Abstract
The effect of water on the solubility of syngas in hydrocarbons has typically been ignored when developing models for Fischer-Tropsch slurry bubble column reactors (SBCR), despite water being a major by-product. Therefore, a generalized correlation was developed to predict water solubility in hydrocarbons at high temperatures, and was used to calculate the effect of water saturation on H2 and CO solubility in hydrocarbons using the Span Wagner equation of state. The presence of water was shown to have a much more significant effect on H2 solubility in hydrocarbons, compared to CO.
1. Introduction
The effect of the presence of water on syngas solubility in Fischer-Tropsch (F-T) Slurry Bubble Column Reactors (SBCR) has received little attention, despite water being a major by-product. Although there have been numerous investigations to measure solubilities and mass transfer parameters of CO and H2 in F-T hydrocarbon fractions and slurries under a variety of operating conditions, the effect of water is typically neglected. Moreover, in most of the F-T SBCR models, liquid properties are typically determined for pure hydrocarbons or hydrocarbon mixtures, while disregarding the effect of water saturation on the physical properties of the hydrocarbons or on the gas solubilities. In fact, there only seems to be one publication concerned with the hydrodynamic effect of water on the F-T mass transfer [1,2]. Thus, the purpose of this work is to determine the solubility of water in various hydrocarbons using thermodynamic models, and to develop a generalized correlation for determining water solubility in hydrocarbons, which can then be used to estimate the effect of water saturation on the solubility of CO and H2 in hydrocarbons.
2. Background
2.1. Empirical Solubility Correlations
Since the behavior of water’s non-ideal behavior could not be explained using classical theoretical arguments, many researchers attempted to develop alternate empirical correlations to account for the temperature dependence of the solubility of hydrocarbons in aqueous systems. Numerous empirical correlations exist for water hydrocarbon mixtures at room temperature. Lindenberg [3] and McAuliff [4] observed an empirical relationship between the logarithms of aqueous solubility of water in hydrocarbons at room temperature and the hydrocarbon molar volume for homologous series. Klevens [5] suggested that the solubility of aromatic hydrocarbons is related to the molar volume, molecular length, and carbon number. Irmann [6] showed that the aqueous solubility can be determined from the sum of atomic and structural parameters for a variety of hydrocarbons. Other investigations have also correlated water solubility with a variety of readily measured or calculated physical properties such as the octanol/water partition coefficient, normal boiling point, chromatographic retention indices, and molecular surface area and volume. Several studies [7,8] demonstrated a mechanistic relationship between the solubility and the molecular surface area of the hydrocarbon solute, similarly, Schatzberg [9] correlated the solubility of water in hydrocarbons ranging from paraffins to n-hexadecane at 298 and 313 K with solute surface tension.
Development of correlations for a wide range of temperatures has been one of the more difficult challenges. Theoretical studies suggest that the logarithm of solubility should be linear in inverse temperature for moderately non-ideal solutions i.e., if the solution operates under the assumption of being a random mixture of spherical particles [10]. Yet, this hypothesis seems to be more applicable to describe the hydrocarbon liquid phase with very low concentrations of the more non ideal liquid molecules (i.e., water), as shown by Hubbard [11]. Correlations for extended temperature ranges have been introduced by Tsonopoulos and Wilson [12] and Heidmann [13], which incorporated more complex dependencies on temperature into their Antoine-type correlations for the solubility of water in C6 to C8 hydrocarbons. Brady et al [14] developed graphical correlations for water solubility in heavy hydrocarbons as a function of temperature and the double bond index using a solution of groups theoretical approach.
Generally, the effect of pressure on binary water hydrocarbon mixtures could be readily estimated in the three phase region (VLLE) as the sum of the pure component vapor pressures at various temperatures approaching the three phase critical point. However, the combined effect of temperature and pressure on the VLE and VLLE of water hydrocarbon mixtures are better correlated in terms of the hydrocarbon or the water volatility, or both. These relationships accounting for both temperature and pressure effects are restricted to temperatures which are far removed from the phase change boundary where the partial molal volume of the solute undergoes considerable change. The high solubility of water in hydrocarbons in general precludes representation of the hydrocarbon rich phase by the means of Henry constant.
