
In Silico Exploration of the Potential Role of Acetaminophen and Pesticides in the Etiology of Autism Spectrum Disorder

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
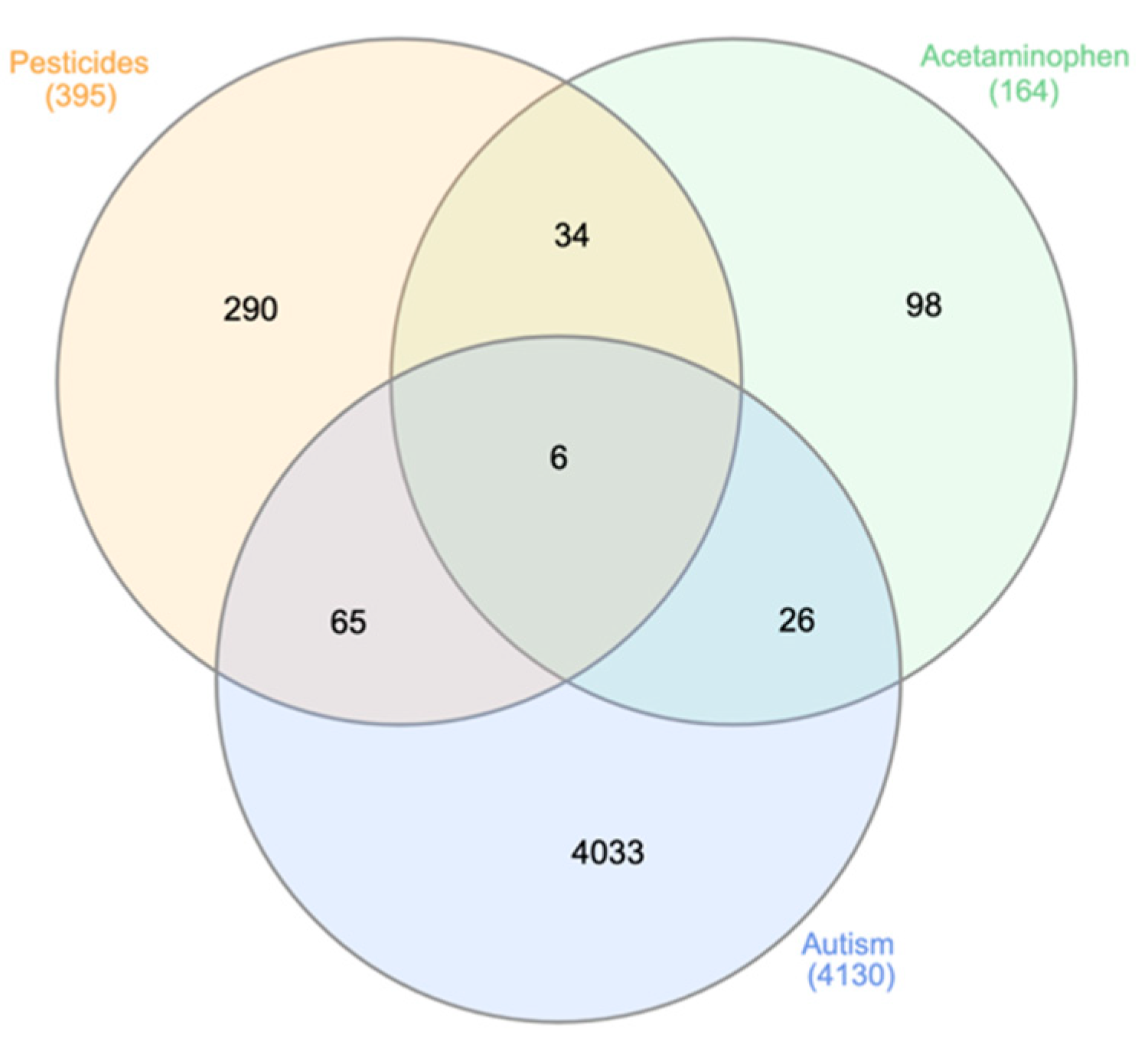
3.1. Molecular Database Search for Genetic Associations
3.1.1. Gene Retrieval from Molecular Databases
3.1.2. Pathway Analysis
3.1.3. Network Analysis
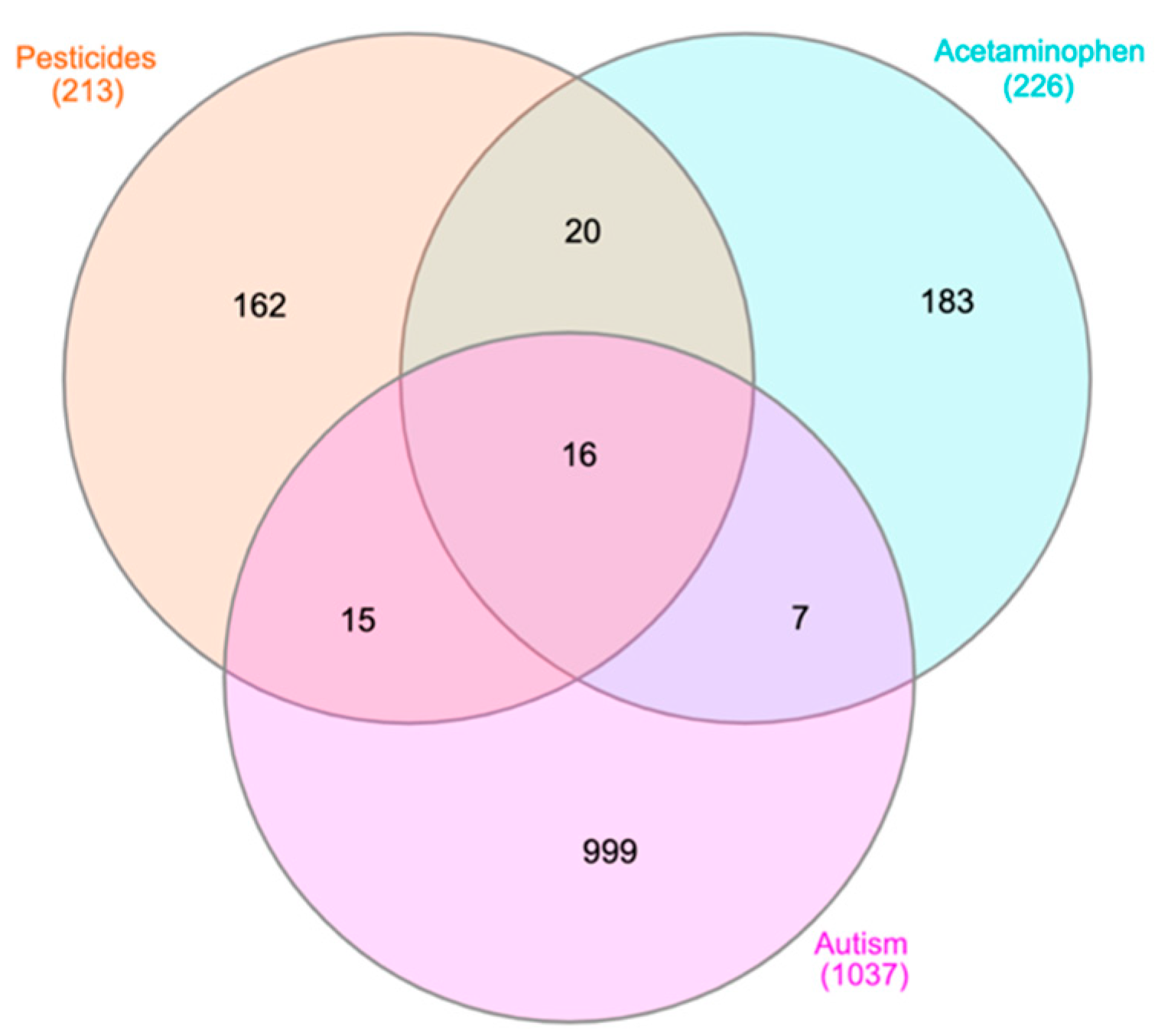
3.2. Literature Search for Genetic Associations
3.2.1. Gene Retrieval from Coremine Medical
3.2.2. Pathway Analysis
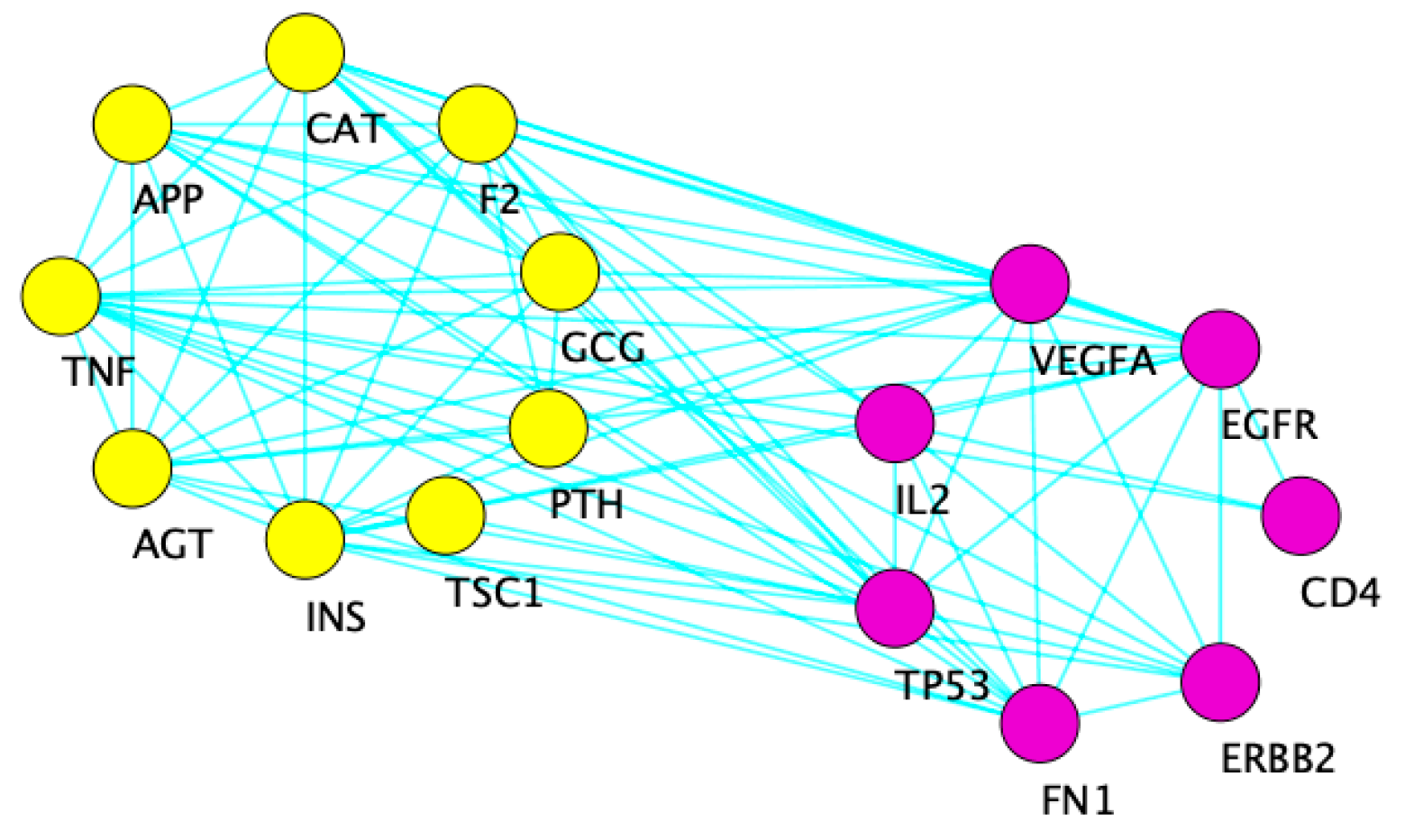
3.2.3. Network Analysis
3.3. Pathway Comparison Analysis
4. Discussion
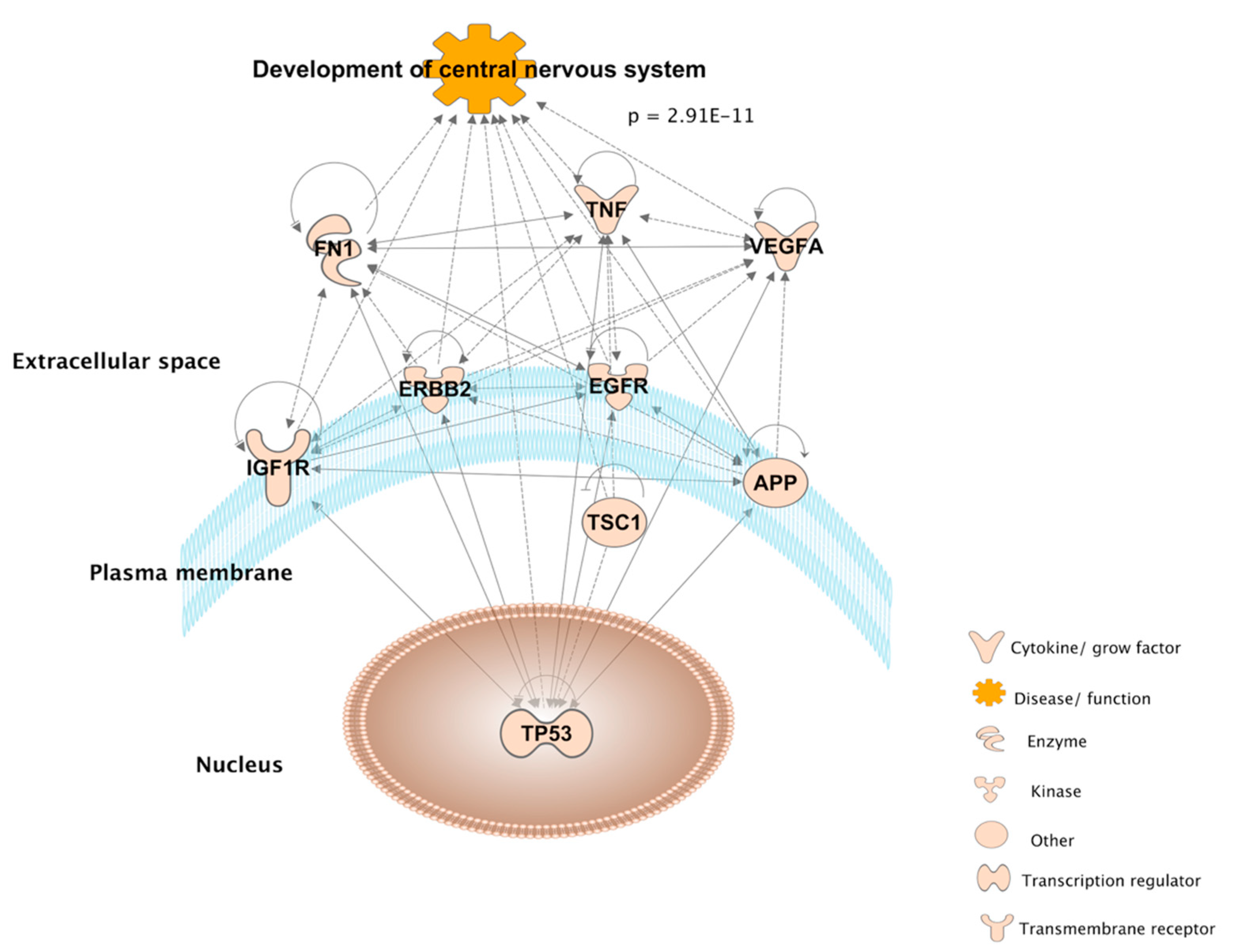
4.1. Main Biological Themes
4.1.1. Apoptosis
4.1.2. Metabolism of ROS
4.1.3. Carbohydrate Metabolism
4.2. Intra-Pathway Interactions
4.3. Strengths and Limitations
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
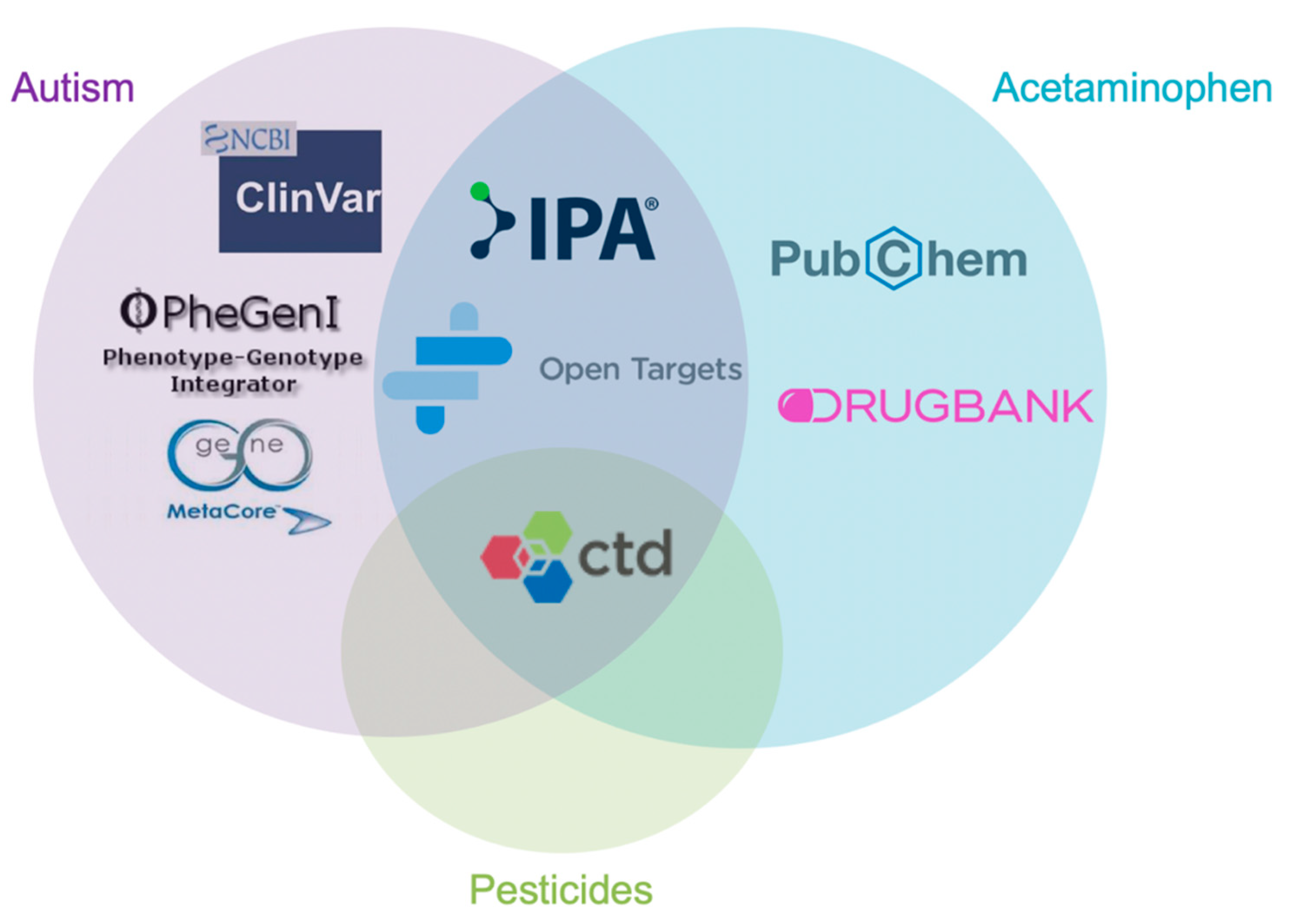{kind=link}
| Database | Information about Database |
|---|---|
| CTD | CTD is a robust, publicly available database that aims to advance understanding about how environmental exposures affect human health. It provides manually curated information about chemical–gene/protein interactions, chemical–disease and gene–disease relationships. These data are integrated with functional and pathway data to aid in development of hypotheses about the mechanisms underlying environmentally influenced diseases. |
| PubChem |
PubChem is an open chemistry database at the National Institutes of Health (NIH). “Open” means that you can put your scientific data in PubChem and that others may use it. Since the launch in 2004, PubChem has become a key chemical information resource for scientists, students, and the general public. Each month our website and programmatic services provide data to several million users worldwide. |
| Open Targets Platform | The Open Targets Platform is a comprehensive and robust data integration for access to and visualization of potential drug targets associated with disease. It brings together multiple data types and aims to assist users to identify and prioritize targets for further investigation. |
| IPA | IPA is an all-in-one, web-based software application that enables analysis, integration, and understanding of data from gene expression, miRNA, and SNP microarrays, as well as metabolomics, proteomics, and RNAseq experiments. IPA can also be used for analysis of small-scale experiments that generate gene and chemical lists. IPA allows searches for targeted information on genes, proteins, chemicals, and drugs, and building of interactive models of experimental systems. Data analysis and search capabilities help in understanding the significance of data, specific targets, or candidate biomarkers in the context of larger biological or chemical systems. The software is backed by the Ingenuity Knowledge Base of highly structured, detail-rich biological and chemical findings. |
| DrugBank | The DrugBank database is a comprehensive, freely accessible, online database containing information on drugs and drug targets. As both a bioinformatics and a cheminformatics resource, DrugBank combines detailed drug (i.e., chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e., sequence, structure, and pathway) information. Because of its broad scope, comprehensive referencing and unusually detailed data descriptions, DrugBank is more akin to a drug encyclopedia than a drug database. As a result, links to DrugBank are maintained for nearly all drugs listed in Wikipedia. DrugBank is widely used by the drug industry, medicinal chemists, pharmacists, physicians, students and the general public. Its extensive drug and drug-target data has enabled the discovery and repurposing of a number of existing drugs to treat rare and newly identified illnesses. |
