
Oxidative Stress and Cocaine Intoxication as Start Points in the Pathology of Cocaine-Induced Cardiotoxicity

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
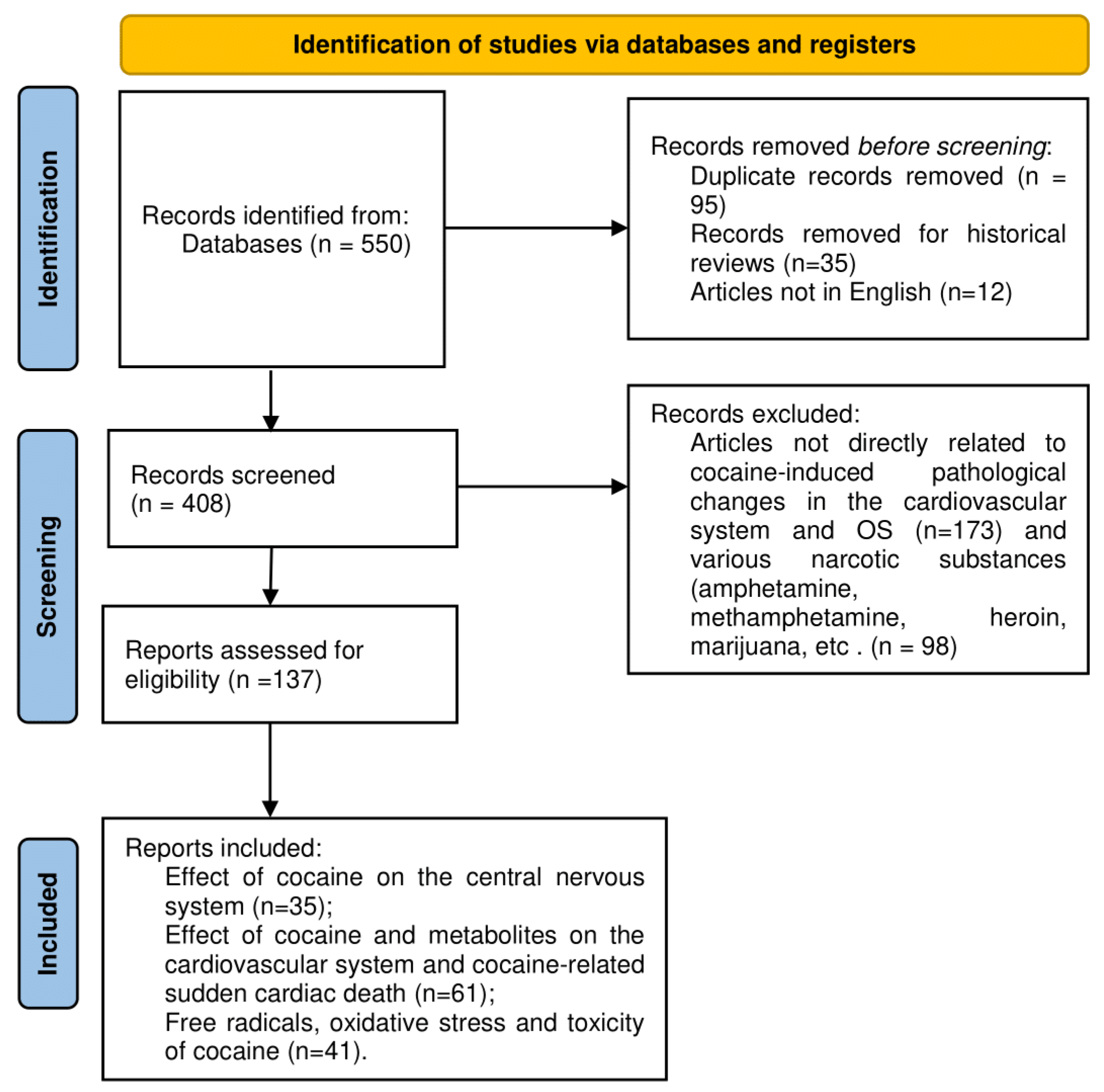
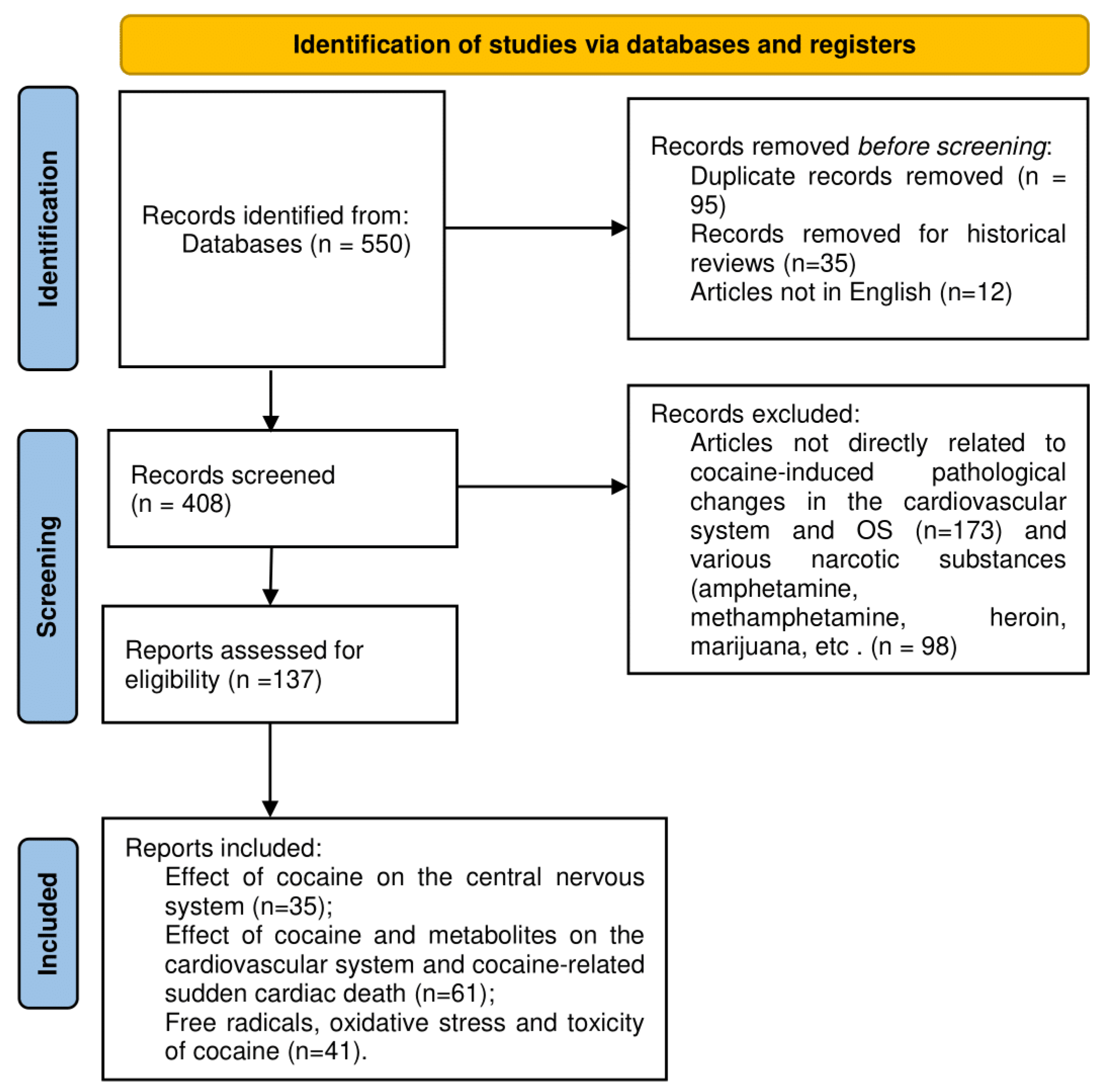
2. Materials and Methods
2.1. Search of the Literature, Materials, and Methods
2.2. Inclusion Criteria
2.3. Exclusion Criteria
3. Effect of Cocaine on Central Neural System and Cardiovascular System
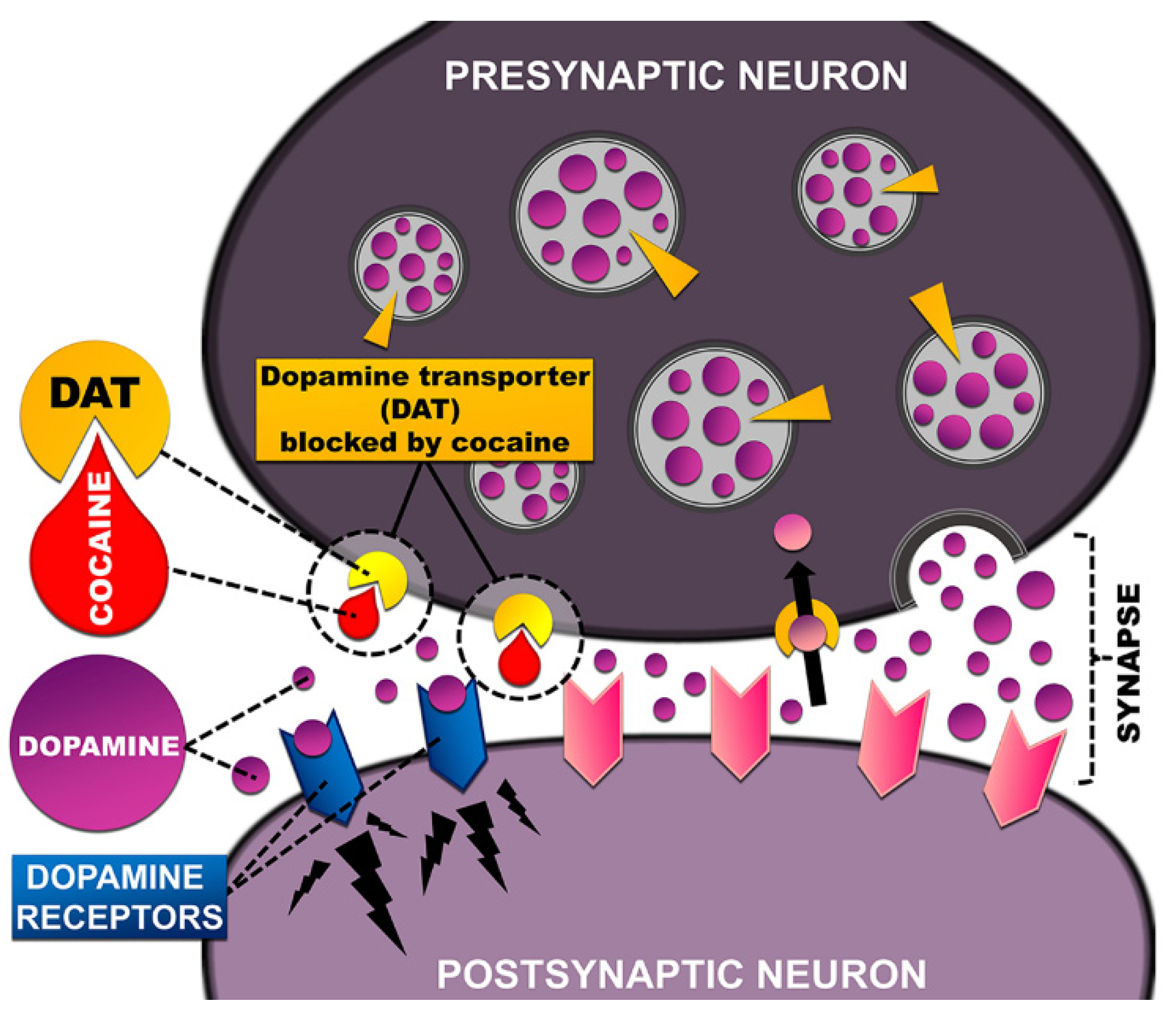
3.1. Cocaine Action on the Brain System
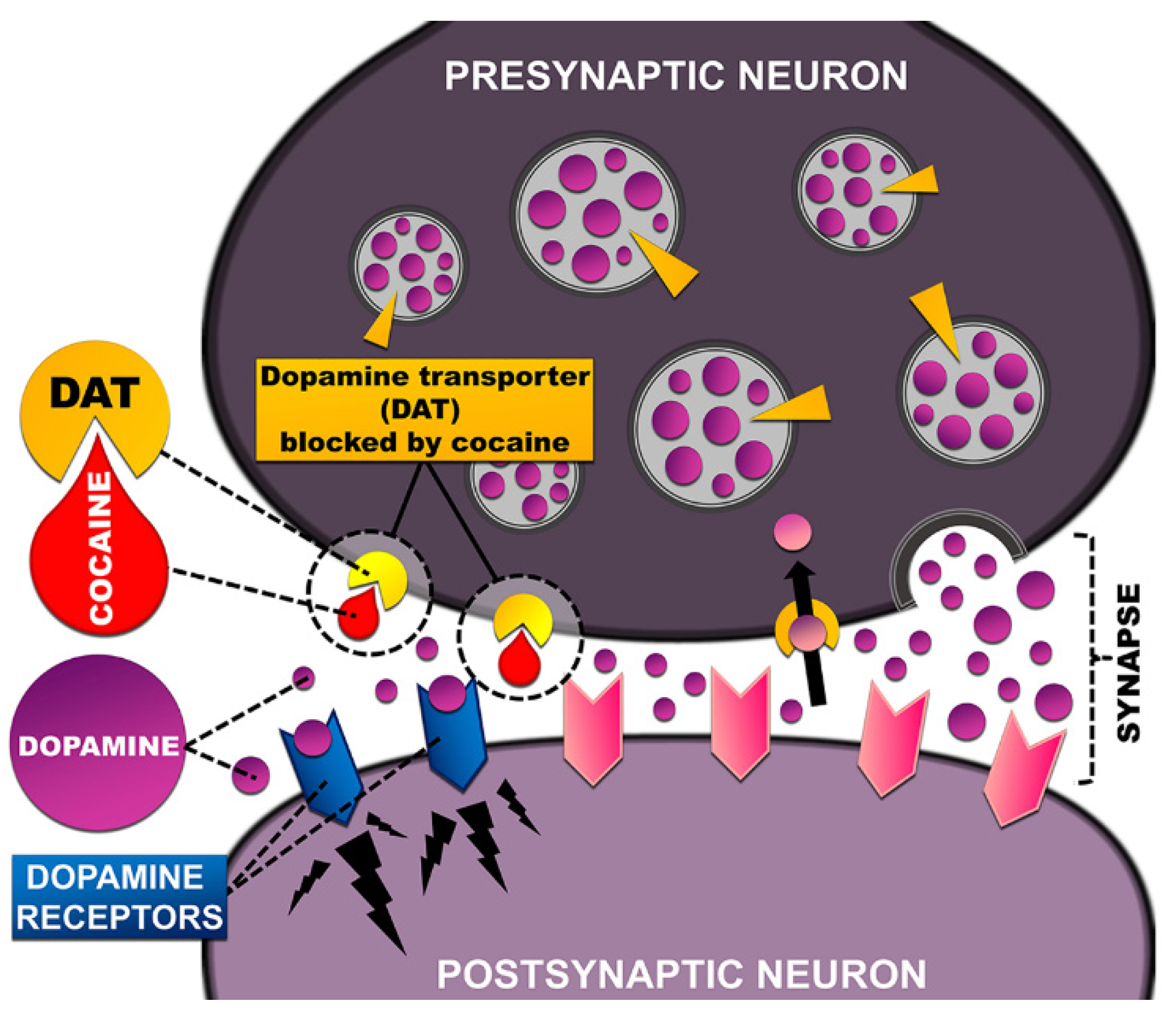
3.2. Dopamine Transporter—The Main Target of Stimulants
4. Cocaine-Related Sudden Cardiac Death
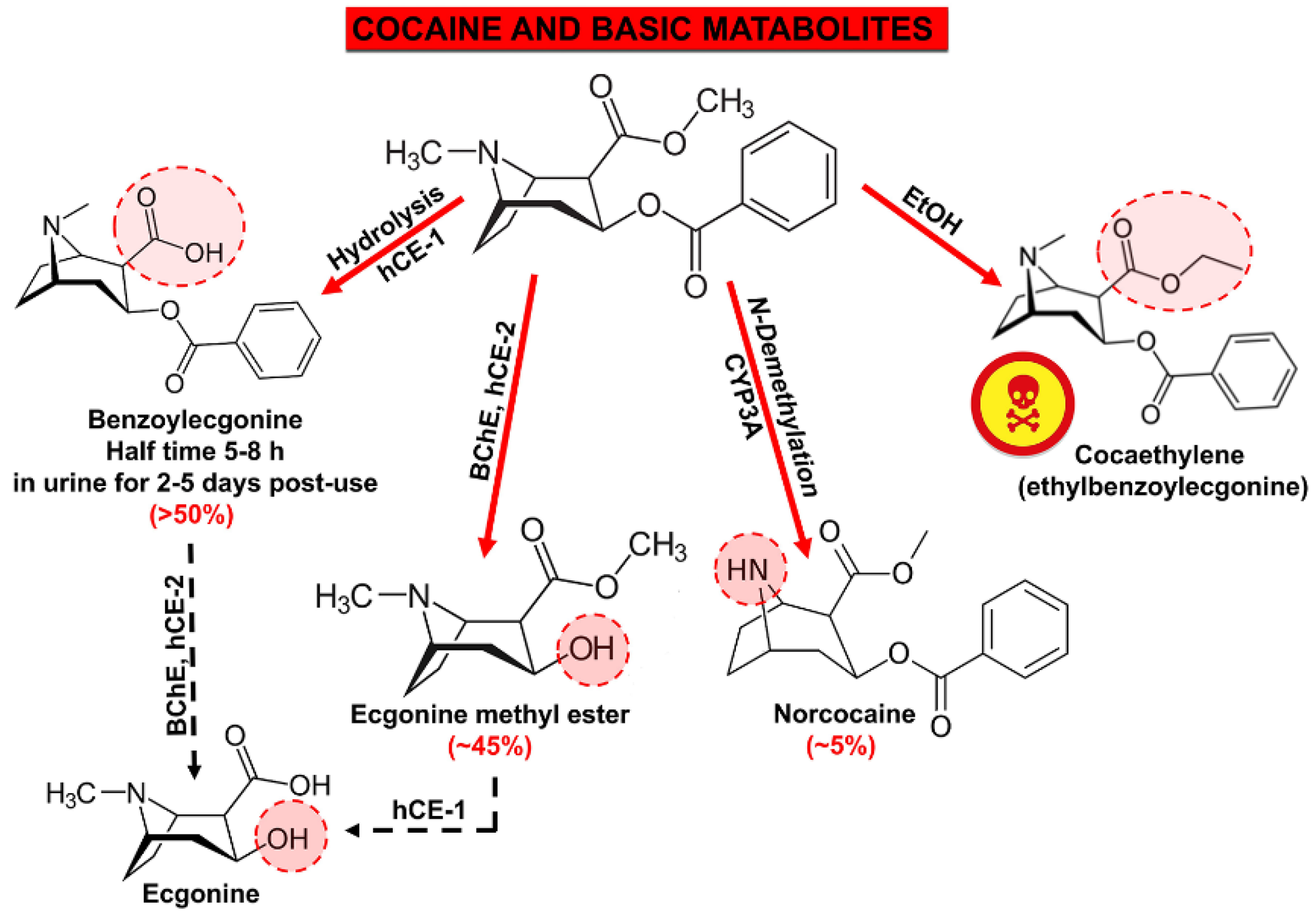
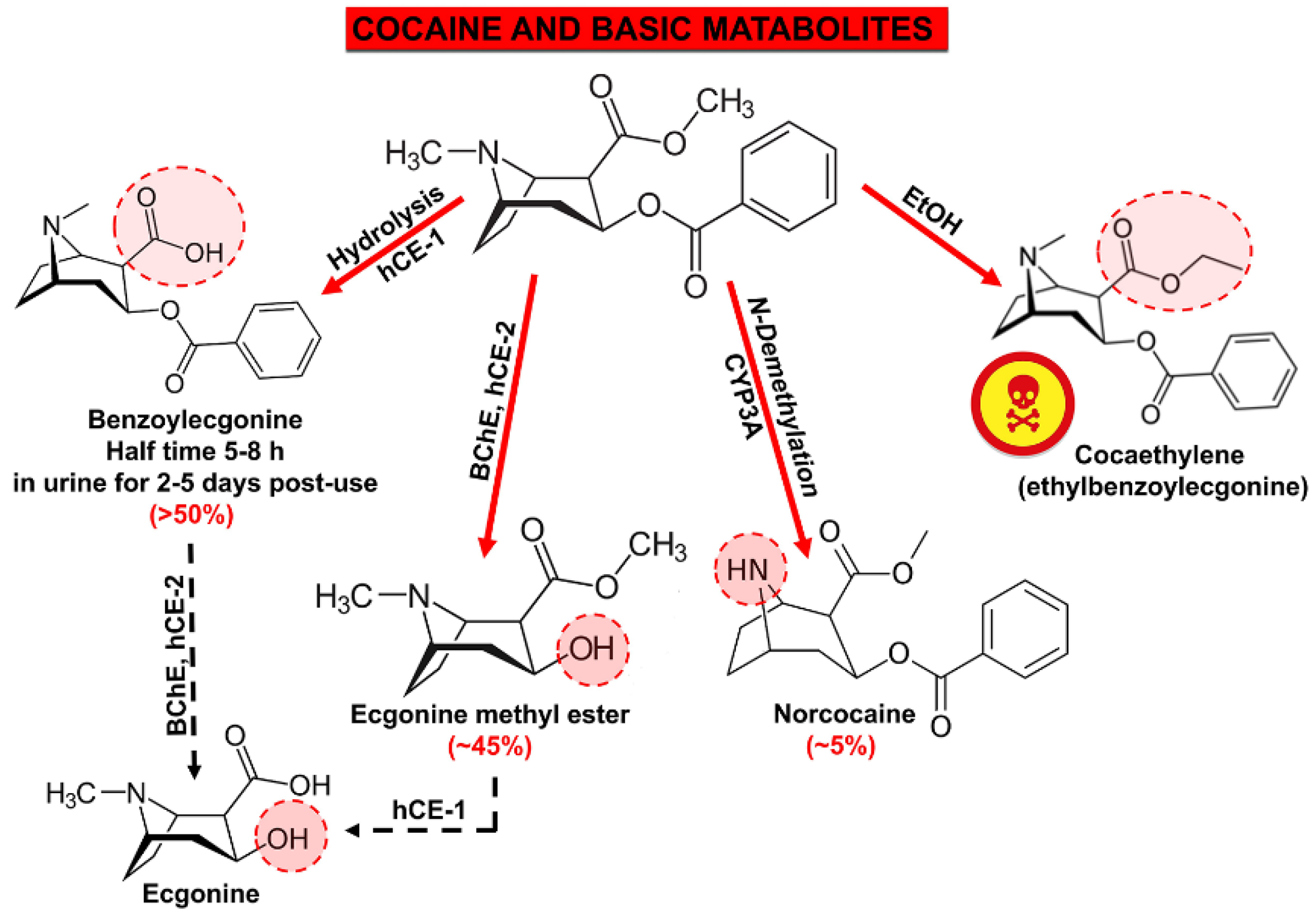
5. Metabolites of Cocaine with Expressed Cardiotoxicity
6. Acute and Chronic Effects of Cocaine Cardiovascular Toxicity and Pathological Changes in the Cardiovascular System
6.1. Acute Toxicity of Cocaine. Myocardial Infarction, Arrhythmias, Sudden Cardiac Death
6.2. Chronic Toxicity of Cocaine
6.2.1. Thrombosis
6.2.2. Atherosclerosis and Myocardial Infarction
6.2.3. Left Ventricular Hypertrophy and Dilated Cardiomyopathy
6.2.4. Myocarditis
6.2.5. Endocardities
6.2.6. Aortic Dissection
7. Free Radicals, Oxidative Stress, and Cocaine-Induced Cardio Toxicity
7.1. The Basic Pathways of Cocaine Cardiotoxicity
7.2. Enzymatic and Non-Enzymatic Pathways of ROS Cardiotoxicity
7.3. Mitochondrial Dysfunction and Nitrosative/Oxidative Stress
7.4. ROS Production from Endothelial Cells
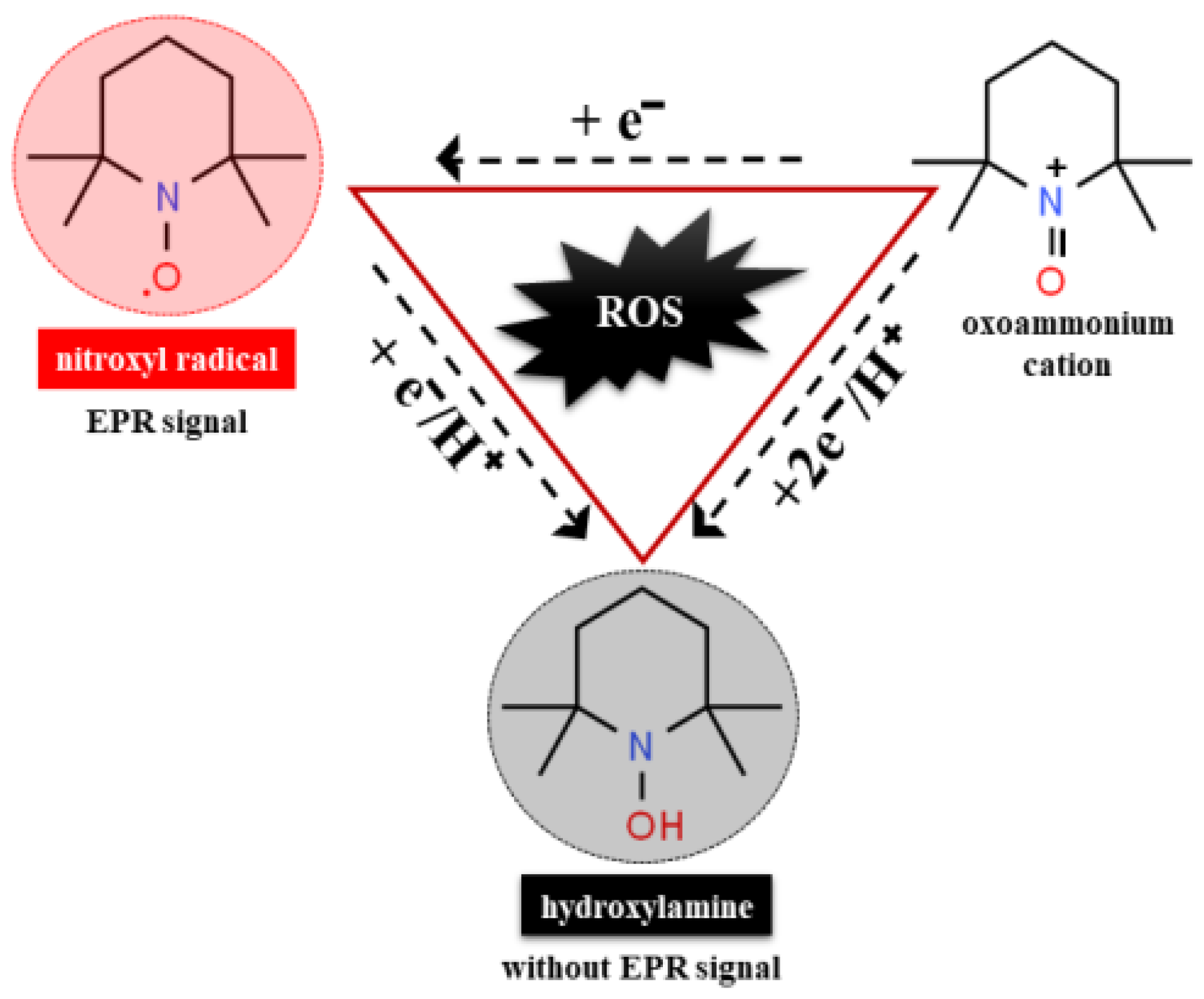
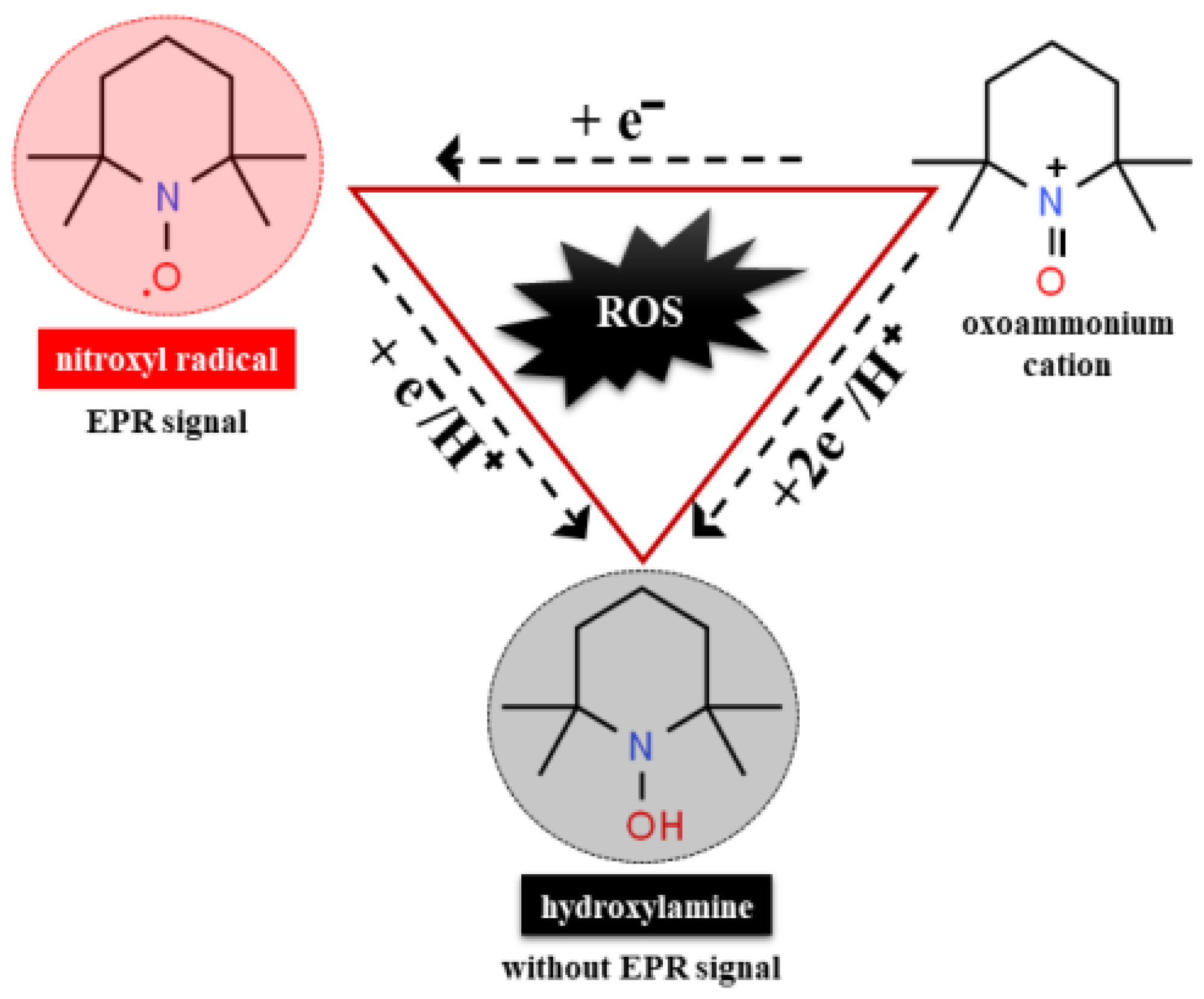
7.5. Nitroxide Radicals as Detector of Oxidative Stress in Human Body
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Declarations
References
