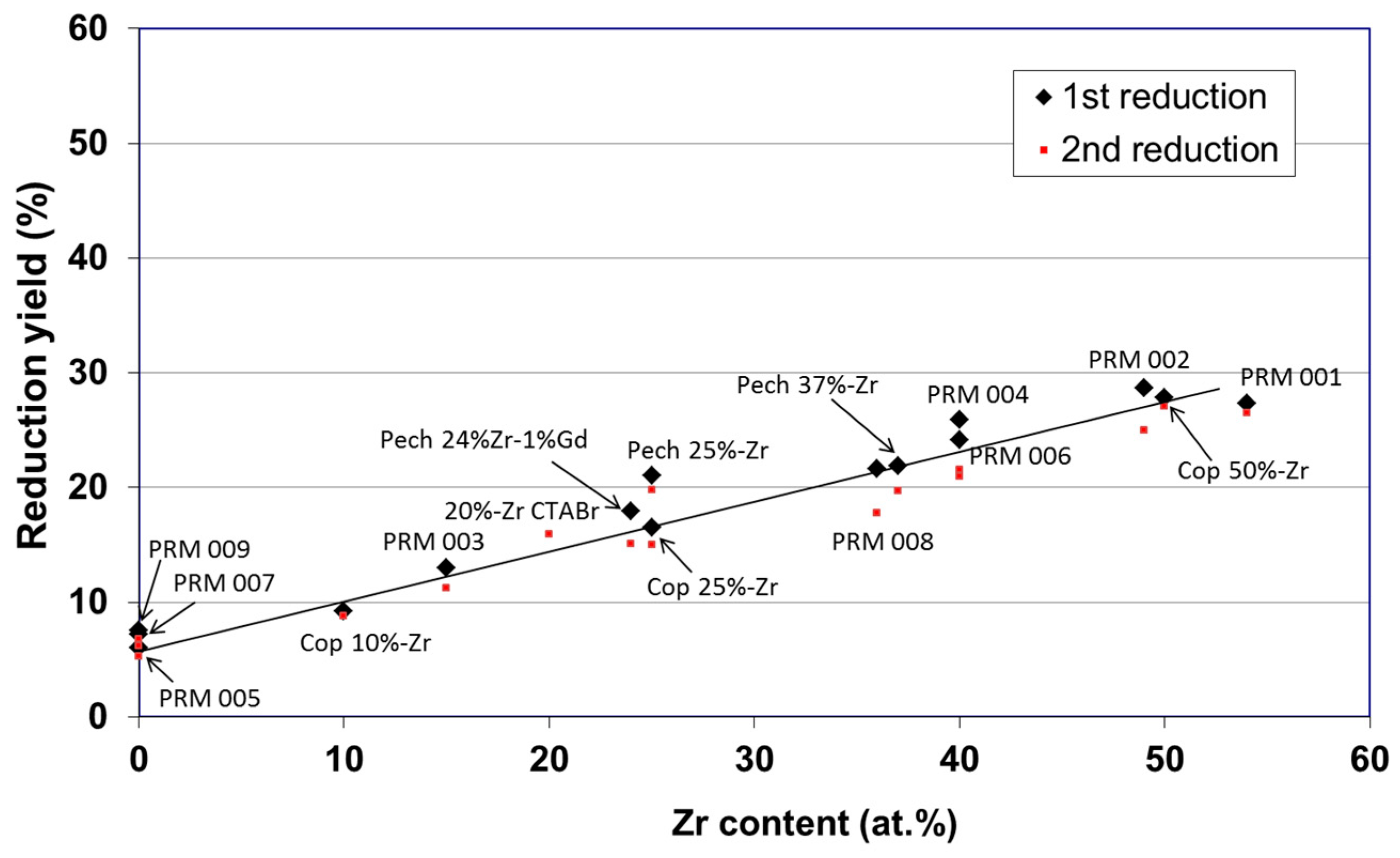
- Le Gal, A.; Abanades, S. Catalytic investigation of ceria-zirconia solid solutions for solar hydrogen production. Int. J. Hydrogen Energy 2011, 36, 4739–4748. [Google Scholar] [CrossRef]
- Le Gal, A.; Abanades, S.; Flamant, G. CO2 and H2O splitting for thermochemical production of solar fuels using non-stoichiometric ceria and ceria/zirconia solid solutions. Energy Fuels 2011, 25, 4836–4845. [Google Scholar] [CrossRef]
- Le Gal, A.; Abanades, S. Dopant incorporation in ceria for enhanced water-splitting activity during solar thermochemical hydrogen generation. J. Phys. Chem. C 2012, 116, 13516–13523. [Google Scholar] [CrossRef]
- Abanades, S.; Le Gal, A. CO2 splitting by thermo-chemical looping based on ZrxCe1-xO2 oxygen carriers for synthetic fuel generation. Fuel 2012, 102, 180–186. [Google Scholar] [CrossRef]
- Le Gal, A.; Abanades, S.; Bion, N.; Le Mercier, T.; Harlé, V. Reactivity of Doped Ceria-Based Mixed Oxides for Solar Thermochemical Hydrogen Generation via Two-Step Water-Splitting Cycles. Energy Fuels 2013, 27, 6068–6078. [Google Scholar] [CrossRef]