| PheGenI | The Phenotype-Genotype Integrator (PheGenI), merges NHGRI genome-wide association study (GWAS) catalog data with several databases housed at the National Center for Biotechnology Information (NCBI), including Gene, dbGaP, OMIM, eQTL and dbSNP |
| MetaCore | MetaCore provides the core capabilities of precise pathway analysis, knowledge mining, simple bioinformatics and effective visualizations in a comprehensive, off-the-shelf package. Use high-quality, 100% manually curated biological pathway data from peer-reviewed literature to accelerate drug development by rapidly generating and validating hypotheses for novel biomarkers, targets and mechanisms of action. |
| Clinvar | ClinVar processes submissions reporting variants found in patient samples, assertions made regarding their clinical significance, information about the submitter, and other supporting data. The alleles described in submissions are mapped to reference sequences, and reported according to the HGVS standard |
| CoreMine Medical | Coremine Medical™, the first domain-specific information community built on top of the COREMINE Platform. It is a free Internet service for searching, updating, and sharing medical information–both search and social network. |
Appendix B

Appendix C
Appendix D
| Ingenuity Canonical Pathways | p-Value | Genes |
|---|---|---|
| Death Receptor Signaling | 2.40 × 10−2 | FAS |
| p53 Signaling | 2.57 × 10−2 | FAS |
| Apoptosis Signaling | 2.57 × 10−2 | FAS |
| IGF-1 Signaling | 2.69 × 10−2 | IGF1R |
| Type I Diabetes Mellitus Signaling | 2.88 × 10−2 | FAS |
| FXR/RXR Activation | 3.31 × 10−2 | ABCB4 |
| Necroptosis Signaling Pathway | 4.07 × 10−2 | FAS |
Appendix E
Appendix F
| Ingenuity Canonical Pathways | p-Value | Genes |
|---|---|---|
| EIF2 Signaling | 1.07 × 10−2 | INS, VEGFA |
| Huntington’s Disease Signaling | 1.17 × 10−2 | EGFR, TP53 |
| Senescence Pathway | 1.58 × 10−2 | CAT, TP53 |
| Xenobiotic Metabolism Signaling | 1.70 × 10−2 | CAT, TNF |
| Neuroinflammation Signaling Pathway | 1.86 × 10−2 | APP, TNF |
| Axonal Guidance Signaling | 4.47 × 10−2 | ERBB2, VEGFA |
References
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281. [Google Scholar] [PubMed]
- Herbert, M.R.; Russo, J.; Yang, S.; Roohi, J.; Blaxill, M.; Kahler, S.; Cremer, L.; Hatchwell, E. Autism and environmental genomics. Neurotoxicology 2006, 27, 671–684. [Google Scholar] [CrossRef]
- Karimi, P.; Kamali, E.; Mousavi, S.M.; Karahmadi, M. Environmental factors influencing the risk of autism. J. Res. Med. Sci. 2017, 22, 27. [Google Scholar] [PubMed]
- Werler, M.M.; Mitchell, A.A.; Hernandez-Diaz, S.; Honein, M.A. Use of over-the-counter medications during pregnancy. Am. J. Obstet. Gynecol. 2005, 193, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.; Hornik, C.D.; Bilbo, S.; Holzknecht, Z.E.; Gentry, L.; Rao, R.; Lin, S.S.; Herbert, M.R.; Nevison, C.D. The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. J. Int. Med. Res. 2017, 45, 407–438. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, S.K.; Harris, M.H.; Gunier, R.B.; Kogut, K.R.; Harley, K.G.; Deardorff, J.; Bradman, A.; Holland, N.; Eskenazi, B. Prenatal Organophosphate Pesticide Exposure and Traits Related to Autism Spectrum Disorders in a Population Living in Proximity to Agriculture. Environ. Health Perspect. 2018, 126, 047012. [Google Scholar] [CrossRef]
- Roberts, J.R.; Dawley, E.H.; Reigart, J.R. Children’s low-level pesticide exposure and associations with autism and ADHD: A review. Pediatr. Res. 2019, 85, 234–241. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef]
