3.1. Toxicokinetic Prediction of Toxicants
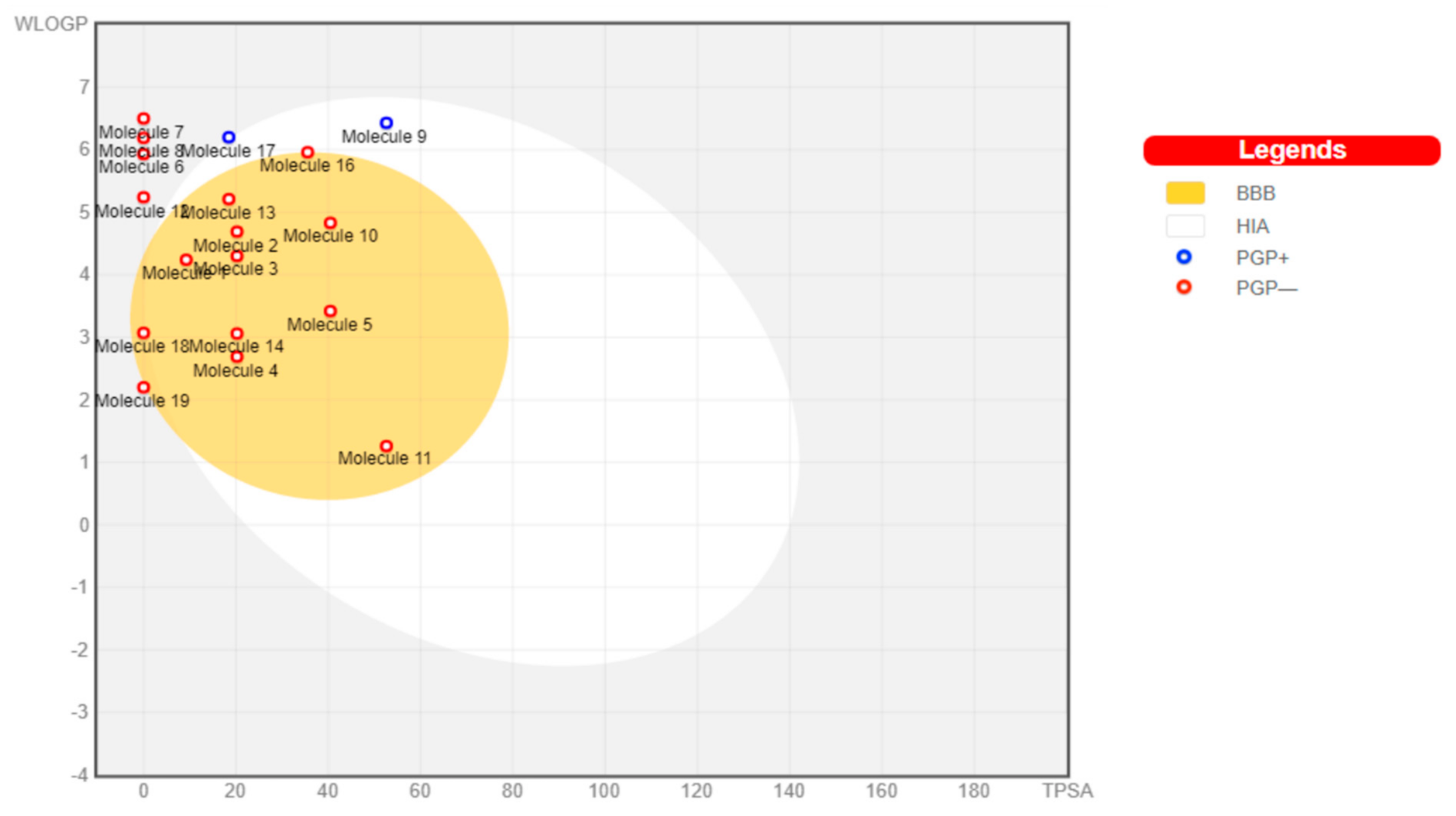
The toxicants’ pharmacokinetic properties are shown in
Figure 1, with perfluorooctanoic acid reported as an outlier due to its WLog P value of 10.75. Dimethyl phthalate, 4-bromodiphenylether, 4-nonylphenol, 4-octylphenol, 4-tert-butylphenol, bisphenol A, diethylstilbestrol, methoxychlor, tetrachloroethylene, and O-phenylphenol were predicted to have a high probability of permeating through the blood-brain barrier (BBB) and passively be absorbed by the gastrointestinal tract (GIT), while diethylhexyl phthalate and permethrin were both predicted to have high GIT but low BBB permeability. All other toxicants were not predicted to be either GIT or BBB permeants. All toxicants were predicted to be nonsubstrates of P-glycoprotein (Pgp) except diethylhexyl phthalate and tetrachlorodibenzodioxin.