2.2. Activity Coefficients for Water Hydrocarbon Mixtures
Saturated aqueous solutions of hydrocarbons at low to moderate temperatures are usually very dilute solutions, therefore hydrocarbon solubilities in water and vice versa could be directly related to the predictions of activity coefficients at infinite dilution. Deal et al. [15,16] presented an extensive correlation for activity coefficients at infinite dilution at 298 K with molecular structure and the interaction of various structural groups in the solution. The resulting correlations are simple and easily applicable to homologous series in water. Tsonopoulos and Prausnitz [17] extended the group contribution method approach to distinct classes of aromatic hydrocarbons and their derivatives. Other studies [18,19] correlated infinite dilution activity coefficients with specific physical properties which reflect the size, shape, and charge distribution of the hydrocarbon solute. Properties examined include carbon number, molecular volume, surface area, molecular connectivity, acentric factor, normal boiling point, melting point, total electronic energy, and dipole moment. These correlations, although specific to single hydrocarbon families and solvents, predicted activity coefficients at infinite dilution with very close agreement to the UNIQUAC and UNIFAC predictions.
Activity coefficient models for more miscible fluids are expected to reflect the thermodynamic effects of increased solubility; therefore, infinite dilution activity coefficients are not very applicable to mixtures of water with hydrocarbon oils or phenols, as they usually exhibit higher solubilities. Moreover, the effect of the solute composition must be incorporated directly into the activity coefficient expression. Black et al. [20] discussed the solubility of water in hydrocarbons using a regular solution model as well as the Flory-Huggins mixture equation of state to account for the discrepancies in the molal volume. Hildebrand [21] combined the two approaches into one model which was widely used in investigations for the modeling of the solubility of water in hydrocarbons at temperatures ranging from 273 to 348 K. More recent activity coefficient models such as the NRTL, UNIQUAC, and UNIFAC models are extremely popular for prediction of mutual solubility data for more miscible aqueous hydrocarbon systems and have been applied in the design of many industrial processes in the oil and gas industry.
Extending activity coefficients to describe VLE and VLLE systems requires the use of an equation of state to describe the vapor phase; Li et al. [22] represented the fugacity coefficient of water in the vapor phase above the three phase crucial pressure using the virial equation of state in reduced form. The activity coefficient of light hydrocarbons in water was graphically correlated as a function of reduced temperature and the solubility parameter, whereas reference state fugacities were graphically correlated as functions of reduced temperatures and pressures. Alternatively, West et al. [23] conducted three phase calculations for light paraffin and water mixtures using the Suave Redlich Kwong equation of state for the vapor phase fugacities and proposed a modified Hilderbrand solubility parameter for the liquid phases.
This approach of mixing activity coefficient correlations with equations of state is mathematically inconvenient. The mathematical flexibility of activity coefficient models enables modeling of systems with very high liquid non-idealities, which is usually counterbalanced by the complex nature of most equations of state. This approach works well, however, for low temperature and moderate pressure systems, beyond which the accuracy and applicability has to closely questioned. Moreover, combining different models for the vapor and liquid phases prevents the correct prediction of the mixtures critical and supercritical properties.
2.3. Equation of State Studies of Water-Hydrocarbon Mixtures
Hiedmann [24] was the first to demonstrate that the phase equilibria of water hydrocarbon mixture could be expressed using a single equation of state by using the Wilson modification of the Redlich-Kwong (RK) equation to predict the VLLE of n-paraffins and water. Using conventional mixing rules, the modified RK equation successfully predicted the water content of both the liquid hydrocarbon and the vapor phases; however, predictions for aqueous hydrocarbon solubilities were only qualitative. This sparked increasing interest into the development of Van der Waal type equations of state and mixing rules for the modeling of the phase equilibriums of hydrocarbon + water systems. However, failure to successfully quantitatively replicate the water hydrocarbon behavior led to the superposition of specific solution models on simple equations of states to represent to the solubility of hydrocarbons in water.