- Roberts, E.M.; English, P.B.; Grether, J.K.; Windham, G.C.; Somberg, L.; Wolff, C. Maternal residence near agricultural pesticide applications and autism spectrum disorders among children in the California Central Valley. Environ. Health Perspect. 2007, 115, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Liew, Z.; Ritz, B.; Rebordosa, C.; Lee, P.-C.; Olsen, J. Acetaminophen use during pregnancy, behavioral problems, and hyperkinetic disorders. JAMA Pediatr. 2014, 168, 313–320. [Google Scholar] [CrossRef]
- Liew, Z.; Ritz, B.; Virk, J.; Olsen, J. Maternal use of acetaminophen during pregnancy and risk of autism spectrum disorders in childhood: AD anish national birth cohort study. Autism Res. 2016, 9, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Avella-Garcia, C.B.; Julvez, J.; Fortuny, J.; Rebordosa, C.; García-Esteban, R.; Galán, I.R.; Tardón, A.; Rodríguez-Bernal, C.L.; Iñiguez, C.; Andiarena, A. Acetaminophen use in pregnancy and neurodevelopment: Attention function and autism spectrum symptoms. Int. J. Epidemiol. 2016, 45, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Brandlistuen, R.E.; Ystrom, E.; Nulman, I.; Koren, G.; Nordeng, H. Prenatal paracetamol exposure and child neurodevelopment: A sibling-controlled cohort study. Int. J. Epidemiol. 2013, 42, 1702–1713. [Google Scholar] [CrossRef]
- Ystrom, E.; Gustavson, K.; Brandlistuen, R.E.; Knudsen, G.P.; Magnus, P.; Susser, E.; Smith, G.D.; Stoltenberg, C.; Surén, P.; Håberg, S.E. Prenatal exposure to acetaminophen and risk of ADHD. Pediatrics 2017, 140, e20163840. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Azuine, R.E.; Zhang, Y.; Hou, W.; Hong, X.; Wang, G.; Riley, A.; Pearson, C.; Zuckerman, B.; Wang, X. Association of cord plasma biomarkers of in utero acetaminophen exposure with risk of attention-deficit/hyperactivity disorder and autism spectrum disorder in childhood. JAMA Psychiatry 2020, 77, 180–189. [Google Scholar] [CrossRef]
- Levy, G.; Garrettson, L.K.; Soda, D.M. Evidence of placental transfer of acetaminophen. Pediatrics 1975, 55, 895. [Google Scholar] [PubMed]
- Horowitz, R.S.; Dart, R.C.; Jarvie, D.R.; Bearer, C.F.; Gupta, U. Placental transfer of N-acetylcysteine following human maternal acetaminophen toxicity. J. Toxicol. Clin. Toxicol. 1997, 35, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Liew, Z.; Ernst, A. Intrauterine Exposure to Acetaminophen and Adverse Developmental Outcomes: Epidemiological Findings and Methodological Issues. Curr. Environ. Health Rep. 2021, 8, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Burman, A.; Garcia-Milian, R.; Wood, M.; DeWitt, N.A.; Vasiliou, V.; Guller, S.; Abrahams, V.M.; Whirledge, S. Acetaminophen Attenuates invasion and alters the expression of extracellular matrix enzymes and vascular factors in human first trimester trophoblast cells. Placenta 2020, 104, 146–160. [Google Scholar] [CrossRef]
- Pristner, M.; Warth, B. Drug—Exposome Interactions: The Next Frontier in Precision Medicine. Trends Pharmacol. Sci. 2020, 41, 994–1005. [Google Scholar] [CrossRef]
- Beger, R.D.; Flynn, T.J. Pharmacometabolomics in drug safety and drug-exposome interactions. Metabolomics 2016, 12, 123. [Google Scholar] [CrossRef]
- Tumiatti, V.; Fimognari, C.; Milelli, A.; Manucra, D. Pollutants and Drugs: Interactions and Human Health. In Clinical Handbook of Air Pollution-Related Diseases; Capello, F., Gaddi, A.V., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 215–230. [Google Scholar] [CrossRef]
- Clayton, T.A.; Baker, D.; Lindon, J.C.; Everett, J.R.; Nicholson, J.K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 14728–14733. [Google Scholar] [CrossRef] [PubMed]
- Fardel, O.; Kolasa, E.; Le Vee, M. Environmental chemicals as substrates, inhibitors or inducers of drug transporters: Implication for toxicokinetics, toxicity and pharmacokinetics. Expert Opin. Drug Metab. Toxicol. 2012, 8, 29–46. [Google Scholar] [CrossRef]