In
Table 3, all the toxicants inhibited one or more isoforms of cytochrome P
450 (CYP
450). Some of these toxicants’ solubility was previously reported, corroborating this finding [
25]. This could further suggest these toxicants can easily be bioaccumulated in tissues where they can induce oxidative stress, insulin resistance, and elicit neurotoxicity [
11]. The inhibition of CYP
450 isoforms by these toxicants will lead to reduced metabolism of drugs, resulting in drug metabolism malfunctioning, drug resistance phenomenon, and elevated toxicity [
20]. The synthetic accessibility score of these toxicants suggests that they are easily synthesised, available, and incorporated into different products. This accounts for the increased incorporation of these compounds in various products used daily by people [
4].
3.3. Molecular Docking of Toxicants
Molecular docking is a conventional structure-based computational method used for analyzing binding interactions between protein–ligand complexes. In toxicity, this method is relevant in predicting the binding poses of toxicants in their various molecular targets’ binding pockets. The predicted activity between sex hormone receptors and environmental toxicants were previously reported in
Table 4. Hence,
Table 5 shows the docking scores of environmental toxicants in binding pockets of their predicted receptors of activity. From the docking scores shown in
Table 5, the sex hormones (α-estradiol, β-estradiol, and dihydrotestosterone (DHT)) had high binding scores in the pocket of their respective receptors (ER-α, ER-β, and AR respectively) with bisphenol A and diethylstilbestrol exhibiting comparable affinity in the binding pocket of all three receptors. However, in AR’s binding pocket, high docking scores were recorded for dichlorodiphenyltrichloroethane, O-phenylphenol, and perfluorooctanoic acid, while in ER-β, a similar observation was made for permethrin (as illustrated in
Table 5).
These high-binding affinities could suggest possible antagonism of these receptors either by competitive or mixed mechanism due to the benzene rings present in these toxicants’ structure [
27]. The binding energy of the endogenous ligands in the receptors was lower than the toxicants as expected due to their role in the hormone system. This finding was similar to that reported by Jeong et al. [
28], where the endogenous ligands had better docking scores than over 95% of the evaluated toxicants. The toxicants that their binding affinities were not determined would probably utilise different mechanisms other than binding to sex hormone receptors to elicit their toxic effect. Triphenyltin and tetrachlorodibenzodioxin were reported to bind to retinoid X receptor, peroxisome proliferator-activated receptor gamma (PPARγ), and aryl hydrocarbon receptor [
29,
30], while others such as tetrachloroethylene, dichlorodiphenyldichloroethane, and dichlorodiphenyldichloroethylene have neuronal nicotinic acetylcholine receptors [
31] PPARγ, PPARα, and ryanodine receptors (type 1 and 2) [
32,
33,
34] as their targets. The LE and LipE of the top-ranking toxicants were ≥0.3 and 3, respectively. However, the LipE of dichlorodiphenyltrichloroethane and perfluorooctanoic acid was less than 3 (as illustrated in
Table 6). These values (0.3 and 3) were identified as the respective ideal threshold cutoff of LE and LipE. These values further buttress these toxicants’ effectiveness to bind to these receptors and elicit their toxic tendencies such as T2DM [
25]. These toxicants were experimentally validated to exhibit estrogenic and androgenic properties in different model systems and cell lines [
35,
36,
37]. In utero [
38], perinatal [
39] and late-life exposure [
40,
41] of rodents to BPA and DDT were reported to result in reduced insulin secretion and impaired glucose tolerance. Studies carried out by Lee et al. [
42] and Wolf et al. [
43] revealed that exposure to these environmental toxicants is associated with increased T2DM risk via endocrine disruption in humans. Some other reports presented a similar association in the elderly [
44,
45] and middle-aged women [
46].