- Howell, L.L.; Kimmel, H.L. Monoamine transporters and psychostimulant addiction. Biochem. Pharmacol. 2008, 75, 196–217. [Google Scholar] [CrossRef]
- Ciccarone, D. Stimulant abuse: Pharmacology, cocaine, methamphetamine, treatment, attempts at pharmacotherapy. Prim. Care Clin. Off. Pract. 2011, 38, 41–58. [Google Scholar] [CrossRef] [Green Version]
- Konova, A.B.; Moeller, S.J.; Tomasi, D.; Goldstein, R.Z. Effects of chronic and acute stimulants on brain functional connectivity hubs. Brain Res. 2015, 1628, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Klimas, J. Drug-Induced Cardiomyopathies; Intech Open Access Publisher: London, UK, 2012; pp. 581–607. [Google Scholar]
- Malacarne, I.T.; De Souza, D.V.; Rosario, B.D.A.; Viana, M.D.B.; Pereira, C.D.S.; Estadella, D.; dos Santos, J.N.; Ribeiro, D.A. Genotoxicity, oxidative stress, and inflammatory response induced by crack-cocaine: Relevance to carcinogenesis. Environ. Sci. Pollut. Res. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.; Luk, A.; Soor, G.S.; Abraham, J.R.; Leong, S.; Butany, J. Cocaine cardiotoxicity: A review of the pathophysiology, pathology, and treatment options. Am. J. Cardiovasc. Drugs 2009, 9, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P.; Wellens, H.J. Sudden cardiac death. In Professor: 33 Years of Cardiology and Arrhythmology: 33 Years of Cardiology and Arrhythmology; Smeets, J.L.R.M., Doevendans, P.A., Josephson, M.E., Kirchhof, C., Vos, M.A., Eds.; Springer: Dordrecht, The Netherlands, 2000. [Google Scholar] [CrossRef]