Moreover, a lot of effort has aimed at the development of conventional quadratic mixing rules for the simultaneous representation of both the hydrocarbon rich and aqueous phases. The introduction of temperature dependent, phase specific interaction constants to develop empirical extensions of quadratic mixing rules, has been deemed undesirable from a theoretical standpoint. The unique phase behavior of water indicates a more complex mechanism and dependency of water’s mixing behavior in the presence of hydrocarbons, as such valuable information is lost in lumping all of these effects into binary interaction parameters. Although these empirical relations work and are easily applicable in phase equilibrium calculations, they fail to converge at two and three phase critical points. Attempts at overcoming the difficulties and limitations of quadratic mixing rules for the characteristic energy of van der Waal type equations of state aimed at the development of mixing rules which are cubic in mole fraction [25]. Other alternate approaches to the development of mixing rules for water hydrocarbon mixtures, include deriving the from complex solution chemistry principles. According these principles, the formations of new associated species such as dimers, is postulated to account for the effects of hydrogen bonding and donor acceptor behavior. The concentrations of all coexisting species are then determined from pure component fugacities using empirically determined chemical equilibrium constants. These methods have been widely used to model low and moderate pressure phase equilibriums of strongly non ideal vapors and liquid mixtures (eg. Acetic acids with alcohols or hydrocarbons), in conjunction with the use of activity coefficients [26]. This approach has also been superimposed on cubic equations of state to represent the phase equilibriums at elevated pressure for polar substances and their mixtures and to model the self-association of water in the presence of hydrocarbon solute, by treating water as a mixture of five polymeric species, the VLE and VLLE of polar and non-polar mixtures of the C1–C8 alkanes with water were successfully modeled using the Schmidt-Wenzel equation.
However, there have been several drawbacks. Primarily, the proposed stoichiometry by which the association of pure water is represented is usually arbitrarily established. Moreover, the representation of the hydrogen bonding behavior of water by higher polymers cannot be readily justified, as the characteristic properties of these polymers cannot be readily determined for extended applications usually associated with equations of state. Other drawbacks include the computational complications associated with initially calculating the chemical equilibria, this complexity increases rapidly with the number of components and the severity of the conditions. At severe conditions, even the simplest of models would require complicated and time consuming computational solutions [27].
Another approach for modeling mixtures containing polar constituents involves the transposition of alternate activity coefficient models for the excess Gibbs free energy of the solution. Huron and Vidal [28] demonstrated that for the Soave equation of state, a simple relation exists between the ratio of energy and co-volume parameters (a,b) in addition to the limit of the Gibbs free energy at infinite pressure. They also suggested the modification of the NRTL model to account for the excess Gibbs free energy of the Soave equation. Although these models successfully represent the phase equilibria of simple systems containing polar components up to the critical point, they have not been tested on water hydrocarbon mixtures and are expected to deviate at the limit of low density due to their empirical nature.
A milestone was achieved by Mollerup [29,30] and Whiting and Prausnitz [31], when they derived density dependent mixing rules for van der Waals type equations of state using local composition averages of the residual internal energy of mixing. Using the analogy of the attractive energies of pure components, the authors expressed the local compositions in terms of bulk composition, temperature, and density. This model was then used to derive the mixing energy, the Helmholtz energy, and the solution pressure, which in turn were used to yield an equation of state for non-random mixtures. This mixing rule for the interaction energy parameter of the mixture represents an interpolation between the successful local composition activity coefficient models at high density and the theoretical quadratic mixing rule for the second virial coefficient in the limit of low density.
The density dependent mixing rules derived from the local composition methods improved on the correlation for the phase equilibria of polar and non-polar mixtures relative to the traditional random-mixing rules. However, this model overestimates the entropic contributions to mixing which making it inapplicable to asymmetric, non-polar mixtures. Dimitrelis and Prausnitz [32] tried to address this overestimation by reformulating the binary interaction parameters as deviations from the ideal values using quasi-chemical theory. Similarly, Mathias and Compeman [29] divided the residual internal energy of mixing into ideal and excess contributions to try and make the best use of the Peng-Robinson EOS’s ability to model asymmetric non-polar mixtures, ultimately developing a truncated model with a low computational load. This simplified model reasonably predicts the VLE of a variety of mixtures as well as the mutual solubilities of water hydrocarbon mixtures.
Local composition mixing rules provide improved modeling and accuracy of mixtures involving polar components, there still remains some discrepancies regarding the formal definition and theoretical basis of the local composition concept based on the results of molecular dynamics and Monte Carlo simulations.
3. Calculating Water Solubility in Hydrocarbons
By taking the solubility of water in hydrocarbons to be linear in fugacity, Henry’s law was used to determine the concentration of the water as shown in Equation (1).
where C1 is the solubility of water, is Henry’s constant, and is the fugacity. Subscripts 1 and 2 refer to water and hydrocarbon respectively. The fugacity was determined from the departure of the molar Gibbs free energy from the ideal gas behavior as shown in Equation (2).