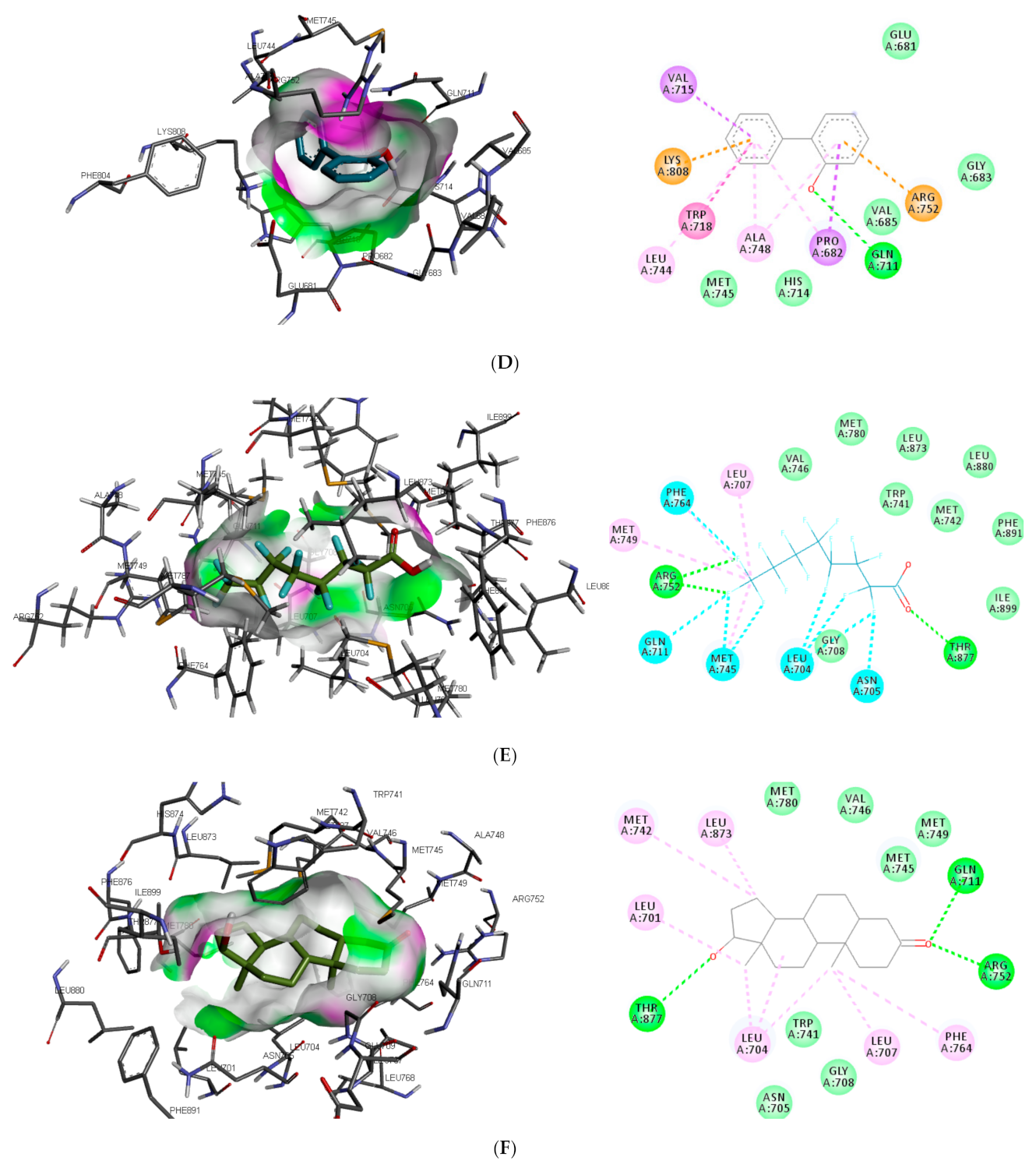
Similar to DHT interaction in AR binding pocket, bisphenol A and O-phenylphenol were stabilised by one hydrogen bond interaction with GLN711, while diethylstilbestrol hydrogen bond interaction was with MET745. On the other hand, dichlorodiphenyltrichloroethane and perfluorooctanoic acid exhibited 0 and 2 (ARG752 and THR877) hydrogen bond interactions, respectively, with AR (as illustrated in
Figure 2).
Also, unlike other toxicants, perfluorooctanoic acid was stabilised by halogen bond interaction with 5 amino acid residues (LEU704, ASN705, GLN711, MET745, and PHE764) in AR binding pocket (as illustrated in
Figure 2E). Halogen bonds are usually short and numerous, forming strong noncovalent interactions [
47]; hence the multiple halogen interaction observed in the perfluorooctanoic acid–AR complex could be attributed to the low-binding energy observed. Halogen interactions were also identified to play a role in substrate specificity [
48]. This could be a reason perfluorooctanoic acid was predicted to target only AR. Some of the amino acid residues such as LEU704, LEU707, MET742, and PHE764 were common π-π alkyl interactions stabilising DHT and the top-ranking toxicants, while ASN705 and GLY708 were the most common observed Van der Waals interactions with AR (as illustrated in
Figure 2). Contrary to other toxicants, the amino acid residues involved in π-π alkyl and Van der Waals interactions between O-phenylphenol and AR were different (as illustrated in
Figure 2D). This study further substantiates previous reports where LEU701, LEU704, ASN705, MET742, and PHE764 were amino acid residues in AR-binding pocket that interacted with toxicants [
49,
50].