- Goodrich, A.J.; Volk, H.E.; Tancredi, D.J.; McConnell, R.; Lurmann, F.W.; Hansen, R.L.; Schmidt, R.J. Joint effects of prenatal air pollutant exposure and maternal folic acid supplementation on risk of autism spectrum disorder. Autism Res. 2018, 11, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Rothman, K.J.; Greenland, S. Causation and causal inference in epidemiology. Am. J. Public Health 2005, 95, S144–S150. [Google Scholar] [CrossRef] [PubMed]
- Von Ehrenstein, O.S.; Ling, C.; Cui, X.; Cockburn, M.; Park, A.S.; Yu, F.; Wu, J.; Ritz, B. Prenatal and infant exposure to ambient pesticides and autism spectrum disorder in children: Population based case-control study. BMJ 2019, 364, l962. [Google Scholar] [CrossRef]
- Aloizou, A.M.; Siokas, V.; Vogiatzi, C.; Peristeri, E.; Docea, A.O.; Petrakis, D.; Provatas, A.; Folia, V.; Chalkia, C.; Vinceti, M.; et al. Pesticides, cognitive functions and dementia: A review. Toxicol. Lett. 2020, 326, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Nicklisch, S.C.; Rees, S.D.; McGrath, A.P.; Gökirmak, T.; Bonito, L.T.; Vermeer, L.M.; Cregger, C.; Loewen, G.; Sandin, S.; Chang, G.; et al. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016, 2, e1600001. [Google Scholar] [CrossRef]
- Manivasagam, T.; Arunadevi, S.; Essa, M.M.; SaravanaBabu, C.; Borah, A.; Thenmozhi, A.J.; Qoronfleh, M.W. Role of Oxidative Stress and Antioxidants in Autism. Adv. Neurobiol. 2020, 24, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef]
- Matysiak, M.; Kruszewski, M.; Jodlowska-Jedrych, B.; Kapka-Skrzypczak, L. Effect of Prenatal Exposure to Pesticides on Children’s Health. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Petric, I.; Ligeti, B.; Gyorffy, B.; Pongor, S. Biomedical hypothesis generation by text mining and gene prioritization. Protein Pept. Lett. 2014, 21, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Audouze, K.; Brunak, S.; Grandjean, P. A computational approach to chemical etiologies of diabetes. Sci. Rep. 2013, 3, 2712. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, X.; Chen, J.Y. Building disease-specific drug-protein connectivity maps from molecular interaction networks and PubMed abstracts. PLoS Comput. Biol. 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The comparative toxicogenomics database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E. Open Targets Platform: New developments and updates two years on. Nucleic Acids Res. 2019, 47, D1056–D1065. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.M.; Hoffman, D.; Junkins, H.A.; Maglott, D.; Phan, L.; Sherry, S.T.; Feolo, M.; Hindorff, L.A. Phenotype—Genotype Integrator (PheGenI): Synthesizing genome-wide association study (GWAS) data with existing genomic resources. Eur. J. Hum. Genet. 2014, 22, 144–147. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhu, J. Entanglement of CCR5 and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 209. [Google Scholar] [CrossRef]
- Famulla, S.; Lamers, D.; Hartwig, S.; Passlack, W.; Horrighs, A.; Cramer, A.; Lehr, S.; Sell, H.; Eckel, J. Pigment epithelium-derived factor (PEDF) is one of the most abundant proteins secreted by human adipocytes and induces insulin resistance and inflammatory signaling in muscle and fat cells. Int. J. Obes. 2011, 35, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Araki, M. Tumor suppressor PTEN: Modulator of cell signaling, growth, migration and apoptosis. J. Cell Sci. 2001, 114, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Koehn, L.M.; Huang, Y.; Habgood, M.D.; Kysenius, K.; Crouch, P.J.; Dziegielewska, K.M.; Saunders, N.R. Effects of paracetamol (acetaminophen) on gene expression and permeability properties of the rat placenta and fetal brain. F1000Research 2020, 9, 573. [Google Scholar] [CrossRef] [PubMed]
