
Anti-Tumor Effects of Ginsenoside 20(S)-Protopanaxadiol and 1,25-Dihydroxyvitamin D3 Combination in Castration Resistant Prostate Cancer

 ,
,  ,
, 
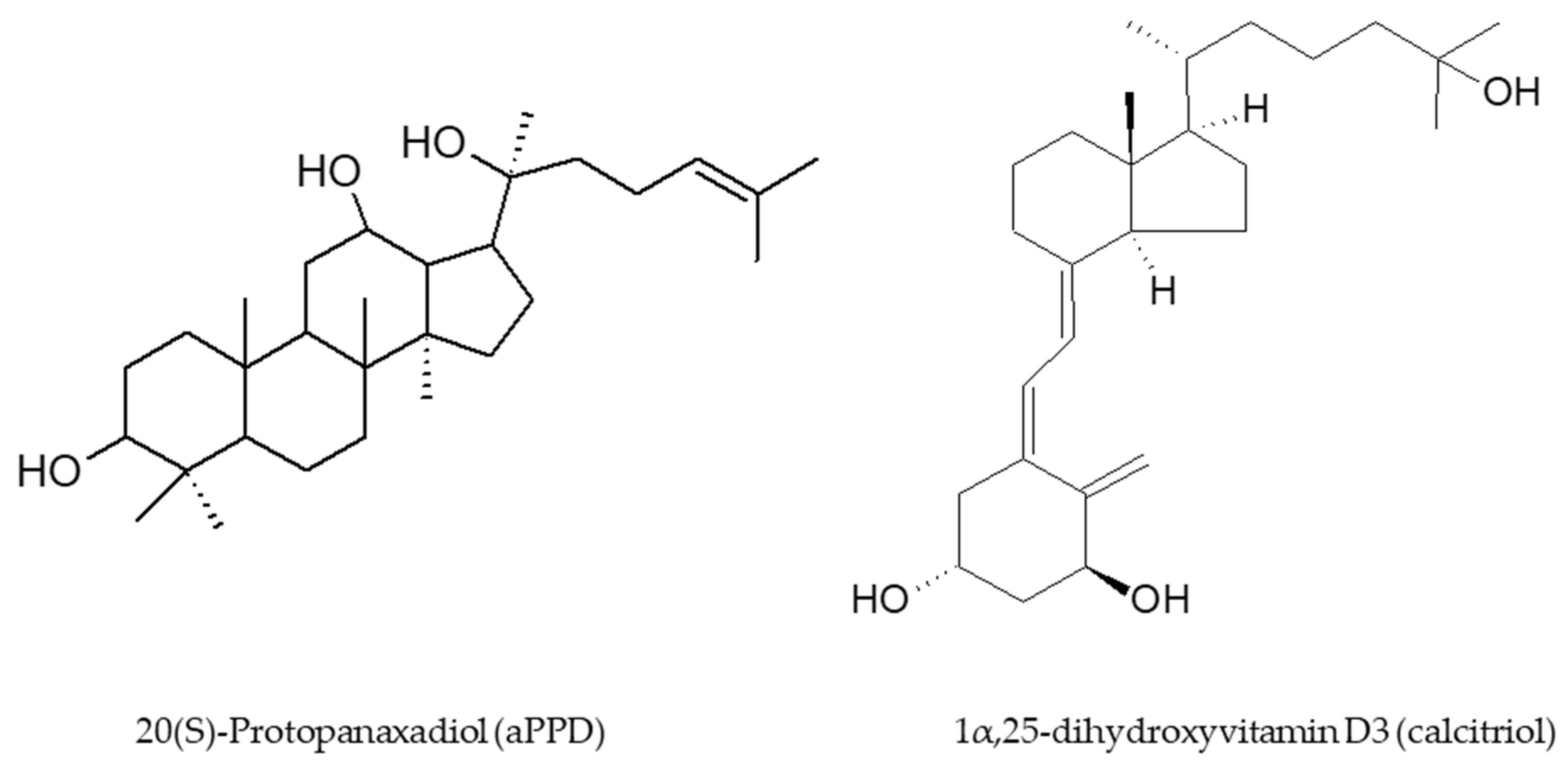{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Test Compounds and Reagents
2.2. Xenograft Preparation and Animal Treatment with Oral Gavage
2.3. Tumor Growth and Toxicity Assessment
2.4. Tumor Collection
2.5. In Silico Docking between aPPD and VDR
2.6. Western Blotting for VDR
2.7. VDR Transactivation Assay
2.8. Assessment of Apoptosis by Immunohistochemistry Analysis
2.9. Statistical Analysis
3. Results
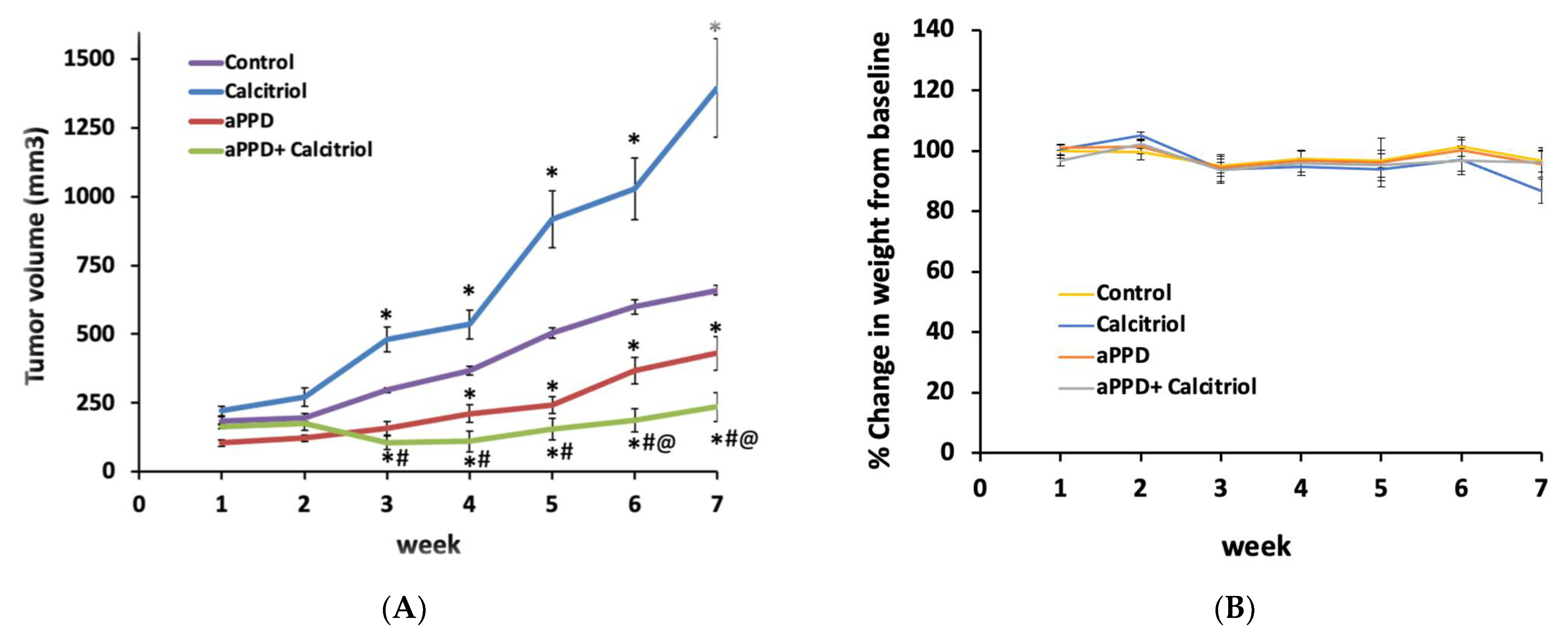
3.1. Calcitriol Sensitizes Castration-Resistant C4-2 Tumors to aPPD Anticancer effects
3.2. Lack of Toxicity from aPPD and Combination Treatment
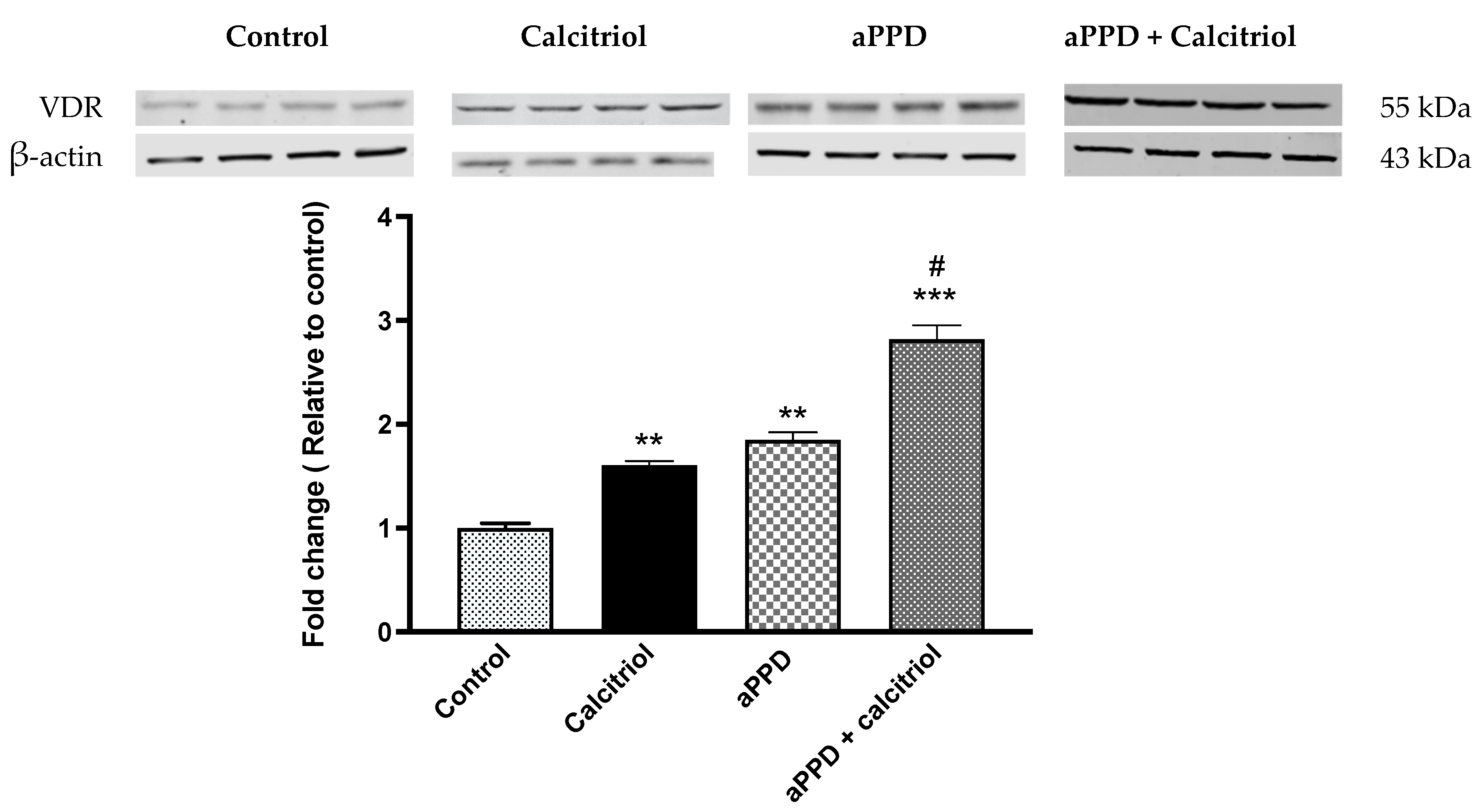
3.3. Induction of VDR Protein Expression by aPPD and Combination
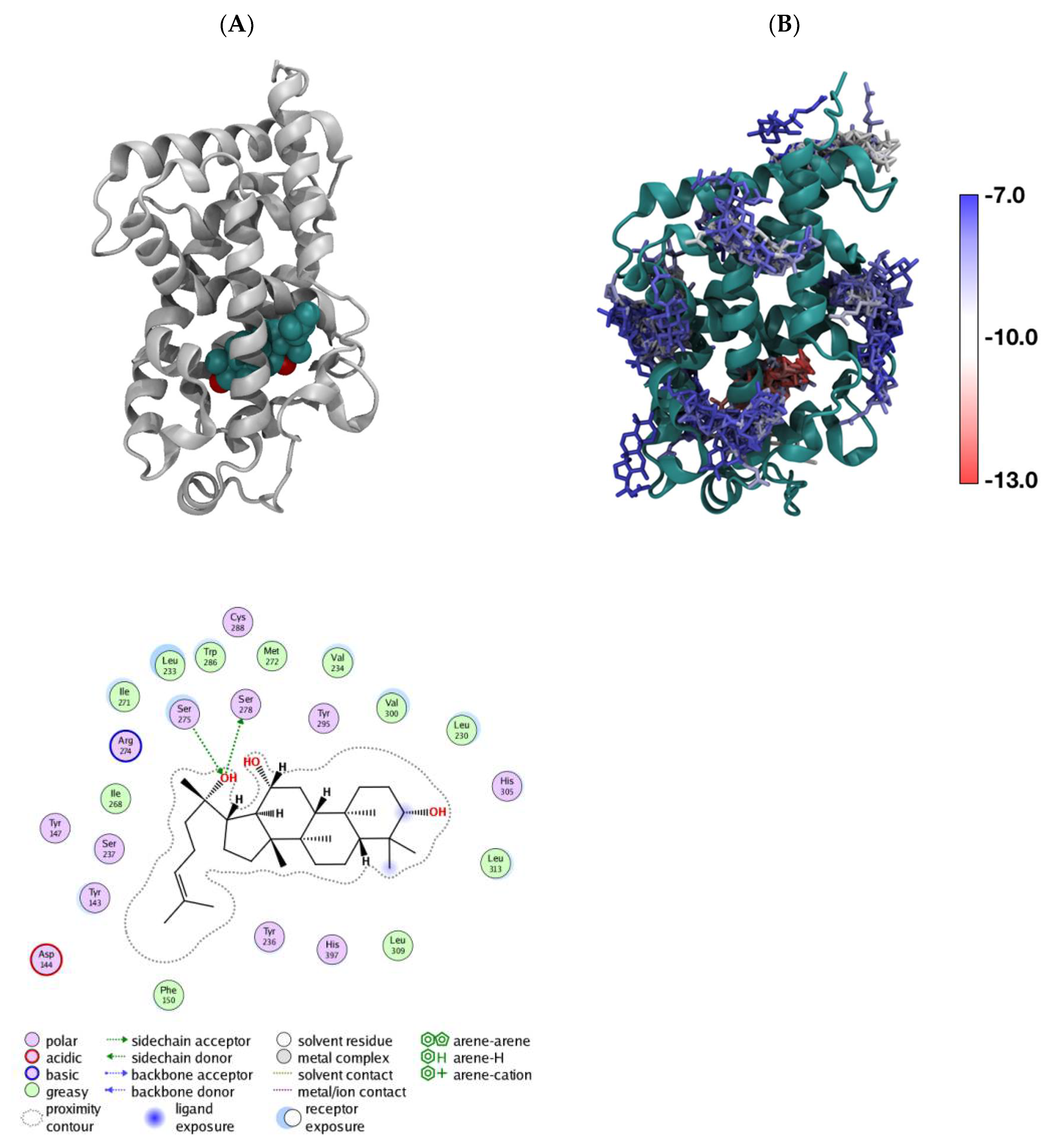
3.4. In Silico aPPD Binds to VDR
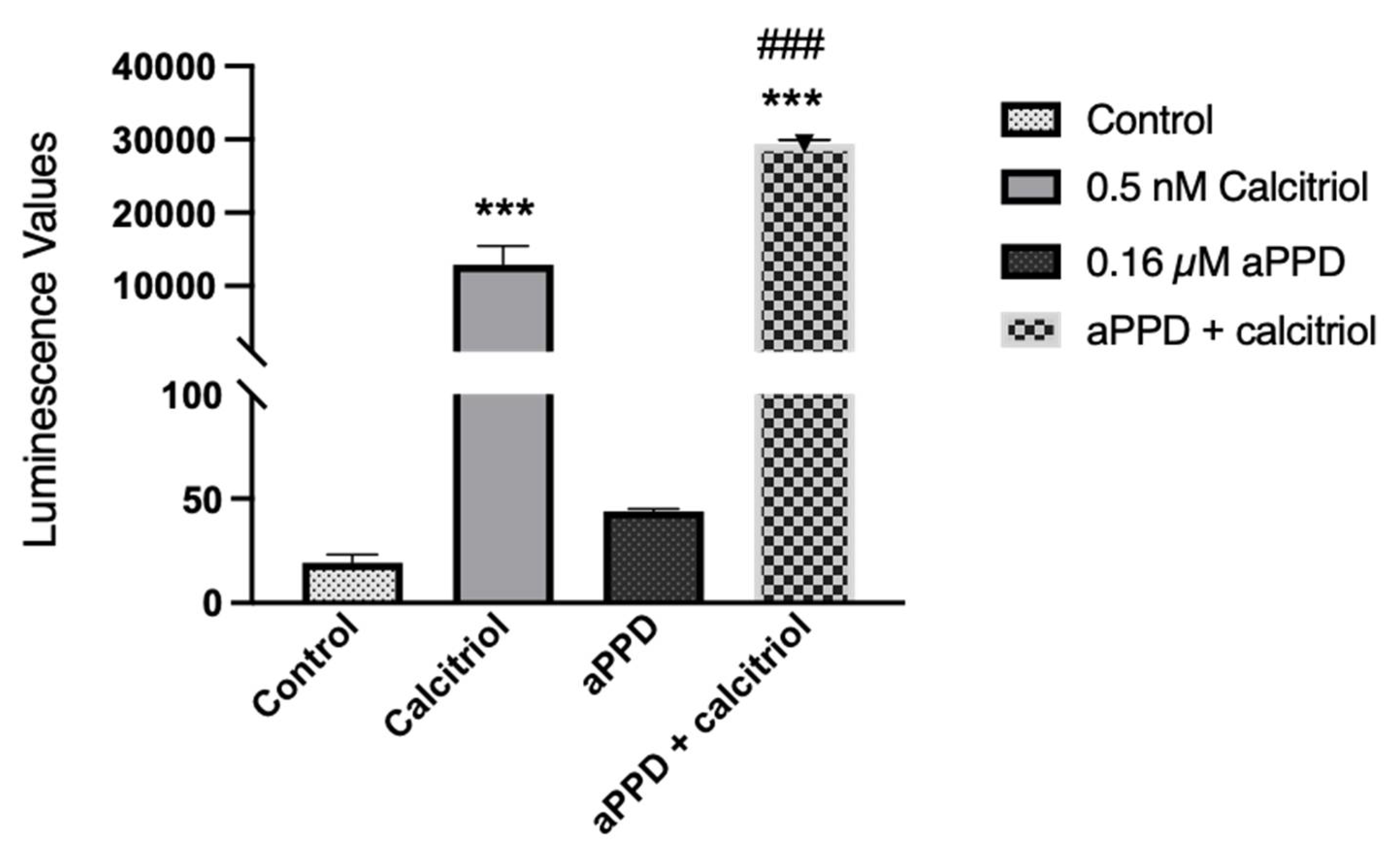
3.5. Combination Enhances VDR Transactivation
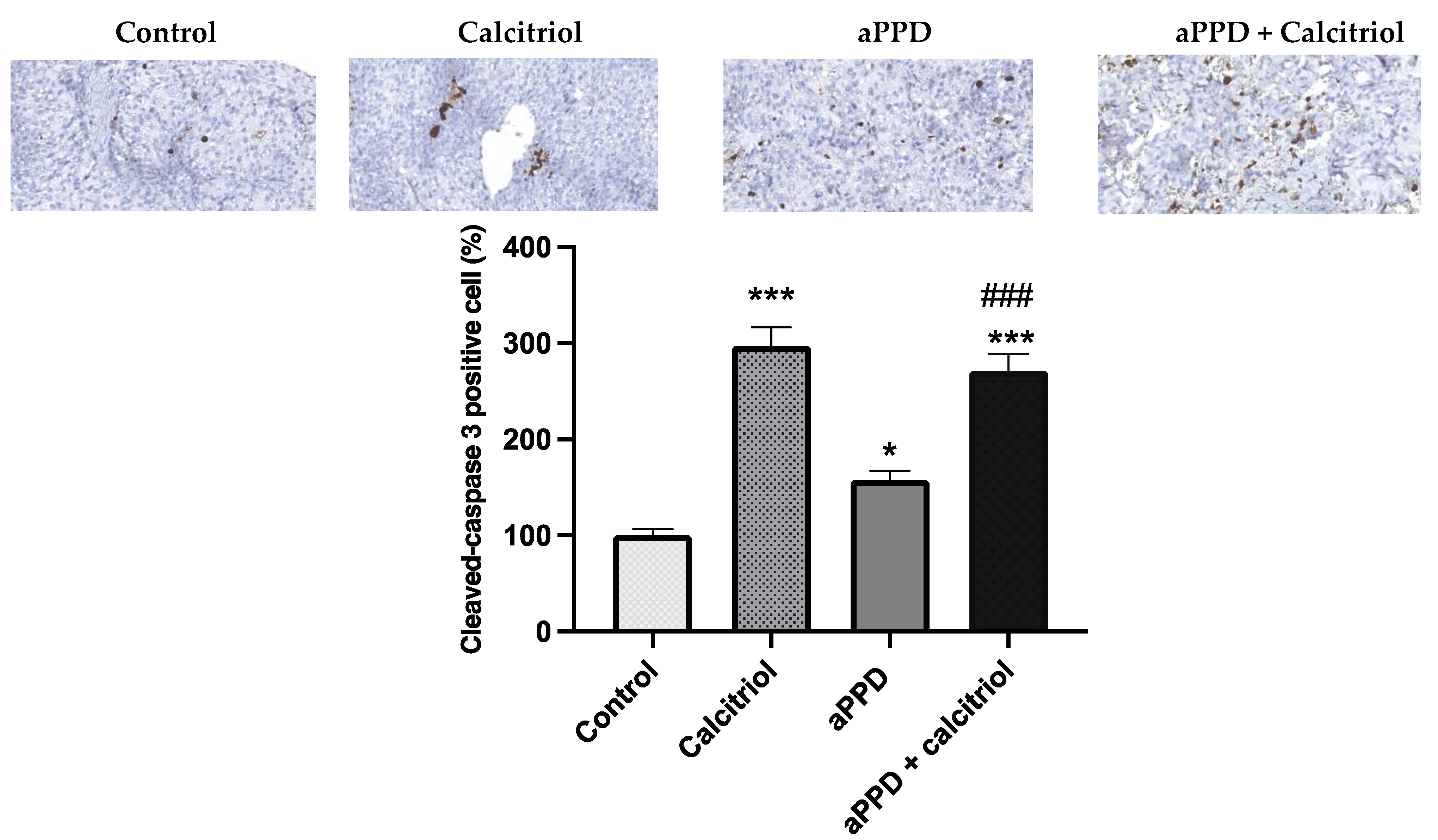
3.6. Induction of Apoptosis by the Combination
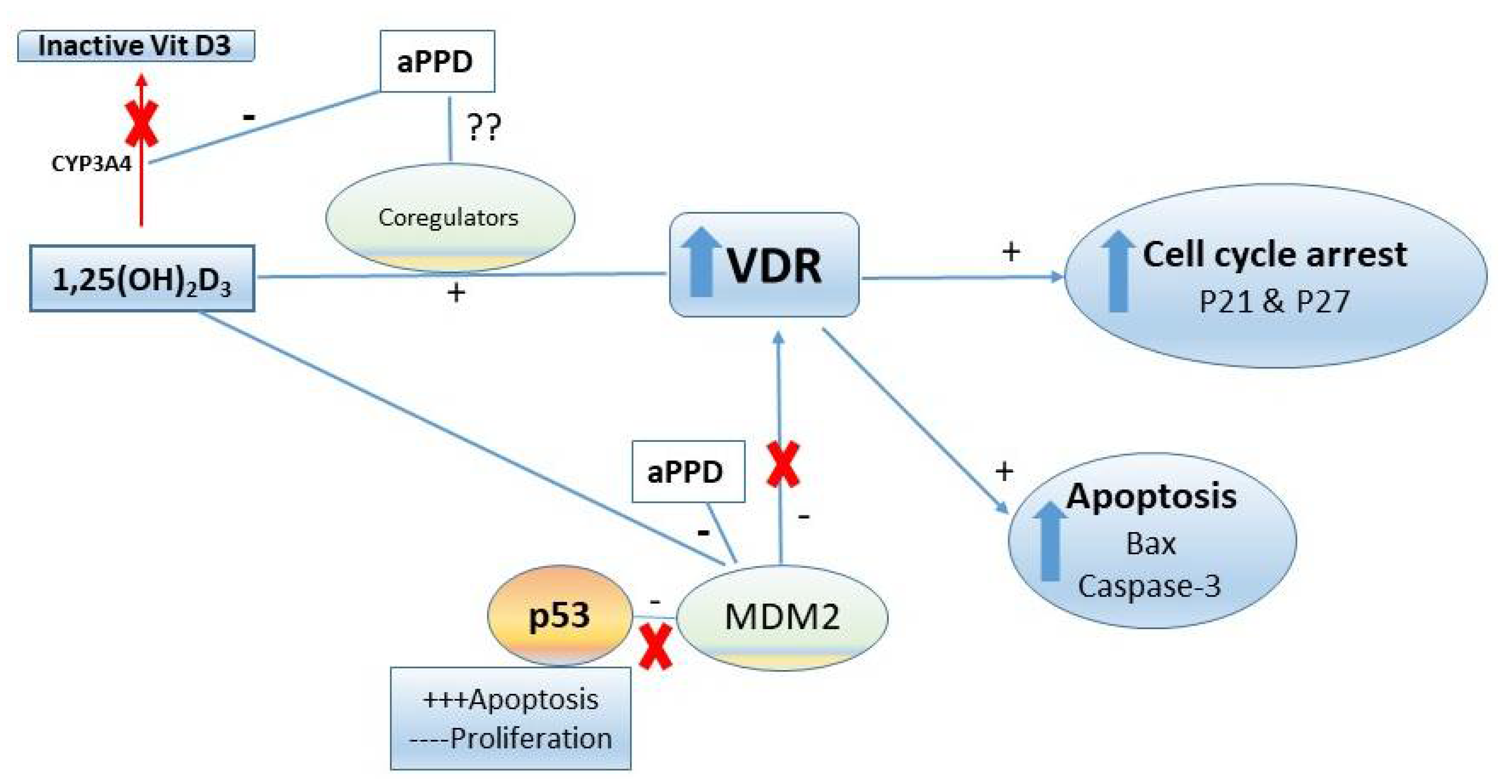
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Gleave, M.E.; Goldenberg, S.L.; Chin, J.L.; Warner, J.; Saad, F.; Klotz, L.H.; Jewett, M.; Kassabian, V.; Chetner, M.; Dupont, C.; et al. Randomized comparative study of 3 versus 8-month neoadjuvant hormonal therapy before radical prostatectomy: Biochemical and pathological effects. J. Urol. 2001, 166, 500–507. [Google Scholar] [CrossRef]
- Bruchovsky, N.; Klotz, L.H.; Sadar, M.; Crook, J.M.; Hoffart, D.; Godwin, L.; Warkentin, M.; Gleave, M.E.; Goldenberg, S.L. Intermittent androgen suppression for prostate cancer: Canadian Prospective Trial and related observations. Mol. Urol. 2000, 4, 191–199. [Google Scholar]
- Goldenberg, S.L.; Gleave, M.E.; Taylor, D.; Bruchovsky, N. Clinical Experience with Intermittent Androgen Suppression in Prostate Cancer: Minimum of 3 Years’ Follow-Up. Mol. Urol. 1999, 3, 287–292. [Google Scholar] [PubMed]
- Goldenberg, S.L.; Bruchovsky, N.; Gleave, M.E.; Sullivan, L.D. Low dose cyproterone acetate plus mini-dose diethylstilbesterol—A protocol for reversible medical castration. Urology 1996, 47, 882–884. [Google Scholar] [CrossRef]