- Call, F.; Roeb, M.; Schmücker, M.; Sattler, C.; Pitz-Paal, R. Ceria Doped with Zirconium and Lanthanide Oxides to Enhance Solar Thermochemical Production of Fuels. J. Phys. Chem. C 2015, 119, 6929–6938. [Google Scholar] [CrossRef]
- Bulfin, B.; Call, F.; Vieten, J.; Roeb, M.; Sattler, C.; Shvets, I.V. Oxidation and Reduction Reaction Kinetics of Mixed Cerium Zirconium Oxides. J. Phys. Chem. C 2016, 120, 2027–2035. [Google Scholar] [CrossRef]
- Muhich, C.; Hoes, M.; Steinfeld, A. Mimicking tetravalent dopant behavior using paired charge compensating dopants to improve the redox performance of ceria for thermochemically splitting H2O and CO2. Acta Materialia 2018, 144, 728–737. [Google Scholar] [CrossRef]
- Davenport, T.C.; Kemei, M.; Ignatowich, M.J.; Haile, S.M. Interplay of material thermodynamics and surface reaction rate on the kinetics of thermochemical hydrogen production. Int. J. Hydrogen Energy 2017, 42, 16932–16945. [Google Scholar] [CrossRef] [Green Version]
- Petkovich, N.D.; Rudisill, G.; Ventrom, L.J.; Bomant, D.B.; Davidson, J.H.; Stein, A. Control of heterogeneity in nanostructured Ce1−xZrxO2 binary oxides for enhanced thermal stability and water splitting activity. J. Phys. Chem. C 2011, 115, 21022–21033. [Google Scholar] [CrossRef]
- Oliveira, F.A.C.; Barreiros, M.A.; Abanades, S.; Caetano, A.P.F.; Novais, R.M.; Pullar, R.C. Solar thermochemical CO2 splitting using cork-templated ceria ecoceramics. J. CO2 Util. 2018, 26, 552–563. [Google Scholar] [CrossRef]
- Furler, P.; Scheffe, J.R.; Steinfeld, A. Syngas production by simultaneous splitting of H2O and CO2 via ceria redox reactions in a high-temperature solar reactor. Energy Environ. Sci. 2012, 5, 6098–6103. [Google Scholar] [CrossRef]
- Gladen, A.C.; Davidson, J.H. The morphological stability and fuel production of commercial fibrous ceria particles for solar thermochemical redox cycling. Sol. Energy 2016, 139, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Furler, P.; Scheffe, J.; Gorbar, M.; Moes, L.; Vogt, U.; Steinfeld, A. Solar thermochemical CO2 splitting utilizing a reticulated porous ceria redox system. Energy Fuels 2012, 26, 7051–7059. [Google Scholar] [CrossRef]