The Gibbs free energy was determined using Equation (3), where the enthalpy and entropy were obtained from the steam tables [33], whereas the ideal gas Gibbs free energy were determined using the same equation, with the enthalpy, and entropy derived from the cubic heat equation as shown in Equations (4) to (6).
The inverse Henry’s law constant was determined using the expression derived by Sanchez and Rodgers [34] for the solubility of gases in liquid polymers. It is based on equating the chemical potential of the absorbed gas to the ideal case, assuming infinite molecular weight of the polymer. A modified version that accounts for the molecular weight of the solvent was used as shown in Equation (7).
where is defined as the bare water-hydrocarbon interaction parameter defined in Equation (8).
is a dimensionless parameter derived by Haschets [35] to account for deviations from the geometric mean for the solubility of water in n-alkanes between C6 to C16, and is determined using Equation (9).
Additionally, the lattice volume equation of state [34] was also used to link the state parameters as shown in Equation (10).
where , and are reduced parameters defined in Equation (11), and , and are reference parameters as shown in Table 1.

Table 1.
Reference state parameters used in calculation [36].
The solubility of water in Hexane, Octane, and Decane was subsequently calculated, the solubility of water in the hydrocarbons increased with increasing temperature and carbon number, with water having the highest solubility in Decane. The solubility of water in hydrocarbons was determined to range anywhere between 0.1 to 0.3 mole fraction, depending on the hydrocarbon. Subsequently, a correlation was fit to determine the equilibrium solubility of water in hydrocarbons over the temperature range of 350 to 650 K, as shown in Equation (12), and as highlighted in Figure 1. It is important to note that there was no significant effect of pressure on the equilibrium solubility of water in hydrocarbons.
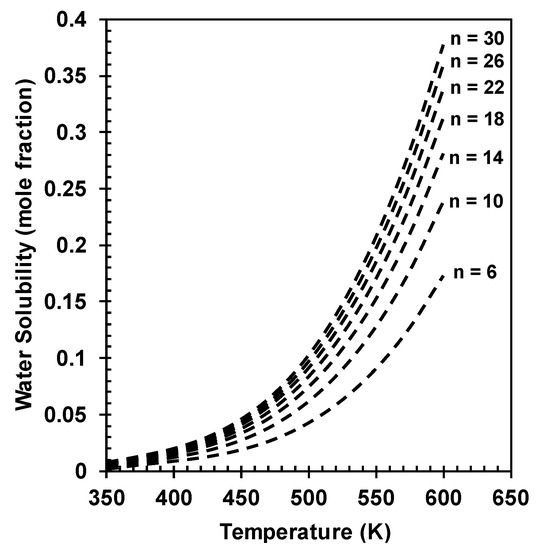
Figure 1.
Solubility of water in C6 to C30 hydrocarbons at 5 MPa.
When comparing the predicted solubilities with experimental data for water solubility in various hydrocarbons ranging from C6 to C12, as summarized by Maączyński et al. [37], the correlation predicted solubilities with errors ranging from 15% to 65%, with the highest deviations observed at higher temperatures. However, there were no qualitative deviations from the experimental data, and therefore the correlation was further used to analyze the effect of water saturation on the solubility of H2 and CO in hydrocarbons.
4. H2 and CO Solubilities in Pure and Water-Saturated Hydrocarbons
Once the solubility of water was determined, simple mixing rules were used to determine the effect of water saturation on H2 and CO solubilities in hydrocarbons. Solubilities were determined Equation (13) [21], which accounts for the non-ideal interactions between the components:
where φ is the Hildebrand solubility parameter, which is derived from the cohesive energy density of the solvent using Equation (14). This parameter provides an estimate of the degree of interaction between different components.
Values for the heat of vaporization were extracted from the NIST Chemistry Web book, whereas values for the molar volumes for each of the components were derived using the reduced Helmholtz energy. Subsequently, the Span and Wagner Equation of State [38], which has the form of a fundamental equation explicit in the Helmholtz Free Energy (HFE) where the function for the residual part of the HFE was fitted to selected data of the fugacity coefficient, as summarized in Equations (15) to (25), was used in an iterative manner to determine the molar volume of each of the components over a wide range of temperatures and pressures. Values of the Helmholtz energy constants for the determining the solubility of H2 was obtained from Leachman [39], whereas those for CO were obtained from Kerley [40].