- Gleave, M.; Goldenberg, S.L.; Bruchovsky, N.; Rennie, P. Intermittent androgen suppression for prostate cancer: Rationale and clinical experience. Prostate Cancer Prostatic Dis. 1998, 1, 289–296. [Google Scholar] [CrossRef]
- Bruchovsky, N.; Sadar, M.; Akakura, K.; Goldenberg, S.L.; Matsuoka, K.; Rennie, P.S. Characterization of 5 a-reducatase gene expression in stroma and epithelium of human prostate. J. Steroid Biochem. Mol. Biol. 1996, 59, 397–404. [Google Scholar] [CrossRef]
- Bhandari, M.S.; Crook, J.; Hussain, M. Should intermittent androgen deprivation be used in routine clinical practice? J. Clin. Oncol. 2005, 23, 8212–8218. [Google Scholar] [CrossRef]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, M.; Rahman, P.K. Medicinal Plants: Their Use in Anticancer Treatment. Int. J. Pharm. Sci. Res. 2015, 6, 4103–4112. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Khor, T.O.; Shu, L.; Su, Z.Y.; Fuentes, F.; Lee, J.H.; Kong, A.N. Plants vs. cancer: A review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Med. Chem. 2012, 12, 1281–1305. [Google Scholar] [CrossRef]
- Qiu, J. ‘Back to the future’ for Chinese herbal medicines. Nat. Rev. Drug Discov. 2007, 6, 506–507. [Google Scholar] [CrossRef]
- Assinewe, V.A.; Baum, B.R.; Gagnon, D.; Arnason, J.T. Phytochemistry of wild populations of Panax quinquefolius L. (North American ginseng). J. Agric. Food Chem. 2003, 51, 4549–4553. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.T. Botanical characteristics, pharmacological effects and medicinal components of Korean Panax ginseng C A Meyer. Acta Pharmacol. Sin. 2008, 29, 1109–1118. [Google Scholar] [CrossRef]
- Chan, T.W.; But, P.P.; Cheng, S.W.; Kwok, I.M.; Lau, F.W.; Xu, H.X. Differentiation and authentication of Panax ginseng, Panax quinquefolius, and ginseng products by using HPLC/MS. Anal. Chem. 2000, 72, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.A.; Han, M.J.; Choo, M.K.; Park, S.Y.; Kim, D.H. Metabolism of 20(S)- and 20(R)-ginsenoside Rg3 by human intestinal bacteria and its relation to in vitro biological activities. Biol. Pharm. Bull. 2002, 25, 58–63. [Google Scholar] [CrossRef]