- Chugh, S.S.; Reinier, K.; Teodorescu, C.; Evanado, A.; Kehr, E.; Al Samara, M.; Mariani, R.; Gunson, K.; Jui, J. Epidemiology of sudden cardiac death: Clinical and research implications. Prog. Cardiovasc. Dis. 2008, 51, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Dugo, E.; Barison, A.; Todiere, G.; Grigoratos, C.; Aquaro, G.D. Cardiac magnetic resonance in cocaine-induced myocardial damage: Cocaine, heart, and magnetic resonance. Heart Fail. Rev. 2020, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Elkattawy, S.; Alyacoub, R.; Al-Nassarei, A.; Younes, I.; Ayad, S.; Habib, M. Cocaine induced heart failure: Report and literature review. J. Community Hosp. Intern. Med. Perspect. 2021, 11, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Ghuran, A.; Nolan, J. The cardiac complications of recreational drug use. West. J. Med. 2000, 173, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pergolizzi, J.V., Jr.; Magnusson, P.; LeQuang, J.A.K.; Breve, F.; Varrassi, G. Cocaine and Cardiotoxicity: A Literature Review. Cureus 2021, 13, 4. [Google Scholar] [CrossRef]
- Reese, J.T. The pharmacology of cocaine. Cocaine: Pharmacology, Effects and Treatment of Abuse. Natl. Inst. Drug Abus. Res. Monogr. 1984, 50, 34–53. [Google Scholar]
- Schindler, C.W.; Tella, S.R.; Erzouki, H.K.; Goldberg, S.R. Pharmacological mechanisms in cocaine’s cardiovascular effects. Drug Alcohol Depend. 1995, 37, 183–191. [Google Scholar] [CrossRef]
- Schindler, C.W.; Zheng, J.W.; Goldberg, S.R. Effects of cocaine and cocaine metabolites on cardiovascular function in squirrel monkeys. Eur. J. Pharmacol. 2001, 431, 53–59. [Google Scholar] [CrossRef]
- Beiser, T.; Yaka, R. The role of oxidative stress in cocaine addiction. J. Neurol. Neuromed. 2019, 4, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, P. Role of oxidative metabolites of cocaine in toxicity and addiction: Oxidative stress and electron transfer. Med. Hypotheses 2005, 64, 350–356. [Google Scholar] [CrossRef]