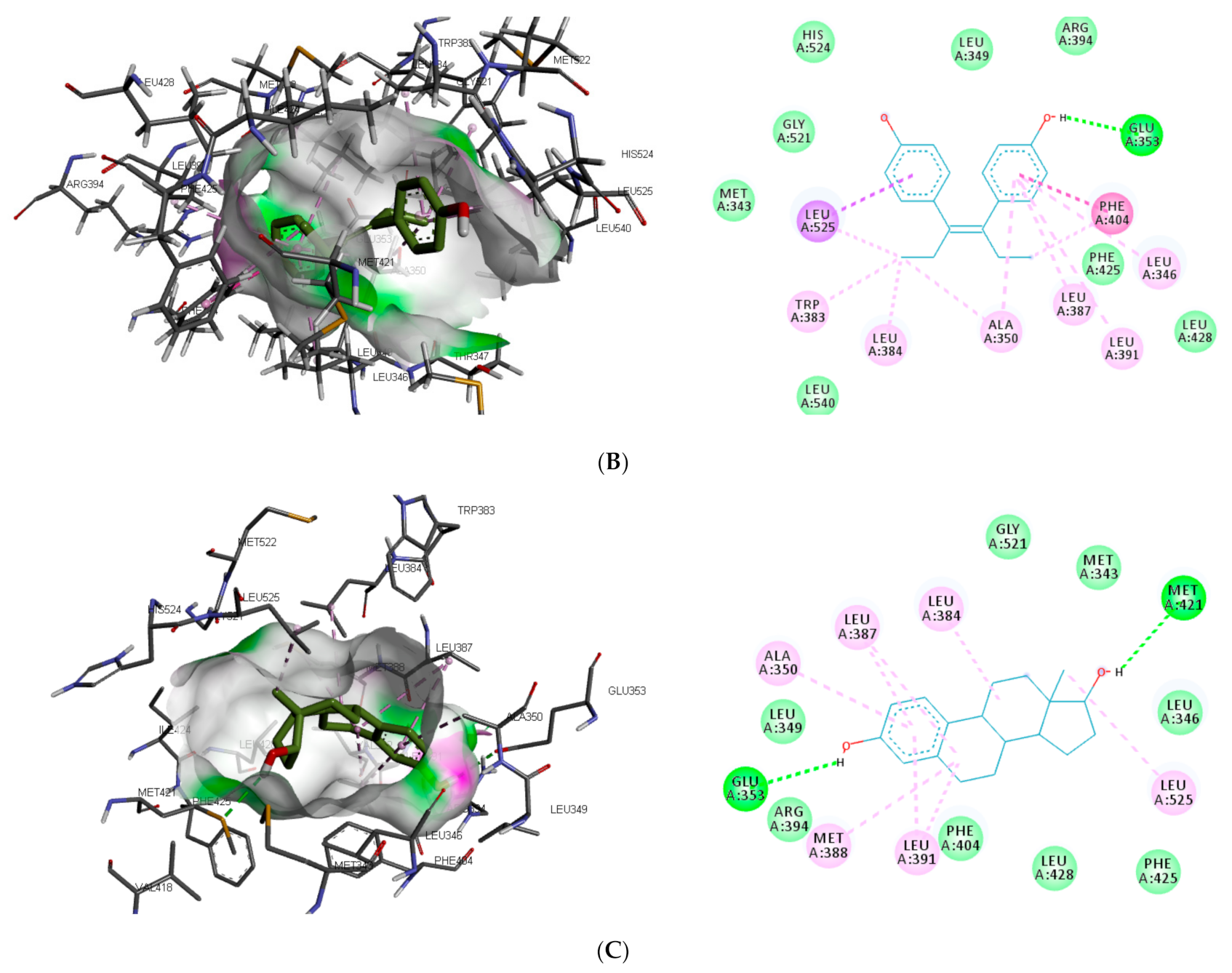
The interaction of the top-ranking toxicants in
Figure 3 depicted bisphenol A, diethylstilbestrol, and α-estradiol all interacting with GLU353 in ER-α binding pocket via hydrogen bond interaction, while ARG394 and MET421 were other hydrogen interactions with bisphenol A and α-estradiol, respectively. Common π-π alkyl (ALA350, LEU384, LEU384, and LEU391) and Van der Waals interactions (PHE425 and LEU428) were observed stabilising α-estradiol and the toxicants. It was also observed that diethylstilbestrol interacted with similar amino acid residues (MET343, LEU349, ARG394, GLY521, and LEU525) as α-estradiol. These interactions were not observed for bisphenol A, which accounted for the low binding energy recorded for diethylstilbestrol (as illustrated in