- Hasegawa, H. Proof of the mysterious efficacy of ginseng: Basic and clinical trials: Metabolic activation of ginsenoside: Deglycosylation by intestinal bacteria and esterification with fatty acid. J. Pharmacol. Sci. 2004, 95, 153–157. [Google Scholar] [CrossRef]
- Ren, H.C.; Sun, J.G.; Wang, G.J.; A, J.Y.; Xie, H.T.; Zha, W.B.; Yan, B.; Sun, F.Z.; Hao, H.P.; Gu, S.H.; et al. Sensitive determination of 20(S)-protopanaxadiol in rat plasma using HPLC-APCI-MS: Application of pharmacokinetic study in rats. J. Pharm. Biomed. Anal. 2008, 48, 1476–1480. [Google Scholar] [CrossRef]
- Ben-Eltriki, M.; Deb, S.; Adomat, H.; Tomlinson Guns, E.S. Calcitriol and 20(S)-protopanaxadiol synergistically inhibit growth and induce apoptosis in human prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2016, 158, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Musende, A.G.; Eberding, A.; Wood, C.A.; Adomat, H.; Fazli, L.; Hurtado-Coll, A.; Jia, W.; Bally, M.B.; Tomlinson Guns, E.S. A novel oral dosage formulation of the ginsenoside aglycone protopanaxadiol exhibits therapeutic activity against a hormone-insensitive model of prostate cancer. Anticancer Drugs 2012, 23, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Popovich, D.G.; Kitts, D.D. Structure-function relationship exists for ginsenosides in reducing cell proliferation and inducing apoptosis in the human leukemia (THP-1) cell line. Arch. Biochem. Biophys. 2002, 406, 1–8. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, Q.; Hang, Y.; Bu, X.; Jia, W. Antiestrogenic effect of 20S-protopanaxadiol and its synergy with tamoxifen on breast cancer cells. Cancer 2007, 109, 2374–2382. [Google Scholar] [CrossRef]
- Ben-Eltriki, M.; Deb, S.; Hassona, M.; Meckling, G.; Fazli, L.; Chin, M.Y.; Lallous, N.; Yamazaki, T.; Jia, W.; Rennie, P.S.; et al. 20(S)-protopanaxadiol regio-selectively targets androgen receptor: Anticancer effects in castration-resistant prostate tumors. Oncotarget 2018, 9, 20965–20978. [Google Scholar] [CrossRef]