- Cerretani, D.; Fineschi, V.; Bello, S.; Riezzo, I.; Turillazzi, E.; Neri, M. Role of oxidative stress in cocaine-induced cardiotoxicity and cocaine-related death. Curr. Med. Chem. 2012, 19, 5619–5623. [Google Scholar] [CrossRef] [PubMed]
- Graziani, M.; Sarti, P.; Arese, M.; Magnifico, M.C.; Badiani, A.; Saso, L. Cardiovascular Mitochondrial Dysfunction Induced by Cocaine: Biomarkers and Possible Beneficial Effects of Modulators of Oxidative Stress. Oxidative Med. Cell. Longev. 2017, 2017, 3034245. [Google Scholar] [CrossRef]
- Graziani, M.; Antonilli, L.; Togna, A.R.; Grassi, M.C.; Badiani, A.; Saso, L. Cardiovascular and Hepatic Toxicity of Cocaine: Potential Beneficial Effects of Modulators of Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 8408479. [Google Scholar] [CrossRef] [Green Version]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA statement. Ann. Intern. Med. 2009, 151, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok, A.H.; Mizuno, Y.; Volkow, N.D.; Howes, O.D. Association of Stimulant Use With Dopaminergic Alterations in Users of Cocaine, Amphetamine, or Methamphetamine: A Systematic Review and Meta-analysis. JAMA Psychiatry 2017, 74, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Yuen, J.; Goyal, A.; Rusheen, A.E.; Kouzani, A.Z.; Berk, M.; Kim, J.H.; Tye, S.J.; Blaha, C.D.; Bennet, K.E.; Jang, D.P.; et al. Cocaine-induced changes in tonic dopamine concentrations measured using multiple-cyclic square wave voltammetry in vivo. Front. Pharmacol. 2021, 12, 705254. [Google Scholar] [CrossRef]
- Harraz, M.M.; Guha, P.; Kang, I.G.; Semenza, E.R.; Malla, A.P.; Song, Y.J.; Reilly, L.; Treisman, I.; Cortés, P.; Coggiano, M.A.; et al. Cocaine-induced locomotor stimulation involves autophagic degradation of the dopamine transporter. Mol. Psychiatry 2021, 26, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Afonso, L.; Mohammad, T.; Thatai, D. Crack whips the heart: A review of the cardiovascular toxicity of cocaine. Am. J. Cardiol. 2007, 100, 1040–1104. [Google Scholar] [CrossRef] [PubMed]