Results for H2 solubility in water saturated and unsaturated hydrocarbons is shown in Figure 2, and as can be seen in this figure, the solubility of H2 is higher in water saturated hydrocarbons, and the effect of water saturation significantly increases with increasing temperatures. Moreover, results for CO solubility in water saturated and unsaturated hydrocarbons is shown in Figure 3, and as can be seen the presence of water does not have a significant effect on CO solubility. The effect of water solubility on H2 solubility compared to CO could be attributed to hydrogen bonding effects.
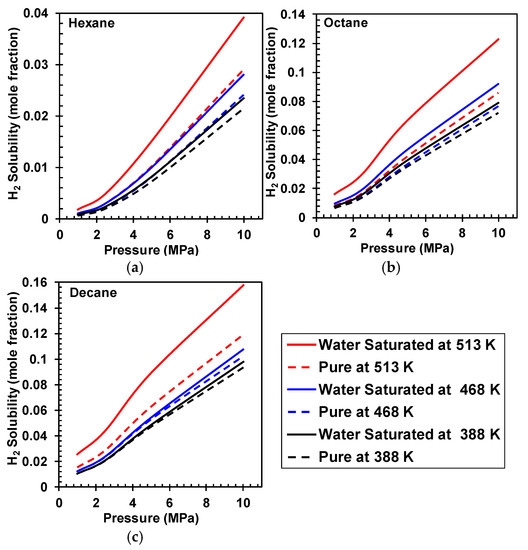
Figure 2.
Comparing H2 solubility in pure and water saturated (a) Hexane, (b) Octane, and (c) Decane at various temperatures.
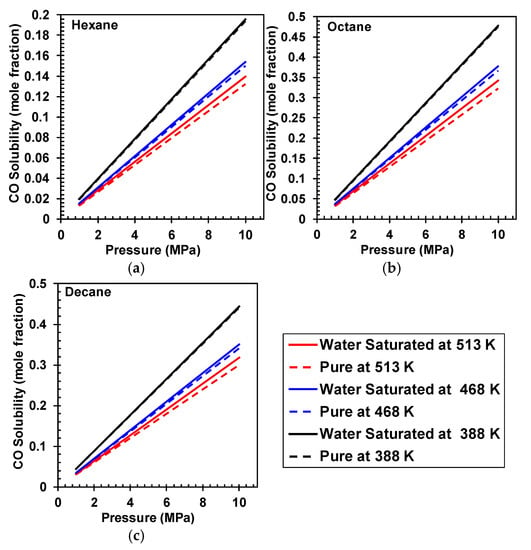
Figure 3.
Comparing CO solubility in pure and water saturated (a) Hexane, (b) Octane, and (c) Decane at various temperatures.
5. Concluding Remarks
In this work, the solubility of water in hydrocarbons was calculated and a correlation was developed to predict the solubility of hydrocarbons over a wide range of temperatures and pressures. Subsequently, the effect of water saturation on H2 and CO solubility in hydrocarbons was investigated. The solubility of water was found to increase with temperature and carbon number, whereas the presence of water in hydrocarbons was found to increase the solubility of H2 and CO in hydrocarbons. However, this effect was significantly higher with H2, which could be attributed to synergistic effect induced by the presence of water.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Karandikar, B.M.; Morsi, B.I.; Shah, Y.T.; Carr, N.L. Effect of water on the solubilities and mass transfer coefficients of gases in a heavy fraction of fischer-tropsch products. Can. J. Chem. Eng. 1987, 65, 973–981. [Google Scholar] [CrossRef]
- Karandikar, B.; Morsi, B.; Shah, Y.; Carr, N. Effect of water on the solubility and mass transfer coefficients of CO and H2 in a Fischer-Tropsch liquid. Chem. Eng. 1986, 33, 157–168. [Google Scholar] [CrossRef]
- Lindenberg, A. Sur une relation simple entre le volume moléculaire et la solubilité dans leau des hydrocarbures et dérivés halogénés. C. R. Hebd. Seances De L Acad. Des Sci. 1956, 243, 2057–2060. [Google Scholar]
- McAuliffe, C. Solubility in Water of Paraffin, Cycloparaffin, Olefin, Acetylene, Cycloolefin, and Aromatic Hydrocarbons1. J. Phys. Chem. 1966, 70, 1267–1275. [Google Scholar] [CrossRef]
- Klevens, H. The The Effect of Added Hydrocarbons upon Critical Concentrations of Soap and Detergent Solutions. J. Phys. Chem. 1950, 54, 1012–1016. [Google Scholar] [CrossRef]
- Irmann, F. Eine einfache Korrelation zwischen Wasserlöslichkeit und Struktur von Kohlenwasserstoffen und Halogenkohlenwasserstoffen. Chemie Ingenieur Technik 1965, 37, 789–798. [Google Scholar] [CrossRef]
- Hermann, R.B. Theory of Hydrophobic Bonding. 2. Correlation of Hydrocarbon Solubility in Water with Solvent Cavity Surface-Area. J. Phys. Chem. 1972, 76, 2754–2759. [Google Scholar] [CrossRef]