- Marxer, D.; Furler, P.; Takacs, M.; Steinfeld, A. Solar thermochemical splitting of CO2 into separate streams of CO and O2 with high selectivity, stability, conversion, and efficiency. Energy Environ. Sci. 2017, 10, 1142–1149. [Google Scholar] [CrossRef]
- Cho, H.S.; Gokon, N.; Kodama, T.; Kang, Y.H.; Lee, H.J. Improved operation of solar reactor for two-step water-splitting H2 production by ceria-coated ceramic foam device. Int. J. Hydrogen Energy. 2015, 40, 114–124. [Google Scholar] [CrossRef]
- Scheffe, J.R.; Steinfeld, A. Oxygen exchange materials for solar thermochemical splitting of H2O and CO2: A review. Mater. Today 2014, 17, 341–348. [Google Scholar] [CrossRef]
- Carrillo, R.J.; Scheffe, J.R. Advances and trends in redox materials for solar thermochemical fuel production. Sol. Energy 2017, 156, 3–20. [Google Scholar] [CrossRef]
- Roeb, M.; Neises, M.; Monnerie, N.; Call, F.; Simon, H.; Sattler, C.; Schmücker, M.; Pitz-Paal, R. Materials-Related Aspects of Thermochemical Water and Carbon Dioxide Splitting: A Review. Materials 2012, 5, 2015–2054. [Google Scholar] [CrossRef] [Green Version]