- Docherty, J.R.; Alsufyani, H.A. Pharmacology of drugs used as stimulants. J. Clin. Pharmacol. 2021, 61, S53–S69. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F. Drugs of abuse: Anatomy, pharmacology and function of reward pathways. Trends Pharmacol. Sci. 1992, 13, 177–184. [Google Scholar] [CrossRef]
- Volkow, N.; Michaelides, M.; Baler, R. The Neuroscience of Drug Reward and Addiction. Physiol. Rev. 2019, 99, 2115–2140. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef] [Green Version]
- Dela Peña, I.; Gevorkiana, R.; Shi, W.X. Psychostimulants affect dopamine transmission through both dopamine transporter-dependent and independent mechanisms. Eur. J. Pharmacol. 2015, 764, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.; Fowler, J.; Wang, G.J.; Swanson, J.M.; Telang, F. Dopamine in drug abuse and addiction: Results from imaging studies and treatment implications. Mol. Psychiatry 2004, 9, 557–569. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Kim, K.; Zheng, X.; Shang, L.; Zhan, C.G.; Zheng, F. Cocaine hydrolase blocks cocaine-induced dopamine transporter trafficking to the plasma membrane. Addict. Biol. 2021, 13089. [Google Scholar] [CrossRef]
- Adinoff, B. Neurobiologic processes in drug reward and addiction. Harv. Rev. Psychiatry 2004, 12, 305–320. [Google Scholar] [CrossRef]
- NIDA. How Does Cocaine Produce Its Effects? Available online: https://www.drugabuse.gov/publications/research-reports/cocaine/how-does-cocaine-produce-its-effects (accessed on 22 November 2021).