- Amidon, G.; Yalkowsky, S.; Anik, S.; Valvani, S. Solubility of nonelectrolytes in polar solvents. V. Estimation of the solubility of aliphatic monofunctional compounds in water using a molecular surface area approach. J. Phys. Chem. 1975, 79, 2239–2246. [Google Scholar] [CrossRef]
- Schatzberg, P. Solubilities of water in several normal alkanes from C7 to C161. J. Phys. Chem. 1963, 67, 776–779. [Google Scholar] [CrossRef]
- Ambrose, D.; Patel, N.C. The correlation and estimation of vapour pressures IV. Extrapolation of vapour pressures and estimation of critical pressures by the principle of corresponding states using two reference fluids with non-spherical molecules. J. Chem. Thermodyn. 1984, 16, 459–468. [Google Scholar] [CrossRef]
- Hubbard, J. Electron Correlations in Narrow Energy Bands. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1963, 276, 238–257. [Google Scholar]
- Tsonopoulos, C.; Wilson, G.M. High-temperature mutual solubilities of hydrocarbons and water. Part I: Benzene, cyclohexane and n-hexane. AIChE J. 1983, 29, 990–999. [Google Scholar] [CrossRef]
- Heidman, J.L.; Tsonopoulos, C.; Brady, C.J.; Wilson, G.M. High-temperature mutual solubilities of hydrocarbons and water. Part II: Ethylbenzene, ethylcyclohexane, and n-octane. AIChE J. 1985, 31, 376–384. [Google Scholar] [CrossRef]
- Brady, C.J.; Cunningham, J.R.; Wilson, G.M. RR-62: Water-Hydrocarbon Liquid-Liquid-Vapor Equilibrium Measurements in 530 Degrees F; Wiltec Research Co.: Provo, UT, USA, 1982. [Google Scholar]
- Deal, C.; Derr, E. Selectivity and solvency in aromatics recovery. Ind. Eng. Chem. Process Des. Dev. 1964, 3, 394–399. [Google Scholar] [CrossRef]
- Deal, C.H.; Derr, E.L. Group contributions in mixtures. Ind. Eng. Chem. 1968, 60, 28–38. [Google Scholar] [CrossRef]
- Tsonopoulos, C.; Prausnitz, J.M. Activity coefficients of aromatic solutes in dilute aqueous solutions. Ind. Eng. Chem. Fundam. 1971, 10, 593–600. [Google Scholar] [CrossRef]
- Spahl, R.; Luft, G. Einfluß von Molmasse und Molekülverzweigungen auf das Entmischungsverhalten von Ethylen/Polyethylen-Systemen unter Hochdruck. Berichte der Bunsengesellschaft für physikalische Chemie 1982, 86, 621–625. [Google Scholar] [CrossRef]
- Hu, Y.; Azevedo, E.; Lüdecke, D.; Prausnitz, J. Thermodynamics of associated solutions: Henry’s constants for nonpolar solutes in water. Fluid Phase Equilib. 1984, 17, 303–321. [Google Scholar] [CrossRef]
- Black, C.; Joris, G.G.; Taylor, A.H.S. The Solubility of Water in Hydrocarbons. J. Chem. Phys. 1948, 16, 537–543. [Google Scholar] [CrossRef]
- Hildebrand, J.H. Solubility of Water in Hydrocarbons. J. Chem. Phys. 1949, 17, 1346–1347. [Google Scholar] [CrossRef]
- Li, C.C.; McKetta, J.J. Vapor-Liquid Equilibrium in the Propylene-Water System. J. Chem. Eng. Data 1963, 8, 271–275. [Google Scholar] [CrossRef]
- West, E.W.; Erbar, J.H. Three Phase Equilibrium Calculations. In Proceedings of the Fifty-Second Annual GPA Convention, 1973. [Google Scholar]
- Heidemann, R.A. Three-phase equilibria using equations of state. AIChE J. 1974, 20, 847–855. [Google Scholar] [CrossRef]