- Dudognon, C.; Pendino, F.; Hillion, J.; Saumet, A.; Lanotte, M.; Segal-Bendirdjian, E. Death receptor signaling regulatory function for telomerase: hTERT abolishes TRAIL-induced apoptosis, independently of telomere maintenance. Oncogene 2004, 23, 7469–7474. [Google Scholar] [CrossRef][Green Version]
- Wortzel, I.; Seger, R. The ERK cascade: Distinct functions within various subcellular organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Posadas, I.; Santos, P.; Blanco, A.; Muñoz-Fernández, M.; Ceña, V. Acetaminophen induces apoptosis in rat cortical neurons. PLoS ONE 2010, 5, e15360. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, C.I.; Pérez, M.J.; Manautou, J.E.; Mottino, A.D. Acetaminophen from liver to brain: New insights into drug pharmacological action and toxicity. Pharmacol. Res. 2016, 109, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Buzanska, L.; Sypecka, J.; Nerini-Molteni, S.; Compagnoni, A.; Hogberg, H.T.; del Torchio, R.; Domanska-Janik, K.; Zimmer, J.; Coecke, S. A human stem cell-based model for identifying adverse effects of organic and inorganic chemicals on the developing nervous system. Stem Cells 2009, 27, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Bohuslavova, R.; Cerychova, R.; Papousek, F.; Olejnickova, V.; Bartos, M.; Görlach, A.; Kolar, F.; Sedmera, D.; Semenza, G.L.; Pavlinkova, G. HIF-1α is required for development of the sympathetic nervous system. Proc. Natl. Acad. Sci. USA 2019, 116, 13414–13423. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Liu, X.; Lu, F.; Zhang, Y.; Jiang, X.; Ferriero, D.M. HIF1α Signaling in the Endogenous Protective Responses after Neonatal Brain Hypoxia-Ischemia. Dev. Neurosci. 2018, 40, 617–626. [Google Scholar] [CrossRef]
- De Souza, J.S.; Laureano-Melo, R.; Herai, R.H.; da Conceição, R.R.; Oliveira, K.C.; da Silva, I.; Dias-da-Silva, M.R.; Romano, R.M.; Romano, M.A.; Maciel, R.M.B.; et al. Maternal glyphosate-based herbicide exposure alters antioxidant-related genes in the brain and serum metabolites of male rat offspring. Neurotoxicology 2019, 74, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.N.; Vaibhav, K.; Bastia, B.; Singh, V.; Ahluwalia, M.; Agrawal, U.; Borgohain, D.; Raisuddin, S.; Jain, A.K. Occupational exposure to pesticides in female tea garden workers and adverse birth outcomes. J. Biochem. Mol. Toxicol. 2020, 35, e22677. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Alberts, I.; Li, X. The apoptotic perspective of autism. Int. J. Dev. Neurosci. 2014, 36, 13–18. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Donahower, B.; Burke, A.S.; McCullough, S.; Hinson, J.A. Induction of the nuclear factor HIF-1α in acetaminophen toxicity: Evidence for oxidative stress. Biochem. Biophys. Res. Commun. 2006, 343, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, A.K.; Szychowski, K.A.; Wnuk, A.; Kajta, M. Dibutyl phthalate (DBP)-induced apoptosis and neurotoxicity are mediated via the aryl hydrocarbon receptor (AhR) but not by estrogen receptor alpha (ERα), estrogen receptor beta (ERβ), or peroxisome proliferator-activated receptor gamma (PPARγ) in mouse cortical neurons. Neurotox. Res. 2017, 31, 77–89. [Google Scholar]
- Kajta, M.; Wójtowicz, A.K.; Maćkowiak, M.; Lasoń, W. Aryl hydrocarbon receptor-mediated apoptosis of neuronal cells: A possible interaction with estrogen receptor signaling. Neuroscience 2009, 158, 811–822. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hatabayashi, K.; Arita, M.; Yajima, N.; Takenaka, C.; Suzuki, T.; Takahashi, M.; Oshima, Y.; Hara, K.; Kagawa, K. Kynurenine signaling through the aryl hydrocarbon receptor maintains the undifferentiated state of human embryonic stem cells. Sci. Signal. 2019, 12, eaaw3306. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.U.; McPherson, Z.E.; Tan, B.; Korecka, A.; Pettersson, S. Host-microbiome interactions: The aryl hydrocarbon receptor and the central nervous system. J. Mol. Med. 2017, 95, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Mandic-Maravic, V.; Mitkovic-Voncina, M.; Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Djordjevic, M.; Pekmezovic, T.; Grujicic, R.; Ercegovac, M.; Simic, T.; Lecic-Tosevski, D.; et al. Autism Spectrum Disorders and Perinatal Complications—Is Oxidative Stress the Connection? Front. Psychiatry 2019, 10, 675. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharm. Genom. 2015, 25, 416. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, K.; Hatano, E.; Iwaisako, K.; Takeiri, M.; Noma, N.; Ohmae, S.; Toriguchi, K.; Tanabe, K.; Tanaka, H.; Seo, S. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio 2014, 4, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Misra, H.P. Reactive oxygen species in in vitro pesticide-induced neuronal cell (SH-SY5Y) cytotoxicity: Role of NFκB and caspase-3. Free Radic. Biol. Med. 2007, 42, 288–298. [Google Scholar] [CrossRef]
- Fotio, A.; Nguepi, M.; Tonfack, L.; Temdie, R.; Nguelefack, T. Acetaminophen induces liver injury and depletes glutathione in mice brain: Prevention by Moringa oleifera extract. S. Afr. J. Bot. 2020, 129, 317–323. [Google Scholar] [CrossRef]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Castora, F.J. Mitochondrial function and abnormalities implicated in the pathogenesis of ASD. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2019, 92, 83–108. [Google Scholar] [CrossRef] [PubMed]
- Steiner, P. Brain fuel utilization in the developing brain. Ann. Nutr. Metab. 2019, 75, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Coleman, M.; Blass, J.P. Autism and lactic acidosis. J. Autism Dev. Disord. 1985, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bonvallot, N.; Canlet, C.; Blas-Y-Estrada, F.; Gautier, R.; Tremblay-Franco, M.; Chevolleau, S.; Cordier, S.; Cravedi, J.-P. Metabolome disruption of pregnant rats and their offspring resulting from repeated exposure to a pesticide mixture representative of environmental contamination in Brittany. PLoS ONE 2018, 13, e0198448. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Bui, L.-C.; Petit, E.; Haddad, I.; Agbulut, O.; Vinh, J.; Dupret, J.-M.; Rodrigues-Lima, F. Molecular mechanisms of allosteric inhibition of brain glycogen phosphorylase by neurotoxic dithiocarbamate chemicals. J. Biol. Chem. 2017, 292, 1603–1612. [Google Scholar] [CrossRef]
- Mencarelli, A.; Fiorucci, S. FXR an emerging therapeutic target for the treatment of atherosclerosis. J. Cell. Mol. Med. 2010, 14, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Lu, Y.; Li, X.Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, K.-J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24. [Google Scholar] [CrossRef] [PubMed]
- Steinman, G.; Mankuta, D. Molecular biology of autism’s etiology—An alternative mechanism. Med. Hypotheses 2019, 130, 109272. [Google Scholar] [CrossRef]
- Geschwind, D.H. Autism: Many genes, common pathways? Cell 2008, 135, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.J.; Blizard, R. Autism genes are selectively targeted by environmental pollutants including pesticides, heavy metals, bisphenol A, phthalates and many others in food, cosmetics or household products. Neurochem. Int. 2016, 101, 83–109. [Google Scholar] [CrossRef] [PubMed]
- Lammert, D.B.; Howell, B.W. RELN mutations in autism spectrum disorder. Front. Cell. Neurosci. 2016, 10, 84. [Google Scholar] [CrossRef] [PubMed]





| Ingenuity Canonical Pathways | p-Value | Genes |
|---|---|---|
| PXR/RXR Activation | 4.47 × 10−7 | ABCB1, CYP1A2, CYP3A4 |
| Xenobiotic Metabolism CAR Signaling Pathway | 1.12 × 10−5 | ABCB1, CYP1A2, CYP3A4 |
| Bupropion Degradation | 1.74 × 10−5 | CYP1A2, CYP3A4 |