- Ben-Eltriki, M.; Deb, S.; Tomlinson Guns, E.S. Calcitriol in combination therapy for prostate cancer: Pharmacokinetic and pharmacodynamic interactions. J. Cancer 2016, 4, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Liu, X.; Li, J.; Liu, S.; Qi, Y.; Xiong, Z.; Zhang, A.; Wiese, T.; Fu, X.; Gu, J.; et al. 20(S)-protopanaxadiol-aglycone downregulation of the full-length and splice variants of androgen receptor. Int. J. Cancer 2013, 132, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Qi, Y.; Yang, Y.; Liu, X.; Xu, D.; Guo, W.; Zhan, Y.; Xiong, Z.; Zhang, A.; Wang, A.R.; et al. 20(S)-protopanaxadiol inhibition of progression and growth of castration-resistant prostate cancer. PLoS ONE 2014, 9, e111201. [Google Scholar] [CrossRef] [PubMed]
- Musende, A.G.; Eberding, A.; Jia, W.; Ramsay, E.; Bally, M.B.; Guns, E.T. Rh2 or its aglycone aPPD in combination with docetaxel for treatment of prostate cancer. Prostate 2010, 70, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Musende, A.G.; Eberding, A.; Wood, C.; Adomat, H.; Fazli, L.; Hurtado-Coll, A.; Jia, W.; Bally, M.B.; Guns, E.T. Pre-clinical evaluation of Rh2 in PC-3 human xenograft model for prostate cancer in vivo: Formulation, pharmacokinetics, biodistribution and efficacy. Cancer Chemother. Pharmacol. 2009, 64, 1085–1095. [Google Scholar] [CrossRef]
- Lu, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef]
- Furukawa, T.; Bai, C.X.; Kaihara, A.; Ozaki, E.; Kawano, T.; Nakaya, Y.; Awais, M.; Sato, M.; Umezawa, Y.; Kurokawa, J. Ginsenoside Re, a main phytosterol of Panax ginseng, activates cardiac potassium channels via a nongenomic pathway of sex hormones. Mol. Pharmacol. 2006, 70, 1916–1924. [Google Scholar] [CrossRef]
- Schuster, I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim. Biophys. Acta 2011, 1814, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W. From vitamin D to hormone D: Fundamentals of the vitamin D endocrine system essential for good health. Am. J. Clin. Nutr. 2008, 88, 491S–499S. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D and cancer: The promise not yet fulfilled. Endocrine 2014. [Google Scholar] [CrossRef] [PubMed]
- Morris, H.A.; Anderson, P.H. Autocrine and paracrine actions of vitamin d. Clin. Biochem. Rev. 2010, 31, 129–138. [Google Scholar]
- Kostner, K.; Denzer, N.; Muller, C.S.; Klein, R.; Tilgen, W.; Reichrath, J. The relevance of vitamin D receptor (VDR) gene polymorphisms for cancer: A review of the literature. Anticancer Res. 2009, 29, 3511–3536. [Google Scholar] [PubMed]