- Scheffe, J.R.; Weibel, D.; Steinfeld, A. Lanthanum-strontium-manganese perovskites as redox materials for solar thermochemical splitting of H2O and CO2. Energy Fuels 2013, 27, 4250–4257. [Google Scholar] [CrossRef]
- McDaniel, A.H.; Miller, E.C.; Arifin, D.; Ambrosini, A.; Coker, E.N.; O’Hayre, R.; Chueh, W.C.; Tong, J. Sr- and Mn-doped LaAlO3-delta for solar thermochemical H2 and CO production. Energy Environ. Sci. 2013, 6, 2424–2428. [Google Scholar] [CrossRef]
- Demont, A.; Abanades, S.; Beche, E. Investigation of perovskite structures as oxygen-exchange redox materials for hydrogen production from thermochemical two-step water-splitting cycles. J. Phys. Chem. C 2014, 118, 12682–12692. [Google Scholar] [CrossRef]
- Yang, C.-K.; Yamazaki, Y.; Aydin, A.; Haile, S.M. Thermodynamic and kinetic assessments of strontium-doped lanthanum manganite perovskites for two-step thermochemical water splitting. J. Mater. Chem. A 2014, 2, 13612–13623. [Google Scholar] [CrossRef] [Green Version]
- Demont, A.; Abanades, S. High redox activity of Sr-substituted lanthanum manganite perovskites for two-step thermochemical dissociation of CO2. RSC Adv. 2014, 4, 54885–54891. [Google Scholar] [CrossRef]
- Demont, A.; Abanades, S. Solar thermochemical conversion of CO2 into fuel via two-step redox cycling of non-stoichiometric Mn-containing perovskite oxides. J. Mater. Chem. A 2015, 3, 3536–3546. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Insights into the Redox Performance of Non-stoichiometric Lanthanum Manganite Perovskites for Solar Thermochemical CO2 Splitting. ChemistrySelect 2016, 1, 4449–4457. [Google Scholar] [CrossRef]
- Nair, M.M.; Abanades, S. Experimental screening of perovskite oxides as efficient redox materials for solar thermochemical CO2 conversion. Sustain. Energy Fuels 2018, 2, 843–854. [Google Scholar] [CrossRef]
- Orfila, M.; Linares, M.; Molina, R.; Botas, J.A.; Sanz, R.; Marugan, J. Perovskite materials for hydrogen production by thermochemical water splitting. Int. J. Hydrogen Energy 2016, 41, 19329–19338. [Google Scholar] [CrossRef]