| Acetone Degradation I (to Methylglyoxal) | 2.51 × 10−5 | CYP1A2, CYP3A4 |
| Xenobiotic Metabolism Signaling | 3.89 × 10−5 | ABCB1, CYP1A2, CYP3A4 |
| Estrogen Biosynthesis | 4.79 × 10−5 | CYP1A2, CYP3A4 |
| Aryl Hydrocarbon Receptor Signaling | 5.75 × 10−4 | CYP1A2, FAS |
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | 9.77 × 10−4 | FAS, IGF1R |
| Xenobiotic Metabolism PXR Signaling Pathway | 1.05 × 10−3 | ABCB1, CYP3A4 |
| LPS/IL-1 Mediated Inhibition of RXR Function | 1.41 × 10−3 | ABCB1, CYP3A4 |
| Ingenuity Canonical Pathways | p-Value | Genes |
|---|---|---|
| Acute Phase Response Signaling | 6.306 × 10−6 | AGT, F2, FN1, TNF |
| Hepatic Cholestasis | 7.244 × 10−6 | GCG, IL2, INS, TNF |
| Telomerase Signaling | 5.494 × 10−5 | EGFR, IL2, TP53 |
| Type I Diabetes Mellitus Signaling | 6.026 × 10−5 | IL2, INS, TNF |
| Estrogen Receptor Signaling | 6.768 × 10−5 | AGT, EGFR, TP53, VEGFA |
| FXR/RXR Activation | 8.91 × 10−5 | AGT, INS, TNF |
| NF-κB Signaling | 2.51 × 10−4 | EGFR, INS, TNF |
| mTOR Signaling | 3.98 × 10−4 | INS, TSC1, VEGFA |
| Hematopoiesis from Pluripotent Stem Cells | 5.37 × 10−4 | CD4, IL2 |
| Myc Mediated Apoptosis Signaling | 5.62 × 10−4 | TNF, TP53 |
| PXR/RXR Activation | 9.33 × 10−4 | INS, TNF |
| Sirtuin Signaling Pathway | 1.02 × 10−3 | APP, TNF, TP53 |
| VDR/RXR Activation | 1.35 × 10−3 | IL2, PTH |
| Agrin Interactions at Neuromuscular Junction | 1.39 × 10−3 | EGFR, ERBB2 |
| Glucocorticoid Receptor Signaling | 1.55 × 10-3 | AGT, IL2, TNF |
| Allograft Rejection Signaling | 1.62 × 10−3 | IL2, TNF |
| Crosstalk between Dendritic Cells and Natural Killer Cells | 1.78 × 10−3 | IL2, TNF |
| OX40 Signaling Pathway | 1.82 × 10-3 | CD4, IL2 |
| ErbB Signaling | 1.95 × 10−3 | EGFR, ERBB2 |
| Neuregulin Signaling | 2.04 × 10−3 | EGFR, ERBB2 |
| Apoptosis Signaling | 2.19 × 10−3 | TNF, TP53 |
| PPAR Signaling | 2.40 × 10−3 | INS, TNF |
| HIF1α Signaling | 2.82 × 10−3 | TP53, VEGFA |
| Neuroprotective Role of THOP1 in Alzheimer’s Disease | 3.02 × 10−3 | AGT, APP |
| Insulin Receptor Signaling | 4.27 × 10−3 | INS, TSC1 |
| Type II Diabetes Mellitus Signaling | 4.37 × 10−3 | INS, TNF |
| Necroptosis Signaling Pathway | 5.37 × 10−3 | TNF, TP53 |
| Mitochondrial Dysfunction | 6.31 × 10−3 | APP, CAT |
| PI3K/AKT Signaling | 6.61 × 10−3 | TP53, TSC1 |
| Canonical Pathways | Molecular Database Analysis p-Value | Literature Analysis p-Value |
|---|---|---|
| Aryl Hydrocarbon Receptor Signaling | 9.79 × 10−4 | 8.86 × 10−5 |
| CCR5 Signaling in Macrophages | 2.69 × 10−2 | 1.69 × 10−2 |
| Death Receptor Signaling | 2.87 × 10−2 | 1.85 × 10−2 |
| FXR/RXR Activation | 2.25 × 10−2 | 4.46 × 10−3 |
| LPS/IL-1 Mediated Inhibition of RXR Function | 9.69 × 10−4 | 6.78 × 10−5 |
| Myc Mediated Apoptosis Signaling | 1.29 × 10−2 | 5.59 × 10−4 |
| NF-κB Signaling | 1.60 × 10−2 | 1.76 × 10−3 |
| p53 Signaling | 3.05 × 10−2 | 4.01 × 10−2 |
| PEDF Signaling | 2.59 × 10−2 | 1.18 × 10−2 |
| PTEN Signaling | 2.38 × 10−2 | 1.05 × 10−2 |
| PXR/RXR Activation | 4.45 × 10−7 | 9.43 × 10−4 |
| Type I Diabetes Mellitus Signaling | 2.15 × 10−2 | 2.17 × 10−3 |
| Xenobiotic Metabolism CAR Signaling Pathway | 1.40 × 10−3 | 1.97 × 10−4 |
| Xenobiotic Metabolism PXR Signaling Pathway | 5.81 × 10−4 | 6.08 × 10−5 |
| Xenobiotic Metabolism Signaling | 1.04 × 10−3 | 1.50 × 10−4 |
| Source of Association | Official Gene Symbol | Name | NCBI Gene Description/Function |
|---|---|---|---|
| Molecular databases | ABCB1 | ATP Binding Cassette subfamily B member 1 | multidrug resistance, and ATP-dependent drug efflux pumps for xenobiotic compounds; transporter in the blood-brain barrier |
| ABCB4 | ATP Binding Cassette subfamily B member 4 | ||
| CYP1A2 | Cytochrome P450 Family 1 Subfamily A Member 2 | catalyzes many reactions involved in drug metabolism and the synthesis of cholesterol, steroids and other lipids | |