- Solomon, J.D.; Heitzer, M.D.; Liu, T.T.; Beumer, J.H.; Parise, R.A.; Normolle, D.P.; Leach, D.A.; Buchanan, G.; DeFranco, D.B. VDR activity is differentially affected by Hic-5 in prostate cancer and stromal cells. Mol. Cancer Res. 2014, 12, 1166–1180. [Google Scholar] [CrossRef]
- Ma, Y.; Trump, D.L.; Johnson, C.S. Vitamin D in combination cancer treatment. J. Cancer 2010, 1, 101–107. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Ben-Eltriki, M.; Hassona, M.; Meckling, G.; Adomat, H.; Deb, S.; Tomlinson Guns, E.S. Pharmacokinetic interaction of calcitriol with 20(S)-protopanaxadiol in mice: Determined by LC/MS analysis. Eur. J. Pharm. Sci. 2019, 130, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Kuruma, H.; Matsumoto, H.; Shiota, M.; Bishop, J.; Lamoureux, F.; Thomas, C.; Briere, D.; Los, G.; Gleave, M.; Fanjul, A.; et al. A novel antiandrogen, Compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.J.; Kim, S.; Pham, S.; Mangalji, A.; Adomat, H.; Guns, E.S.; Zoubeidi, A.; Moore, W.; Gleave, M.E. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol. Cancer Ther. 2015, 14, 59–69. [Google Scholar] [CrossRef]
- Fokidis, H.B.; Yieng Chin, M.; Ho, V.W.; Adomat, H.H.; Soma, K.K.; Fazli, L.; Nip, K.M.; Cox, M.; Krystal, G.; Zoubeidi, A.; et al. A low carbohydrate, high protein diet suppresses intratumoral androgen synthesis and slows castration-resistant prostate tumor growth in mice. J. Steroid Biochem. Mol. Biol. 2015, 150, 35–45. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Zardan, A.; Beraldi, E.; Fazli, L.; Sowery, R.; Rennie, P.; Nelson, C.; Gleave, M. Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 2007, 67, 10455–10465. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment. Available online: https://www.chemcomp.com/ (accessed on 20 May 2018).
- Muindi, J.R.; Yu, W.D.; Ma, Y.; Engler, K.L.; Kong, R.X.; Trump, D.L.; Johnson, C.S. CYP24A1 inhibition enhances the antitumor activity of calcitriol. Endocrinology 2010, 151, 4301–4312. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Kirk, J.S.; Karasik, E.; Gillard, B.; Moser, M.T.; Johnson, C.S.; Trump, D.L.; Foster, B.A. Early growth inhibition is followed by increased metastatic disease with vitamin D (calcitriol) treatment in the TRAMP model of prostate cancer. PLoS ONE 2014, 9, e89555. [Google Scholar] [CrossRef]