- Dackis, C.A.; Gold, M.S. New concepts in cocaine addiction: The dopamine depletion hypothesis. Neurosci. Biobehav. Rev. 1985, 9, 469–477. [Google Scholar] [CrossRef]
- Heard, K.; Palmer, R.; Zahniser, N.R. Mechanisms of acute cocaine toxicity. Open Pharmacol. J. 2008, 2, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Grewen, K.; Burchinal, M.; Vachet, C.; Gouttard, S.; Gilmore, J.H.; Lin, W.; Johns, J.; Elam, M.; Gerig, G. Prenatal cocaine effects on brain structure in early infancy. Neuroimage 2014, 101, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pramanik, P.; Vidua, R.K. Cocaine Cardiac Toxicity: Revisited. In Cardiotoxicity; IntechOpen: Rijeka, Croatia, 2018. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Wang, G.J.; Fischman, M.W.; Foltin, R.; Fowler, J.S.; Franceschi, D.; Franceschi, M.; Logan, J.; Gatley, S.J.; Wong, C.; et al. Effects of route of administration on cocaine induced dopamine transporter blockade in the human brain. Life Sci. 2000, 67, 1507–1515. [Google Scholar] [CrossRef]
- Grace, A.A. The tonic/phasic model of dopamine system regulation and its implications for understanding alcohol and psychostimulant craving. Addiction 2000, 95, S119–S128. [Google Scholar] [CrossRef]
- Calipari, E.S.; Ferris, M.J. Amphetamine mechanisms and actions at the dopamine terminal revisited. J. Neurosci. 2013, 33, 8923–8925. [Google Scholar] [CrossRef] [Green Version]
- Olguin, J.H.; Guzman, C.D.; Garcia, H.E.; Mejia, B.G. The role of dopamine and its dysfunction as a consequence of oxidative stress. Oxid. Med. Cell. Longev. 2016, 2016, 9730467. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, A.E.; Volz, T.J.; Riddle, E.L.; Gibb, J.W.; Hanson, G.R. New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 681–698. [Google Scholar] [CrossRef]
- Reith, M.E.A.; Gnegy, M.E. Molecular Mechanisms of Amphetamines. Handb. Exp. Pharmacol. 2020, 258, 265–297. [Google Scholar] [CrossRef] [PubMed]
- Bachi, K.; Mani, V.; Jeyachandran, D.; Fayad, Z.A.; Goldstein, R.Z.; Alia-Klein, N. Vascular disease in cocaine addiction. Atherosclerosis 2017, 262, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel, I.; Amorim, M.; Gonçalves, A.; Sousa, C.; Bettencourt, P.; Maciel, M.J. Toxic Dilated Cardiomyopathy: Recognizing a Potentially Reversible Disease. Arq. Bras. Cardiol. 2014, 102, e37–e39. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Orme Merve, A.; Remeškevičius, V.; Sobiecka, P.; Taylor, L.; Lawton, S.; Jones, B.P.; Polycarpou, E.; Bennett, J.; Rooney, B. Cocaine induces cytoskeletal changes in cardiac myocytes: Implications for cardiac morphology. Int. J. Mol. Sci. 2021, 22, 2263. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Park, T. Acute and Chronic Effects of Cocaine on Cardiovascular Health. Int. J. Mol. Sci. 2019, 20, 584. [Google Scholar] [CrossRef] [Green Version]
- Zaitsu, K.; Hayashi, Y.; Kusano, M.; Tsuchihashi, H.; Ishii, A. Application of metabolomics to toxicology of drugs of abuse: A mini review of metabolomics approach to acute and chronic toxicity studies. Drug Metab. Pharmacokinet. 2016, 31, 21–26. [Google Scholar] [CrossRef]
- Darke, S.; Kaye, S.; Duflou, J. Comparative cardiac pathology among deaths due to cocaine toxicity, opioid toxicity and non-drug-related causes. Addiction 2006, 101, 1771–1777. [Google Scholar] [CrossRef]
- Obeng-Okyere, P.; Sun, M.; Alcorn, K.L.; Haydel, J.M.; Mukerji, V. Cardiac Effects of Cocaine Use. J. Community Med. 2021, 4, 1030. [Google Scholar]
- Schwartz, B.G.; Rezkalla, S.; Kloner, R.A. Cardiovascular effects of cocaine. Circulation 2010, 122, 2558–2569. [Google Scholar] [CrossRef] [Green Version]
- Bolla, K.I.; Cadet, J.L.; London, E.D. The neuropsychiatry of chronic cocaine abuse. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 280–289. [Google Scholar] [CrossRef]
- Clergue-Duval, V.; Nicolas-Sacy, L.; Karsinti, E.; Zerdazi, E.H.; Laplanche, J.L.; Brousse, G.; Marees, A.T.; Derks, E.M.; Henry, P.; Bellivier, F.; et al. Risk and Protective Factors of Lifetime Cocaine-Associated Chest Pain. Front. Psychiatry 2021, 12, 704276. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Ding, Y.S. Cardiotoxic properties of cocaine: Studies with positron emission tomography. NIDA Res. Monogr. 1996, 163, 159–174. [Google Scholar] [PubMed]
- Hollander, J.E.; Hoffman, R.S. Cocaine-induced myocardial infarction: An analysis and review of the literature. J. Emerg. Med. 1992, 10, 169–177. [Google Scholar] [CrossRef]
- Crumb, W.J., Jr.; Clarkson, C.W. Characterization of cocaine-induced block of cardiac sodium channels. Biophys. J. 1990, 57, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Kloner, R.A.; Hale, S.; Alker, K.; Rezkalla, S. The effects of acute and chronic cocaine use on the heart. Circulation 1992, 85, 407–419. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, M.E.; Hancox, J.C. Role of voltage-gated sodium, potassium and calcium channels in the development of cocaine-associated cardiac arrhythmias. Br. J. Clin. Pharmacol. 2010, 69, 427–442. [Google Scholar] [CrossRef]
- Lange, R.A.; Hillis, L.D. Cardiovascular complications of cocaine use. N. Engl. J. Med. 2001, 345, 351–358. [Google Scholar] [CrossRef]
- Isner, J.M.; Chokshi, S.K. Cardiovascular complications of cocaine. Curr. Probl. Cardiol. 1991, 16, 89–123. [Google Scholar] [CrossRef]
- Mittleman, M.A.; Mintzer, D.; Maclure, M.; Tofler, G.H.; Sherwood, J.B.; Muller, J.E. Triggering of myocardial infarction by cocaine. Circulation 1999, 99, 2737–2741. [Google Scholar] [CrossRef] [Green Version]
- Minor, R.L.; Scott, B.D.; Brown, D.D.; Winniford, M.D. Cocaine-induced myocardial infarction in patients with normal coronary arteries. Ann. Intern. Med. 1991, 115, 797–806. [Google Scholar] [CrossRef]
- Richard, A.; Lange, L.; Hillis, D. Sudden death in cocaine abusers. Eur. Heart J. 2010, 31, 271–273. [Google Scholar] [CrossRef]
- Abuse, S. Mental Health Services Administration. Results 2013, 2, 13. [Google Scholar]
- McCord, J.; Jneid, H.; Hollander, J.E.; de Lemos, J.A.; Cercek, B.; Hsue, P.; Gibler, W.B.; Ohman, E.M.; Drew, B.; Philippides, G.; et al. Management of cocaine-associated chest pain and myocardial infarction: A scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation 2008, 117, 1897–1907. [Google Scholar] [CrossRef] [Green Version]
- Dahl, A.R.; Hadley, W.M. Formaldehyde production promoted by rat nasal cytochrome P-450-dependent monooxygenases with nasal decongestants, essences, solvents, air pollutants, nicotine, and cocaine as substrates. Toxicol. Appl. Pharmacol. 1983, 67, 200–205. [Google Scholar] [CrossRef]
- Verstraete, A.G. Detection times of drugs of abuse in blood, urine, and oral fluid. Ther. Drug Monit. 2004, 26, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacic, P.; Ames, J.R.; Jawdosiuk, M.; Ryan, M.D. Electron Transfer Mechanism for Cocaine Action. In Redox Chemistry and Interfacial Behavior of Biological Molecules; Dryhurst, G., Niki, K., Eds.; Springer: Boston, MA, USA, 1988; pp. 323–331. [Google Scholar] [CrossRef]
- Zheng, X.; Shang, L.; Zhan, C.G.; Zheng, F. In vivo characterization of toxicity of norcocaethylene and norcocaine identified as the most toxic cocaine metabolites in male mice. Drug Alcohol Depend. 2019, 204, 107462. [Google Scholar] [CrossRef]
- Mahlakaarto, J.; Ruskoaho, H.; Huttunen, P.; MacDonald, E.; Pasanen, M. Norcocaine is a potent modulator of haemodynamic responses, plasma catecholamines and cardiac hormone release in conscious rats. Toxicology 1998, 128, 101–111. [Google Scholar] [CrossRef]