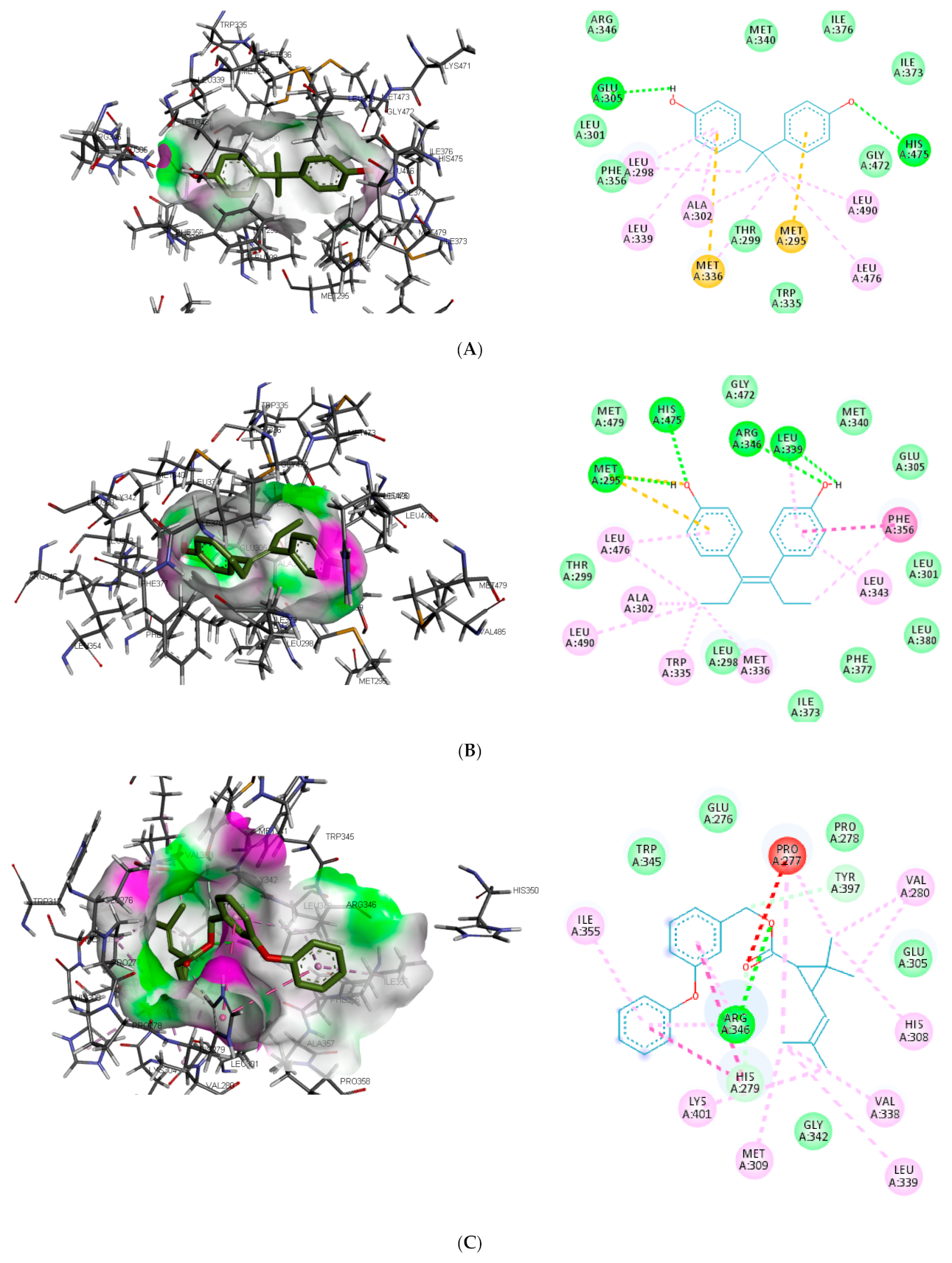
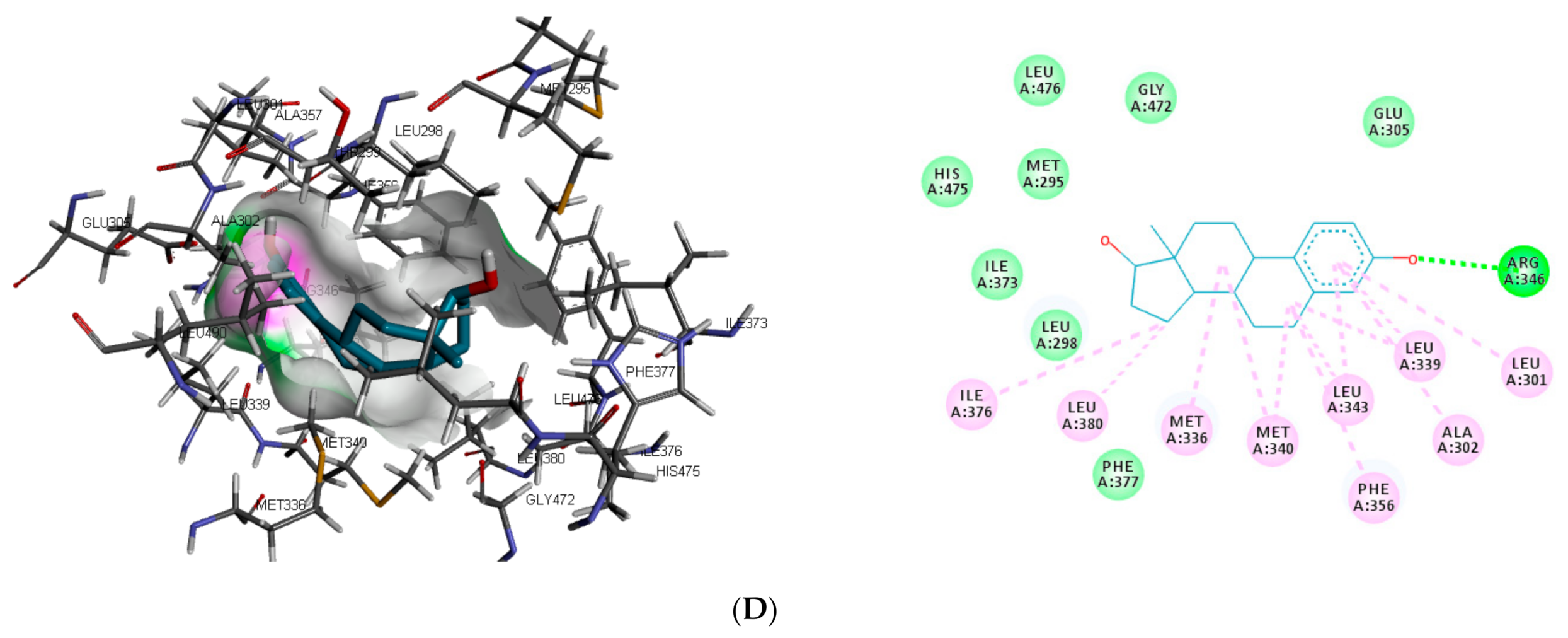
Figure 3). In the binding pocket of ER-β, ARG346 was depicted as the common hydrogen bond interaction between the receptor, β-estradiol, and two toxicants (diethylstilbestrol and permethrin). However, other hydrogen interactions were observed for bisphenol A (GLU305 and HIS475) and diethylstilbestrol (MET295, LEU339, and HIS475).
Despite the difference in the amino acid residues involved in other binding interactions between β-estradiol and the toxicants, similar π-π alkyl (ALA302 and LEU343) and Van der Waals interactions (LEU298, GLU305, ILE373, PHE377 and GLY472) were identified in the binding pose of β-estradiol and diethylstilbestrol in ER-β (as illustrated in
Figure 4). These common similarities and the increased number of hydrogen bond interaction with ER-β binding pocket amino acid residues could be attributed to the slightly lower binding energy observed for diethylstilbestrol. Comparing the binding pose of the toxicants and β-estradiol, permethrin had the least similarity. This observation might be ascribed to its structure as it can be seen overlapping out of ER-β binding pocket.
Different studies reported similar amino acid residues such as GLU353, ARG394, PHE404, and PHE425 that stabilise the binding of toxicants and EDCs in the active site of ER, which corroborate this study [
25,
51,
52]. The side chains of GLU and ARG ensures upon orientation that these amino acid residues form hydrogen bonds with the ligands, while that of PHE is nonpolar, which makes it suitable for nonpolar interactions.