- Anisiewicz, A.; Pawlik, A.; Filip-Psurska, B.; Turlej, E.; Dzimira, S.; Milczarek, M.; Gdesz, K.; Papiernik, D.; Jarosz, J.; Klopotowska, D.; et al. Unfavorable effect of calcitriol and its low-calcemic analogs on metastasis of 4T1 mouse mammary gland cancer. Int. J. Oncol. 2018, 52, 103–126. [Google Scholar] [CrossRef]
- Alagbala, A.A.; Moser, M.T.; Johnson, C.S.; Trump, D.L.; Foster, B.A. Characterization of Vitamin D insensitive prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2007, 103, 712–716. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hendrickson, W.K.; Flavin, R.; Kasperzyk, J.L.; Fiorentino, M.; Fang, F.; Lis, R.; Fiore, C.; Penney, K.L.; Ma, J.; Kantoff, P.W.; et al. Vitamin D receptor protein expression in tumor tissue and prostate cancer progression. J. Clin. Oncol. 2011, 29, 2378–2385. [Google Scholar] [CrossRef]
- Huss, L.; Butt, S.T.; Borgquist, S.; Elebro, K.; Sandsveden, M.; Rosendahl, A.; Manjer, J. Vitamin D receptor expression in invasive breast tumors and breast cancer survival. Breast Cancer Res. 2019, 21, 84. [Google Scholar] [CrossRef] [PubMed]
- Mooso, B.; Madhav, A.; Johnson, S.; Roy, M.; Moore, M.E.; Moy, C.; Loredo, G.A.; Mehta, R.G.; Vaughan, A.T.; Ghosh, P.M. Androgen Receptor regulation of Vitamin D receptor in response of castration-resistant prostate cancer cells to 1alpha-Hydroxyvitamin D5—A calcitriol analog. Genes Cancer 2010, 1, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.A.; Qin, J.J.; Wang, W.; Wang, M.H.; Wang, H.; Zhang, R. Ginsenosides as Anticancer Agents: In vitro and in vivo Activities, Structure-Activity Relationships, and Molecular Mechanisms of Action. Front. Pharmacol. 2012, 3, 25. [Google Scholar] [CrossRef]
- Khedkar, S.A.; Samad, M.A.; Choudhury, S.; Lee, J.Y.; Zhang, D.; Thadhani, R.I.; Karumanchi, S.A.; Rigby, A.C.; Kang, P.M. Identification of Novel Non-secosteroidal Vitamin D Receptor Agonists with Potent Cardioprotective Effects and devoid of Hypercalcemia. Sci. Rep. 2017, 7, 8427. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W.; Mizwicki, M.T.; Norman, D.P. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat. Rev. Drug Discov. 2004, 3, 27–41. [Google Scholar] [CrossRef]