- Babapoor-Farrokhran, S.; Kalla, A.; Gill, D.; Gulab, A.; Banka, S.; Kalra, S. Peripheral Administration of Nitroglycerin in Pulseless Ventricular Tachycardia due to Cocaine-Induced Coronary Vasospasm. Cardiovasc. Toxicol 2021, 21, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Rezkalla, S.H.; Kloner, R.A. Cocaine-Induced Acute Myocardial Infarction. Clin. Med. Res. 2007, 5, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Bencharit, S.; Morton, C.L.; Xue, Y.; Potter, P.M.; Redinbo, M.R. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat. Struct. Biol. 2003, 10, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, E.T.; Jackson, G.F.; Paul, B.D. Cocaine, Crack Cocaine, and Ethanol: A Deadly Mix. In Critical Issues in Alcohol and Drugs of Abuse Testing; Academic Press: Cambridge, MA, USA, 2019; pp. 215–224. [Google Scholar] [CrossRef]
- Farooq, M.U.; Bhatt, A.; Patel, M.B. Neurotoxic and cardiotoxic effects of cocaine and ethanol. J. Med. Toxicol. 2009, 5, 134–138. [Google Scholar] [CrossRef]
- Henning, R.J.; Wilson, L.D.; Glauser, J.M. Cocaine plus ethanol is more cardiotoxic than cocaine or ethanol alone. Crit. Care Med. 1994, 22, 1896–1906. [Google Scholar] [CrossRef]
- Moliterno, D.J.; Willard, J.E.; Lange, R.A.; Negus, B.H.; Boehrer, J.D.; Glamann, D.B.; Landau, C.; Rossen, J.D.; Winniford, M.D.; Hillis, L.D. Coronary-artery vasoconstriction induced by cocaine, cigarette smoking, or both. N. Engl. J Med. 1994, 330, 454–459. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, A.; Fabbian, F.; Pala, M.; Bonetti, F.; Babini, I.; Bagnaresi, I.; Manfredini, F.; Portaluppi, F.; Mikhailidis, D.P.; Manfredini, R. Cocaine and acute vascular diseases. Curr. Drug Abuse Rev. 2012, 5, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Turillazzi, E.; Bello, S.; Neri, M.; Pomara, C.; Riezzo, I.; Fineschi, V. Cardiovascular effects of cocaine: Cellular, ionic and molecular mechanisms. Curr. Med. Chem. 2012, 19, 5664–5676. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, G.; Catapano, F.; Cundari, G.; Ascione, A.; Galea, N.; Catalano, C.; Francone, M. Cocaine Abuse: An Attack to the Cardiovascular System—Insights from Cardiovascular MRI. Radiol. Cardiothorac. Imaging 2019, 1, e180031. [Google Scholar] [CrossRef] [PubMed]
- Morentin, B.; Callado, L.F.; García-Hernández, S.; Bodegas, A.; Lucena, J. The role of toxic substances in sudden cardiac death. Span. J. Leg. Med. 2018, 44, 13–21. [Google Scholar] [CrossRef]
- Michaud, K.; Basso, C.; d’Amati, G.; Giordano, C.; Kholová, I.; Preston, S.D.; Rizzo, S.; Sabatasso, S.; Sheppard, M.N.; Vink, A.; et al. Diagnosis of myocardial infarction at autopsy: AECVP reappraisal in the light of the current clinical classification. Virchows Archiv. 2020, 476, 179–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, W.R.; Lange, R.A.; Cigarroa, J.E.; Hillis, L.D. Cocaine-induced myocardial ischemia and infarction: Pathophysiology, recognition, and management. Prog. Cardiovasc. Dis. 1997, 40, 65–76. [Google Scholar] [CrossRef]
- Hoffman, R.S. Treatment of patients with cocaine-induced arrhythmias: Bringing the bench to the bedside. Br. J. Clin. Pharmacol. 2010, 69, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Kloner, R.A.; Rezkalla, S.H. Cocaine and the heart. N. Engl. J. Med. 2003, 348, 487–488. [Google Scholar] [CrossRef]
- Fischman, M.W.; Schuster, C.R.; Resnekov, L.; Shick, J.F.E.; Krasnegor, N.A.; Fennell, W.; Freedman, D.X. Cardiovascular and subjective effects of intravenous cocaine administration in humans. Arch. Gen. Psychiatry 1976, 33, 983–989. [Google Scholar] [CrossRef]
- O’Leary, M.E. Inhibition of HERG potassium channels by cocaethylene: A metabolite of cocaine and ethanol. Cardiovasc. Res. 2002, 53, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Steffel, J.; Iseli, S.; Arnet, C.; Lüscher, T.F.; Tanner, F.C. Cocaine unbalances endothelial tissue factor and tissue factor pathway inhibitor expression. J. Mol. Cell. Cardiol. 2006, 40, 746–749. [Google Scholar] [CrossRef]
- Yeo, K.K.; Wijetunga, M.; Ito, H.; Efird, J.T.; Tay, K.; Seto, T.B.; Alimineti, K.; Kimata, C.; Schatz, I.J. The association of methamphetamine use and cardiomyopathy in young patients. Am. J. Med. 2007, 120, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Havakuk, O.; Rezkalla, S.H.; Kloner, R.A. The cardiovascular effects of cocaine. J. Am. Coll. Cardiol. 2017, 70, 101–113. [Google Scholar] [CrossRef]
- Benzaquen, B.S.; Cohen, V.; Eisenberg, M.J. Effects of cocaine on the coronary arteries. Am. Heart J. 2001, 142, 402–410. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Virmani, R.; Cornhill, J.F.; Herderick, E.E.; Smialek, J. Increase in atherosclerosis and adventitial mast cells in cocaine abusers: An alternative mechanism of cocaine-associated coronary vasospasm and thrombosis. J. Am. Coll. Cardiol. 1991, 17, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Brickner, M.E.; Willard, J.E.; Eichhorn, E.J.; Black, J.; Grayburn, P.A. Left ventricular hypertrophy associated with chronic cocaine abuse. Circulation 1991, 84, 1130–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, S.L.; Alker, K.J.; Rezkalla, S.H.; Eisenhauer, A.C.; Kloner, R.A. Nifedipine protects the heart from the acute deleterious effects of cocaine if administered before but not after cocaine. Circulation 1991, 83, 1437–1443. [Google Scholar] [CrossRef] [Green Version]
- Maceira, A.M.; Ripoll, C.; Cosin-Sales, J.; Igual, B.; Gavilan, M.; Salazar, J.; Belloch, V.; Pennell, D.J. Long term effects of cocaine on the heart assessed by cardiovascular magnetic resonance at 3T. J. Cardiovasc. Magn. Reson. 2014, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Restrepo, C.S.; Rojas, C.A.; Martinez, S.; Riascos, R.; Marmol-Velez, A.; Carrillo, J.; Vargas, D. Cardiovascular complications of cocaine: Imaging findings. Emerg. Radiol. 2009, 16, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Dettmeyer, R.B. Forensic Histopathology: Fundamentals and Perspectives; Springer: New York, NY, USA, 2018. [Google Scholar]
- White, S.M.; Lambe, C.J. The pathophysiology of cocaine abuse. J. Clin. Forensic Med. 2003, 10, 27–39. [Google Scholar] [CrossRef]
- Frontera, J.A.; Gradon, J.D. Right-side endocarditis in injection drug users: Review of proposed mechanisms of pathogenesis. Clin. Infect. Dis. 2000, 30, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Eagle, K.A.; Isselbacher, E.M.; DeSanctis, R.W.; International Registry for Aortic Dissection (IRAD) Investigators. Cocaine-related aortic dissection in perspective. Circulation 2002, 105, 1529–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogan, W.C., 3rd; Lange, R.A.; Glamann, D.B.; Hillis, L.D. Recurrent coronary vasoconstriction caused by intranasal cocaine: Possible role for metabolites. Ann. Intern. Med. 1992, 116, 556–561. [Google Scholar] [CrossRef]
- Ye, Z.W.; Zhang, J.; Townsend, D.M.; Tew, K.D. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim. Biophys. Acta 2015, 1850, 1607–1621. [Google Scholar] [CrossRef] [Green Version]
- Uys, J.D.; Mulholland, P.J.; Townsend, D.M. Glutathione and redox signaling in substance abuse. Biomed. Pharmacother. 2014, 68, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhl, G.R.; Grow, R.W. The burden of complex genetics in brain disorders. Arch. Gen. Psychiatry 2004, 61, 223–229. [Google Scholar] [CrossRef]