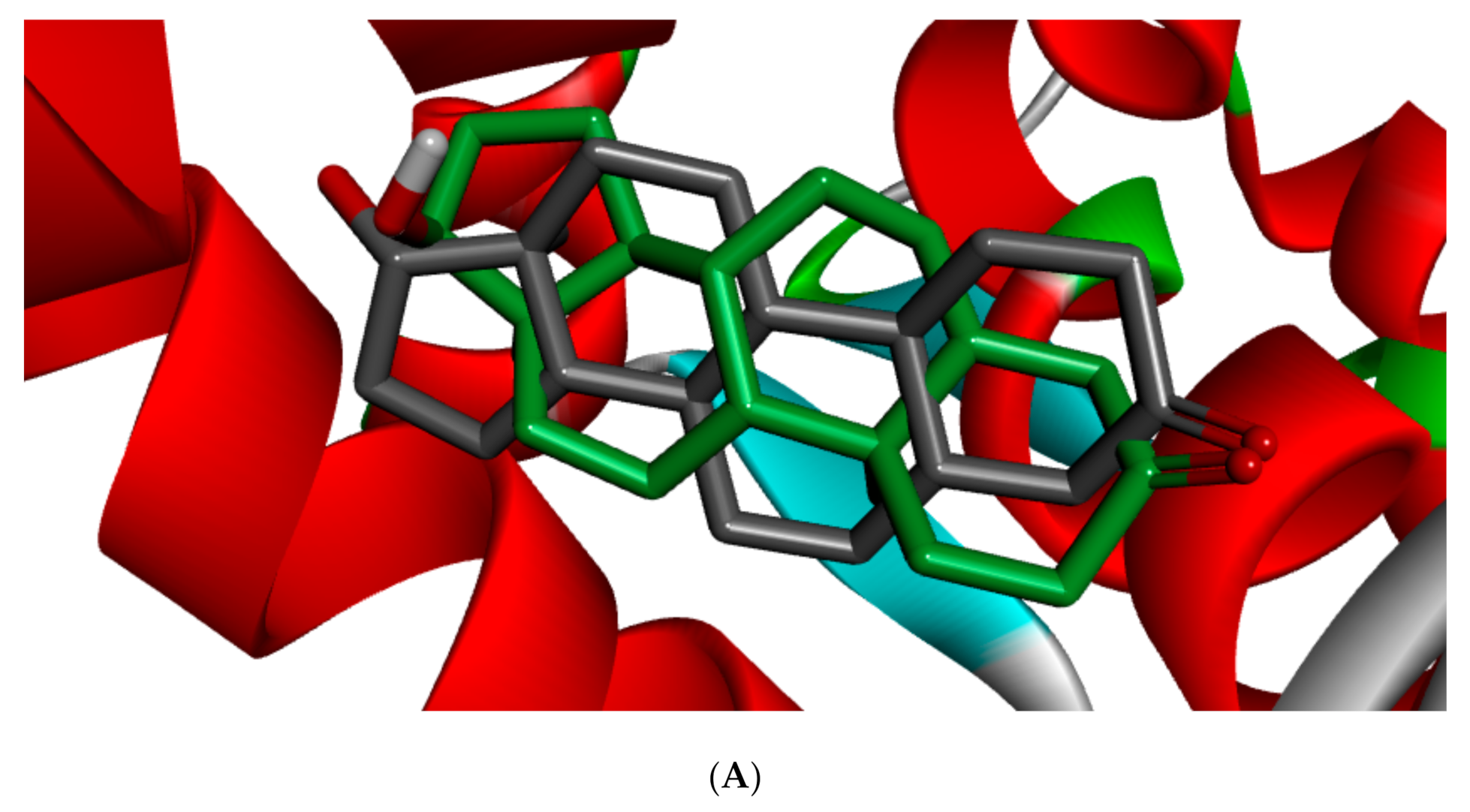
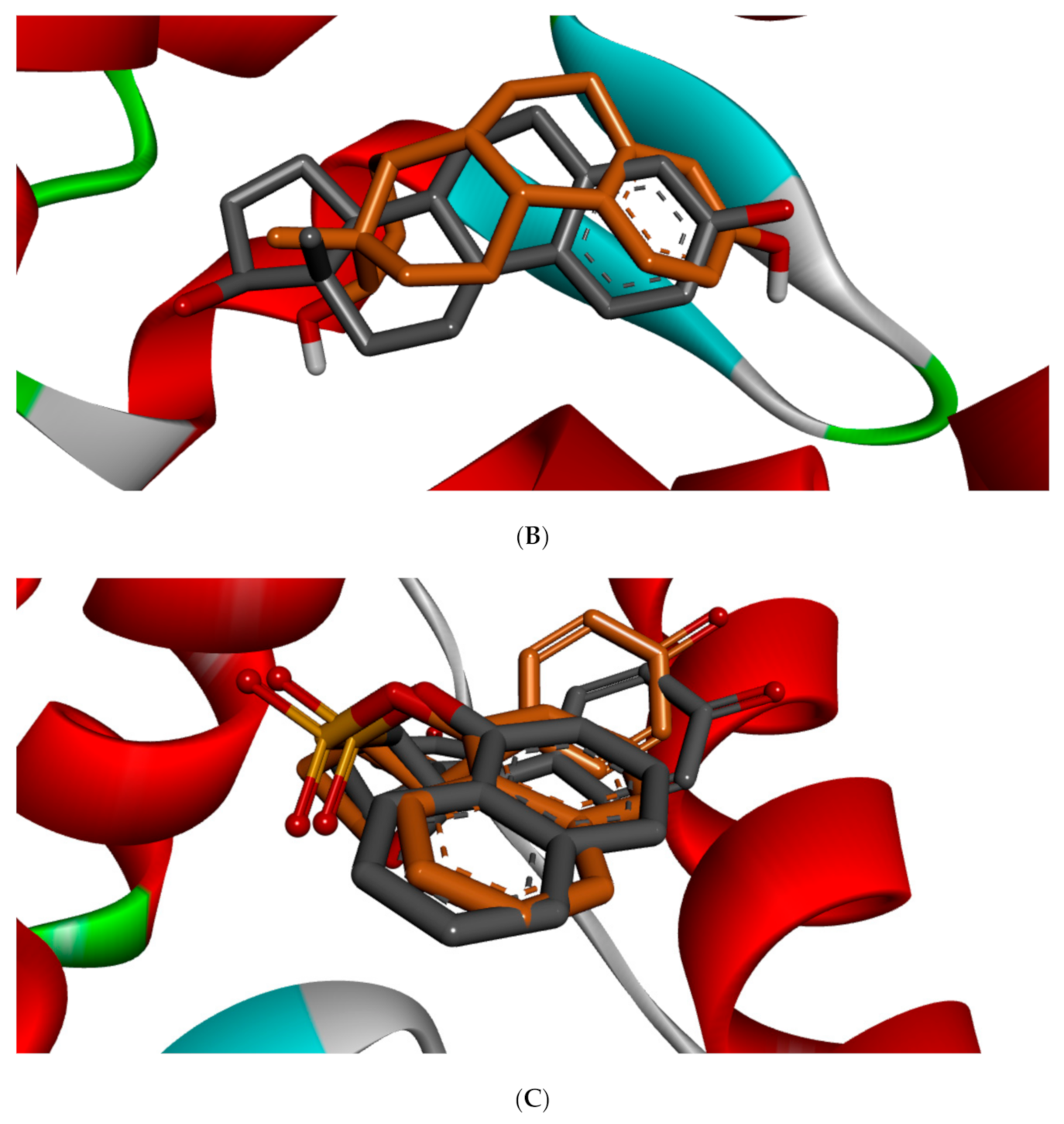
Figure 5 shows the redocked ligands in comparison with that of the experimental pose after superimposition. The RMSD of the experimental and docked ligands in AR, ER-α, and ER-β pocket was 0.77, 0.85, and 0.61 Å, respectively. This low deviation value (<2.0 Å) credits the docking parameters rationality and docking method reliability used in this study. It