- Mizwicki, M.T.; Keidel, D.; Bula, C.M.; Bishop, J.E.; Zanello, L.P.; Wurtz, J.M.; Moras, D.; Norman, A.W. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2-vitamin D3 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12876–12881. [Google Scholar] [CrossRef]
- Dampf Stone, A.; Batie, S.F.; Sabir, M.S.; Jacobs, E.T.; Lee, J.H.; Whitfield, G.K.; Haussler, M.R.; Jurutka, P.W. Resveratrol potentiates vitamin D and nuclear receptor signaling. J. Cell. Biochem. 2015, 116, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Mizwicki, M.T.; Norman, A.W. The vitamin D sterol-vitamin D receptor ensemble model offers unique insights into both genomic and rapid-response signaling. Sci. Signal. 2009, 2, re4. [Google Scholar] [CrossRef]
- Jager, R.; Zwacka, R.M. The enigmatic roles of caspases in tumor development. Cancers 2010, 2, 1952–1979. [Google Scholar] [CrossRef]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Donato, A.L.; Huang, Q.; Liu, X.; Li, F.; Zimmerman, M.A.; Li, C.Y. Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J. Investig. Dermatol. 2014, 134, 1686–1692. [Google Scholar] [CrossRef]
- Deeb, K.K.; Trump, D.L.; Johnson, C.S. Vitamin D signalling pathways in cancer: Potential for anticancer therapeutics. Nat. Rev. Cancer 2007, 7, 684–700. [Google Scholar] [CrossRef]
- Diaz, L.; Diaz-Munoz, M.; Garcia-Gaytan, A.C.; Mendez, I. Mechanistic Effects of Calcitriol in Cancer Biology. Nutrients 2015, 7, 5020–5050. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, C.J.; Welsh, J. Role of mitochondria and caspases in vitamin D-mediated apoptosis of MCF-7 breast cancer cells. J. Biol. Chem. 2001, 276, 9101–9107. [Google Scholar] [CrossRef]
- Chen, H.; Reed, G.; Guardia, J.; Lakhan, S.; Couture, O.; Hays, E.; Chandar, N. Vitamin D directly regulates Mdm2 gene expression in osteoblasts. Biochem. Biophys. Res. Commun. 2013, 430, 370–374. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.J.; Li, X.; Tao, G.; Wang, Q.; Wu, X.; Zhou, J.; Zi, X.; Zhang, R. Pevention of prostate cancer by natural product MDM2 inhibitor GS25: In vitro and in vivo activities and molecular mechanisms. Carcinogenesis 2018, 39, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Gu, W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012, 3, 240–248. [Google Scholar] [CrossRef]