- Macedo, D.S.; deVasconcelos, S.M.; Andrate-Neto, M.; Belchior, L.D.; Honorino, J.E.R.; Goncalves, D.O.; Fonteles, M.M.F.; Silva, M.I.G.; Aguiar, L.M.V.; Viana, G.S.B.; et al. Cocaine-induced status epilepticus and death generate oxidative stress in prefrontal cortex and striatum of mice. Neurochem. Int. 2010, 56, 183–187. [Google Scholar] [CrossRef]
- Turillazzi, E.; Cerretani, D.; Cantatore, S.; Fiaschi, A.I.; Frati, P.; Micheli, L.; Neri, M.; Cipolloni, L.; Di Paolo, M.; Pinchi, E.; et al. Myocardial oxidative damage is induced by cardiac Fas-dependent and mitochondria-dependent apoptotic pathways in human cocaine-related overdose. Sci. Rep. 2017, 7, 44262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, L.; Li, Q.; He, L.; Sun, W.; Jia, Z.; Ma, H. Protective Effect of Tempol Against Hypoxia-Induced Oxidative Stress and Apoptosis in H9c2 Cells. Med. Sci. Monit. Basic Res. 2017, 23, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.A.; Cui, S.X.; Waters, C.D.; Sano, S.; Wang, Y.; Doviak, H.; Leor, J.; Walsh, K.; French, B.A.; Epstein, F.H. Nitroxide-enhanced MRI of cardiovascular oxidative stress. NMR Biomed. 2020, 33, e4359. [Google Scholar] [CrossRef]
- Womersley, J.S.; Townsend, D.M.; Kalivas, P.W.; Uys, J.D. Targeting redox regulation to treat substance use disorder using N-acetylcysteine. Eur. J. Neurosci. 2019, 50, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.M.; Carvalho, F.; Bastos, M.L.; Carvalho, R.A.; Carvalho, M.; Remião, F. Contribution of catecholamine reactive intermediates and oxidative stress to the pathologic features of heart diseases. Curr. Med. Chem. 2011, 18, 2272–2314. [Google Scholar] [CrossRef] [PubMed]
- Pomierny-Chamioło, L.; Moniczewski, A.; Wydra, K.; Suder, A.; Filip, M. Oxidative stress biomarkers in some rat brain structures and peripheral organs underwent cocaine. Neurotox. Res. 2013, 23, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Cunha-Oliveira, T.; Cristina Rego, A.R.; Oliveira, C. Oxidative stress and drugs of abuse: An update. Mini-Rev. Org. Chem. 2013, 10, 321–334. [Google Scholar] [CrossRef]
- Kalivas, P.W.; Volkow, N.D. The neural basis of addiction: A pathology of motivation and choice. Am. J. Psychiatry 2005, 162, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Fineschi, V.; Baroldi, G.; Centini, F.; Cerretani, D.; Fiaschi, A.I.; Micheli, L.; Parolini, M.; Turillazzi, E.; Giorgi, G. Markers of cardiac oxidative stress and altered morphology after intraperitoneal cocaine injection in a rat model. Int. J. Legal Med. 2001, 114, 323–330. [Google Scholar] [CrossRef]
- Moritz, F.; Monteil, C.; Isabelle, M.; Bauer, F.; Renet, S.; Mulder, P.; Richard, V.; Thuillez, C. Role of reactive oxygen species in cocaine-induced cardiac dysfunction. Cardiovasc. Res. 2003, 59, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Stankowski, R.V.; Kloner, R.A.; Rezkalla, S.H. Cardiovascular consequences of cocaine use. Trends Cardiovasc. Med. 2015, 25, 517–526. [Google Scholar] [CrossRef]
- Frustaci, A.; Russo, M.A.; Morgante, E.; Scopelliti, F.; Aquilano, K.; Ciriolo, M.R.; Grande, C.; Verardo, R.; Chimenti, C. Oxidative myocardial damage in human cocaine-related cardiomyopathy. Eur. J. Heart Fail. 2015, 17, 283–290. [Google Scholar] [CrossRef]
- Fan, L.; Sawbridge, D.; George, V.; Teng, L.; Bailey, A.; Kitchen, I.; Li, J.M. Chronic cocaine-induced cardiac oxidative stress and mitogen-activated protein kinase activation: The role of Nox2 oxidase. J. Pharmacol. Exp. Ther. 2009, 328, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial dysfunction and redox imbalance as a diagnostic marker of “free radical diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar] [PubMed] [Green Version]
- Liaudet, L.; Calderari, B.; Pacher, P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail. Rev. 2014, 19, 815–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, F.; Wilson, W.D.C. Excretion of cocaine and its metabolites in man. J. Pharm. Pharmacol. 1996, 21, 135S–138S. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS-Induced) ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD (P) H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Hyodo, F.; Anzai, K.; Utsumi, H.; Mitchell, J.B.; Krishna, M.C. Brain redox imaging. Methods Mol. Biol. 2011, 711, 397–419. [Google Scholar] [CrossRef]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007, 42, 1632–1650. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Hyodo, F.; Matsumoto, A.; Koretsky, A.P.; Sowers, A.L.; Mitchell, J.B.; Krishna, M.C. High-resolution mapping of tumor redox status by magnetic resonance imaging using nitroxides as redox-sensitive contrast agents. Clin. Cancer Res. 2006, 12, 2455–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuppusamy, P.; Chzhan, M.; Vij, K.; Shteynbuk, M.; Lefer, D.J.; Giannella, E.; Zweier, J.L. Three-dimensional spectral-spatial EPR imaging of free radicals in the heart: A technique for imaging tissue metabolism and oxygenation. Proc. Natl. Acad. Sci. USA 1994, 91, 3388–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.M.; Matsumoto, S.; Bernardo, M.; Sowers, A.; Matsumoto, K.I.; Krishna, M.C.; Mitchell, J.B. Magnetic resonance imaging of organic contrast agents in mice: Capturing the whole-body redox landscape. Free Radic. Biol. Med. 2011, 50, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.M.; Mitchell, J.B.; Krishna, M.C. Nitroxides as cancer imaging agents. Anticancer Agents Med. Chem. 2011, 11, 347–358. [Google Scholar] [CrossRef]
- Matsumoto, K.I.; Nakanishi, I.; Zhelev, Z.; Bakalova, R.; Aoki, I. Nitroxyl Radical as a Theranostic Contrast Agent in Magnetic Resonance Redox Imaging. Antioxid. Redox Signal. 2021, 1–27. [Google Scholar] [CrossRef]
- Kato, N.; Yanaka, K.; Hyodo, K.; Homma, K.; Nagase, S.; Nose, T. Stable nitroxide Tempol ameliorates brain injury by inhibiting lipid peroxidation in a rat model of transient focal cerebral ischemia. Brain Res. 2003, 25, 188–193. [Google Scholar] [CrossRef]
- Johanson, C.E.; Fischman, M.W. The pharmacology of cocaine related to its abuse. Pharmacol. Rev. 1989, 41, 3–52. [Google Scholar] [PubMed]
- Liang, Q.; Smith, A.D.; Pan, S.; Tyurin, V.A.; Kagan, V.E.; Hastings, T.G.; Schor, N.F. Neuroprotective effects of TEMPOL in central and peripheral nervous system models of Parkinson’s disease. Biochem. Pharmacol. 2005, 70, 1371–1381. [Google Scholar] [CrossRef]
- Kévin, M.; Dufayet, L.; Nicolas, S.; Charlotte, G.; Dion, E. Aortic dissection in a body packer: Did cocaine play a part? Forensic Sci. Int. 2021, 327, 110963. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgieva, E.; Karamalakova, Y.; Miteva, R.; Abrashev, H.; Nikolova, G. Oxidative Stress and Cocaine Intoxication as Start Points in the Pathology of Cocaine-Induced Cardiotoxicity. Toxics 2021, 9, 317. https://doi.org/10.3390/toxics9120317
Georgieva E, Karamalakova Y, Miteva R, Abrashev H, Nikolova G. Oxidative Stress and Cocaine Intoxication as Start Points in the Pathology of Cocaine-Induced Cardiotoxicity. Toxics. 2021; 9(12):317. https://doi.org/10.3390/toxics9120317
Chicago/Turabian StyleGeorgieva, Ekaterina, Yanka Karamalakova, Radostina Miteva, Hristo Abrashev, and Galina Nikolova. 2021. "Oxidative Stress and Cocaine Intoxication as Start Points in the Pathology of Cocaine-Induced Cardiotoxicity" Toxics 9, no. 12: 317. https://doi.org/10.3390/toxics9120317
APA StyleGeorgieva, E., Karamalakova, Y., Miteva, R., Abrashev, H., & Nikolova, G. (2021). Oxidative Stress and Cocaine Intoxication as Start Points in the Pathology of Cocaine-Induced Cardiotoxicity. Toxics, 9(12), 317. https://doi.org/10.3390/toxics9120317