- Haeussler, A.; Abanades, S.; Jouannaux, J.; Julbe, A. Non-stoichiometric redox active perovskite materials for solar thermochemical fuel production: A review. Catalysts 2018, 8, 611. [Google Scholar] [CrossRef]



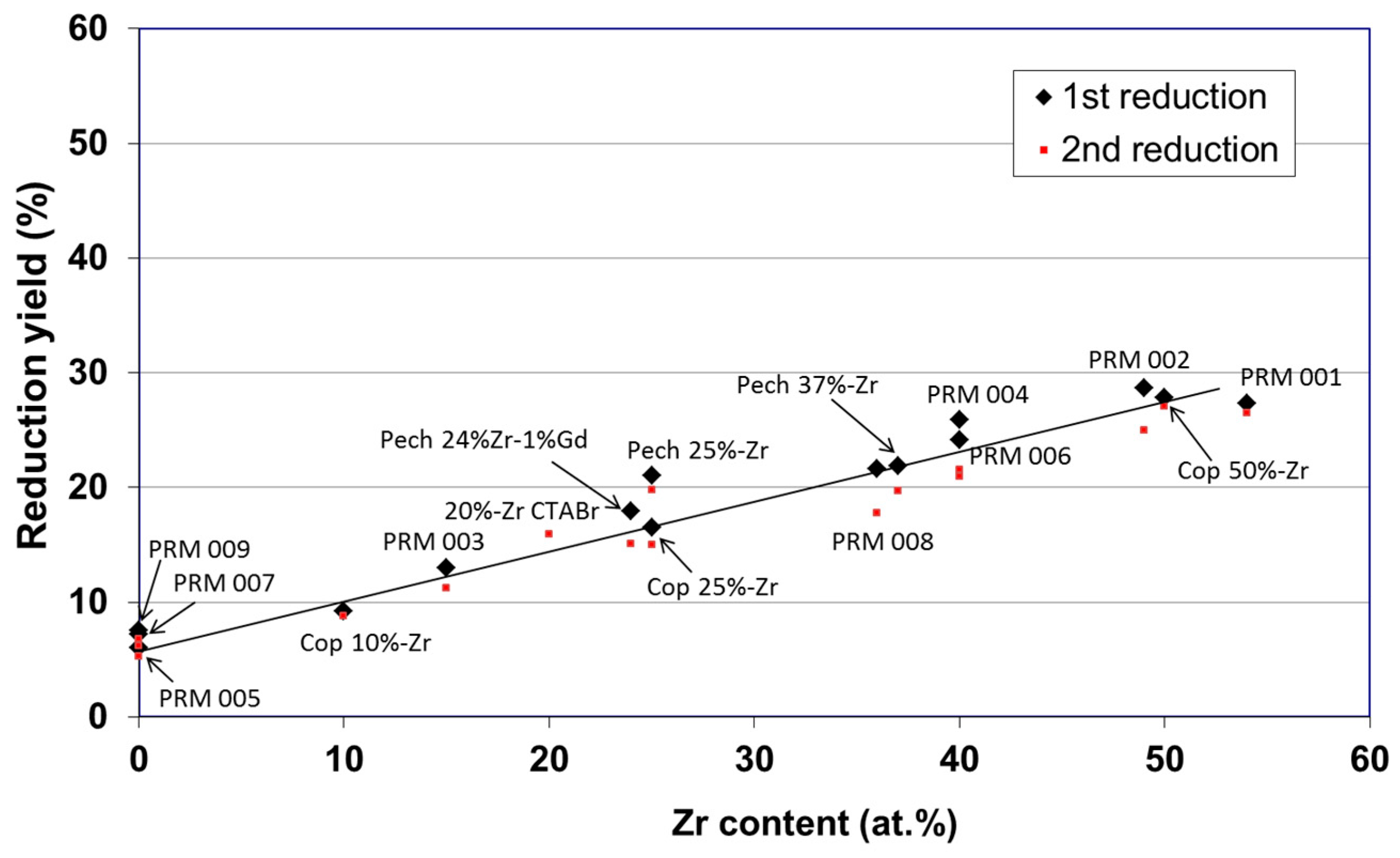
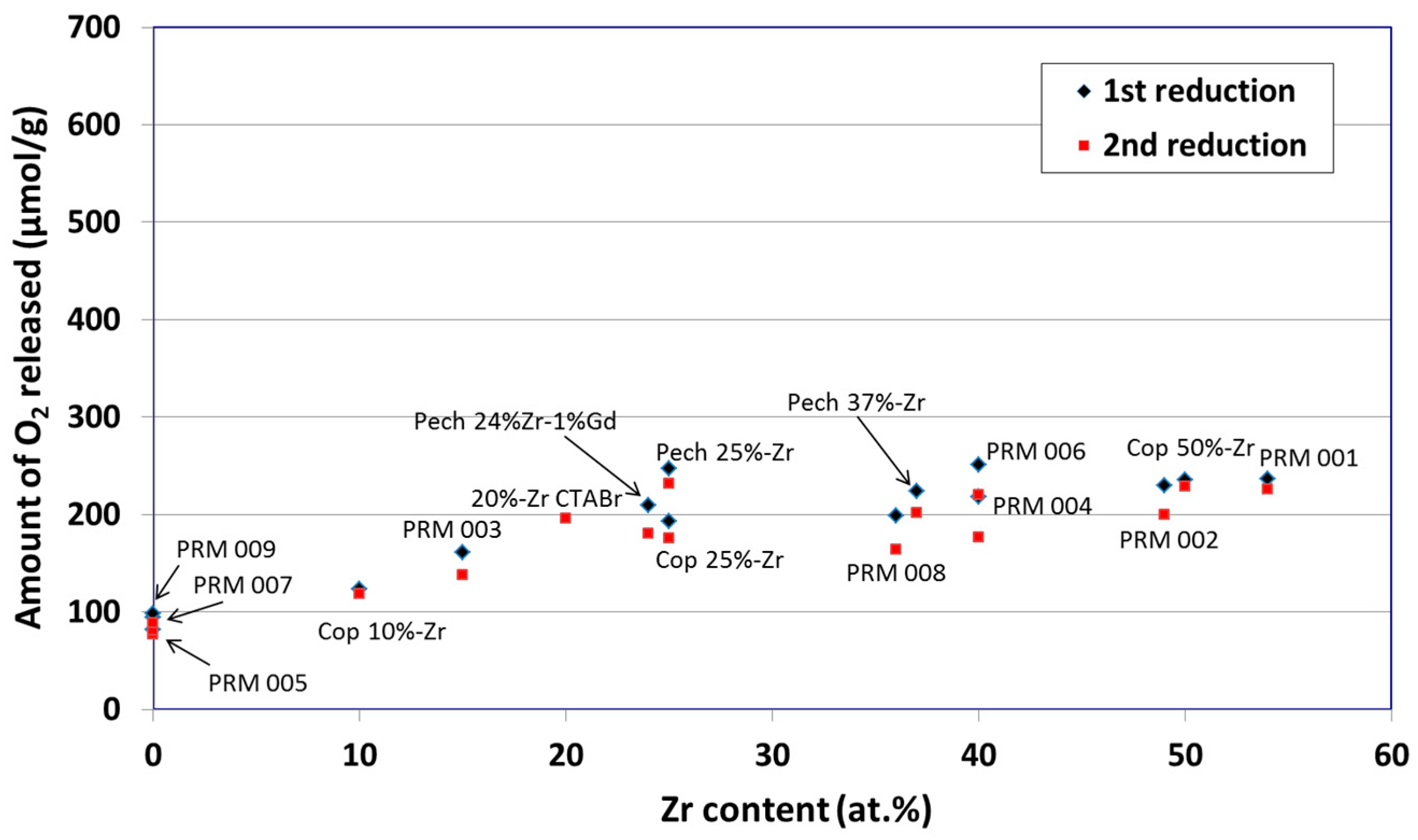
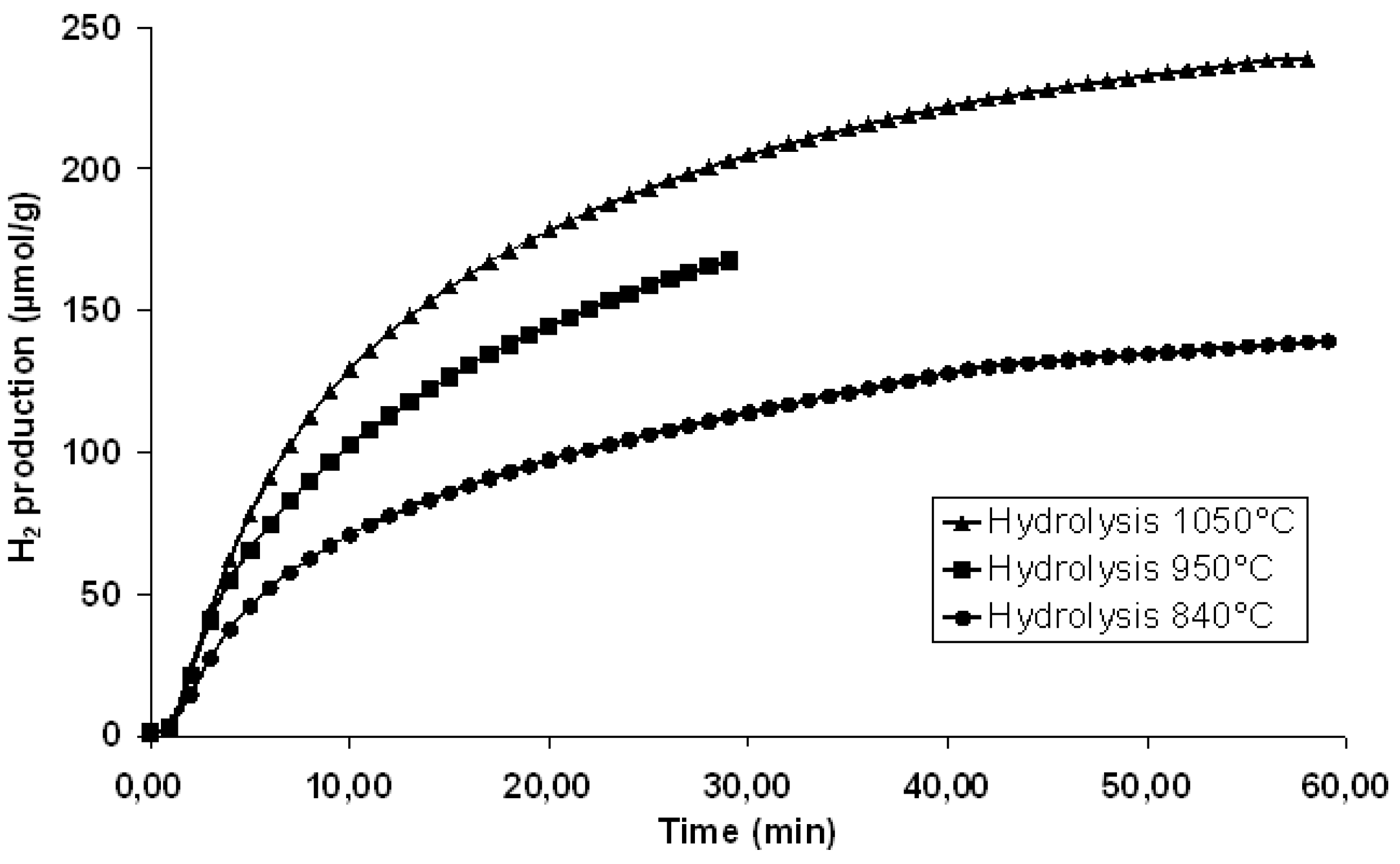
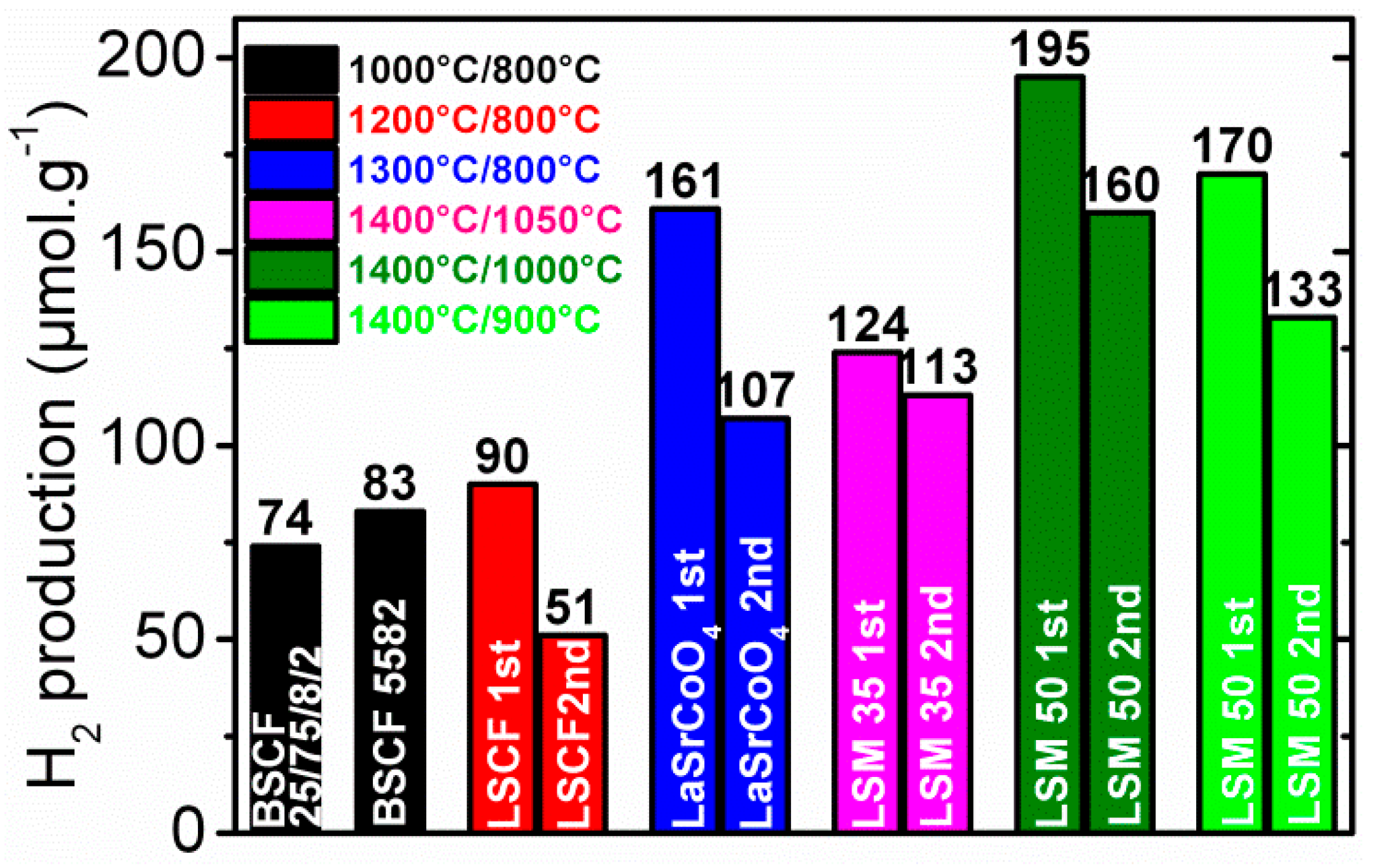
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abanades, S. Metal Oxides Applied to Thermochemical Water-Splitting for Hydrogen Production Using Concentrated Solar Energy. ChemEngineering 2019, 3, 63. https://doi.org/10.3390/chemengineering3030063
Abanades S. Metal Oxides Applied to Thermochemical Water-Splitting for Hydrogen Production Using Concentrated Solar Energy. ChemEngineering. 2019; 3(3):63. https://doi.org/10.3390/chemengineering3030063
Chicago/Turabian StyleAbanades, Stéphane. 2019. "Metal Oxides Applied to Thermochemical Water-Splitting for Hydrogen Production Using Concentrated Solar Energy" ChemEngineering 3, no. 3: 63. https://doi.org/10.3390/chemengineering3030063