- Reid, R.C.; Prausnitz, J.M.; Sherwood, T.K. The Properties of Gases and Liquids; McGraw-Hill Education: New York, NY, USA, 1977. [Google Scholar]
- Ambrose, D. Thermodynamic properties of organic oxygen compounds XLIX. The vapour pressure of solid acetic acid. J. Chem. Thermodyn. 1979, 11, 183–185. [Google Scholar] [CrossRef]
- Leet, W.A. Fluid-phase equilibria in mixtures of water and heavy hydrocarbons at elevated temperatures and pressures. Ph.D. Thesis, Purdue University, West Lafayette, IN, USA, 1986. [Google Scholar]
- Huron, M.J.; Vidal, J. New Mixing Rules in Simple Equations of State for Representing Vapor-Liquid-Equilibria of Strongly Non-Ideal Mixtures. Fluid Phase Equilib. 1979, 3, 255–271. [Google Scholar] [CrossRef]
- Mathias, P.M.; Copeman, T.W. Extension of the Peng-Robinson equation of state to complex mixtures: Evaluation of the various forms of the local composition concept. Fluid Phase Equilib. 1983, 13, 91–108. [Google Scholar] [CrossRef]
- Mollerup, J. Correlation of Thermodynamic Properties of Mixtures Using A Random-Mixture Reference State. Fluid Phase Equilib. 1983, 15, 189–207. [Google Scholar] [CrossRef]
- Whiting, W.B.; Prausnitz, J.M. Equations of state for strongly nonideal fluid mixtures: Application of local compositions toward density-dependent mixing rules. Fluid Phase Equilib. 1982, 9, 119–147. [Google Scholar] [CrossRef]
- Dimitrelis, D.; Prausnitz, J.M. Comparison of 2 Hard-Sphere Reference Systems for Perturbation Theories for Mixtures. Fluid Phase Equilib. 1986, 31, 1–21. [Google Scholar] [CrossRef]
- Wagner, W.; Pruß, A. The IAPWS Formulation 1995 for the Thermodynamic Properties of Ordinary Water Substance for General and Scientific Use. J. Phys. Chem. Ref. Data 2002, 31, 387–535. [Google Scholar] [CrossRef]
- Sanchez, I.; Rodgers, P. Solubility of gases in polymers. Pure Appl. Chem. 1990, 62, 2107–2114. [Google Scholar] [CrossRef]
- Haschets, C.W.C.; Shine, A.D.A.; Secor, R.M.R. Prediction of water solubilities in hydrocarbons and polyethylene at elevated temperatures and pressures. Ind. Eng. Chem. Res. 1994, 33, 1040–1046. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology (NIST). National Institute of Standards and Technology Property Database; NIST: Gaithersburg, MD, USA, 2015. [Google Scholar]
- Maączyński, A.; Góral, M.; Wiśniewska-Gocłowska, B.; Skrzecz, A.; Shaw, D. Mutual solubilities of water and alkanes. Monatshefte für Chemie/Chem. Mon. 2003, 134, 633–653. [Google Scholar] [CrossRef]
- Span, R.; Wagner, W. A new equation of state for carbon dioxide covering the fluid region from the triple-point temperature to 1100 K at pressures up to 800 MPa. J. Phys. Chem. Ref. Data 1996, 25, 1509. [Google Scholar] [CrossRef]
- Leachman, J.W.; Jacobsen, R.T.; Penoncello, S.G.; Lemmon, E.W. Fundamental Equations of State for Parahydrogen, Normal Hydrogen, and Orthohydrogen. J. Phys. Chem. Ref. Data 2009, 38, 721. [Google Scholar] [CrossRef]
- Kerley, G.I.; Abdallah, J., Jr. Theoretical equations of state for molecular fluids: Nitrogen, oxygen, and carbon monoxide. J. Chem. Phys. 1980, 73, 5337–5350. [Google Scholar] [CrossRef]
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).