| CYP3A4 | Cytochrome P450 Family 3 Subfamily A Member 4 | metabolizes steroids as well as carcinogens, involved in the metabolism of approximately half of all drugs currently in use | |
| FAS | Fas Cell Surface Death Receptor | contains a death domain, plays a central role in the physiological regulation of programmed cell death, involved in transducing the proliferating signals in normal diploid fibroblast and T cells | |
| IGF1R | Insulin like Growth Factor Receptor 1 | binds insulin-like growth factor, highly overexpressed in most malignant tissues, functions as anti-apoptotic agent by enhancing cell survival | |
| Literature | CAT | Catalase | enzyme that protects cells from ROS-induced oxidative damage |
| CD4 | Cluster of differentiation 4 | membrane glycoprotein of T lymphocytes | |
| TNF | Tumor Necrosis Factor | triggers activation of the MLKL cascade which is critical in the generation of ROS | |
| VEGFA | Vascular Endothelial Growth Factor A | encodes heparin-binding protein, induces proliferation and migration of vascular endothelial cells | |
| EGFR | Epidermal Growth Factor Receptor | encodes for a transmembrane glycoprotein that is a member of the protein kinase superfamily | |
| INS | Insulin | peptide hormone, plays major role in regulating carbohydrate and lipid metabolism | |
| AGT | Angiotensinogen | codes for a liver protein involved in maintaining blood pressure | |
| IL2 * | Interleukin 2 | encodes for a protein important in the proliferation of B and T lymphocytes | |
| FN1 | Fibronectin 1 | codes for fibronectin, a protein involved in cell adhesion and migration processes including embryogenesis | |
| PTH | Parathyroid Hormone | encodes a preproprotein that is proteolytically processed to a protein involved in parathyroid hormone signaling | |
| TP53 | Tumor Protein p53 | tumor repressor protein involved in cellular stress responses that can induce cell cycle arrest, apoptosis, senescence, DNA repair, and changes in metabolism | |
| ERBB2 | erb-b2 Receptor Tyrosine Kinase 2 | encodes a member of the epidermal growth factor receptor family | |
| GCG | Glucagon | stimulates glycogenolysis and gluconeogenesis | |
| TSC1 | TSC Complex Subunit 1 | encodes hamartin, a growth inhibitory protein | |
| APP | Amyloid Beta Precursor Protein | serves as a cell surface receptor and transmembrane protein that is cleaved to form several types of peptides | |
| F2 | Coagulation Factor II, Thrombin | encodes the prothrombin protein that is cleaved in several steps to generate thrombin, a protein that plays a role in cell proliferation, tissue repair, and maintaining vasculature during perinatal development |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furnary, T.; Garcia-Milian, R.; Liew, Z.; Whirledge, S.; Vasiliou, V. In Silico Exploration of the Potential Role of Acetaminophen and Pesticides in the Etiology of Autism Spectrum Disorder. Toxics 2021, 9, 97. https://doi.org/10.3390/toxics9050097
Furnary T, Garcia-Milian R, Liew Z, Whirledge S, Vasiliou V. In Silico Exploration of the Potential Role of Acetaminophen and Pesticides in the Etiology of Autism Spectrum Disorder. Toxics. 2021; 9(5):97. https://doi.org/10.3390/toxics9050097
Chicago/Turabian StyleFurnary, Tristan, Rolando Garcia-Milian, Zeyan Liew, Shannon Whirledge, and Vasilis Vasiliou. 2021. "In Silico Exploration of the Potential Role of Acetaminophen and Pesticides in the Etiology of Autism Spectrum Disorder" Toxics 9, no. 5: 97. https://doi.org/10.3390/toxics9050097
APA StyleFurnary, T., Garcia-Milian, R., Liew, Z., Whirledge, S., & Vasiliou, V. (2021). In Silico Exploration of the Potential Role of Acetaminophen and Pesticides in the Etiology of Autism Spectrum Disorder. Toxics, 9(5), 97. https://doi.org/10.3390/toxics9050097