- Heyne, K.; Heil, T.C.; Bette, B.; Reichrath, J.; Roemer, K. MDM2 binds and inhibits vitamin D receptor. Cell Cycle 2015, 14, 2003–2010. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deb, S.; Chin, M.Y.; Adomat, H.; Guns, E.S. Ginsenoside-mediated blockade of 1alpha,25-dihydroxyvitamin D inactivation in human liver and intestine in vitro. J. Steroid Biochem. Mol. Biol. 2014, 141, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Pandey, M.; Adomat, H.; Guns, E.S. Cytochrome P450 3A-mediated microsomal biotransformation of 1alpha,25-dihydroxyvitamin D3 in mouse and human liver: Drug-related induction and inhibition of catabolism. Drug Metab. Dispos. 2012, 40, 907–918. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben-Eltriki, M.; Deb, S.; Shankar, G.; Meckling, G.; Hassona, M.; Yamazaki, T.; Fazli, L.; Chin, M.Y.; Tomlinson Guns, E.S. Anti-Tumor Effects of Ginsenoside 20(S)-Protopanaxadiol and 1,25-Dihydroxyvitamin D3 Combination in Castration Resistant Prostate Cancer. Medicines 2021, 8, 28. https://doi.org/10.3390/medicines8060028
Ben-Eltriki M, Deb S, Shankar G, Meckling G, Hassona M, Yamazaki T, Fazli L, Chin MY, Tomlinson Guns ES. Anti-Tumor Effects of Ginsenoside 20(S)-Protopanaxadiol and 1,25-Dihydroxyvitamin D3 Combination in Castration Resistant Prostate Cancer. Medicines. 2021; 8(6):28. https://doi.org/10.3390/medicines8060028
Chicago/Turabian StyleBen-Eltriki, Mohamed, Subrata Deb, Gehana Shankar, Gray Meckling, Mohamed Hassona, Takeshi Yamazaki, Ladan Fazli, Mei Yieng Chin, and Emma S. Tomlinson Guns. 2021. "Anti-Tumor Effects of Ginsenoside 20(S)-Protopanaxadiol and 1,25-Dihydroxyvitamin D3 Combination in Castration Resistant Prostate Cancer" Medicines 8, no. 6: 28. https://doi.org/10.3390/medicines8060028
APA StyleBen-Eltriki, M., Deb, S., Shankar, G., Meckling, G., Hassona, M., Yamazaki, T., Fazli, L., Chin, M. Y., & Tomlinson Guns, E. S. (2021). Anti-Tumor Effects of Ginsenoside 20(S)-Protopanaxadiol and 1,25-Dihydroxyvitamin D3 Combination in Castration Resistant Prostate Cancer. Medicines, 8(6), 28. https://doi.org/10.3390/medicines8060028