further infers the toxicants’ strong affinity in the receptors’ pockets [
53].
3.4. Molecular Dynamics of Toxicants
Protein dynamics is an important technique that gives greater insight into the protein’s dynamics at the atomic level and its relationship with the structure. In ligand-driven protein dynamics, the effect of the protein’s internal dynamics variation on ligand binding is the outcome [
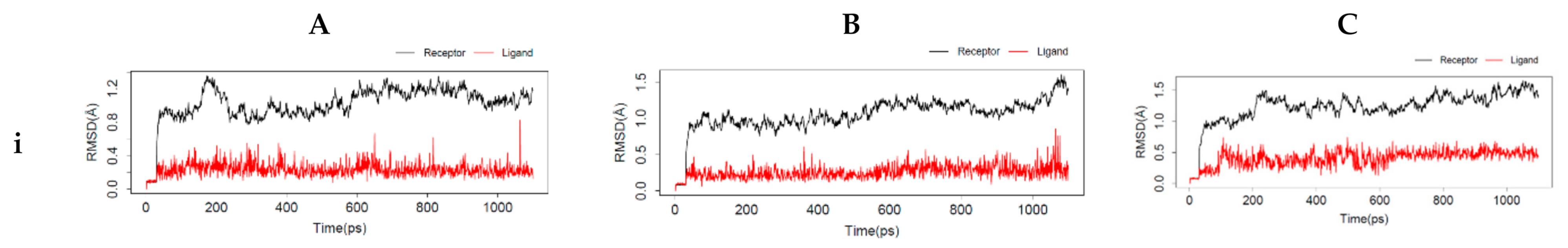
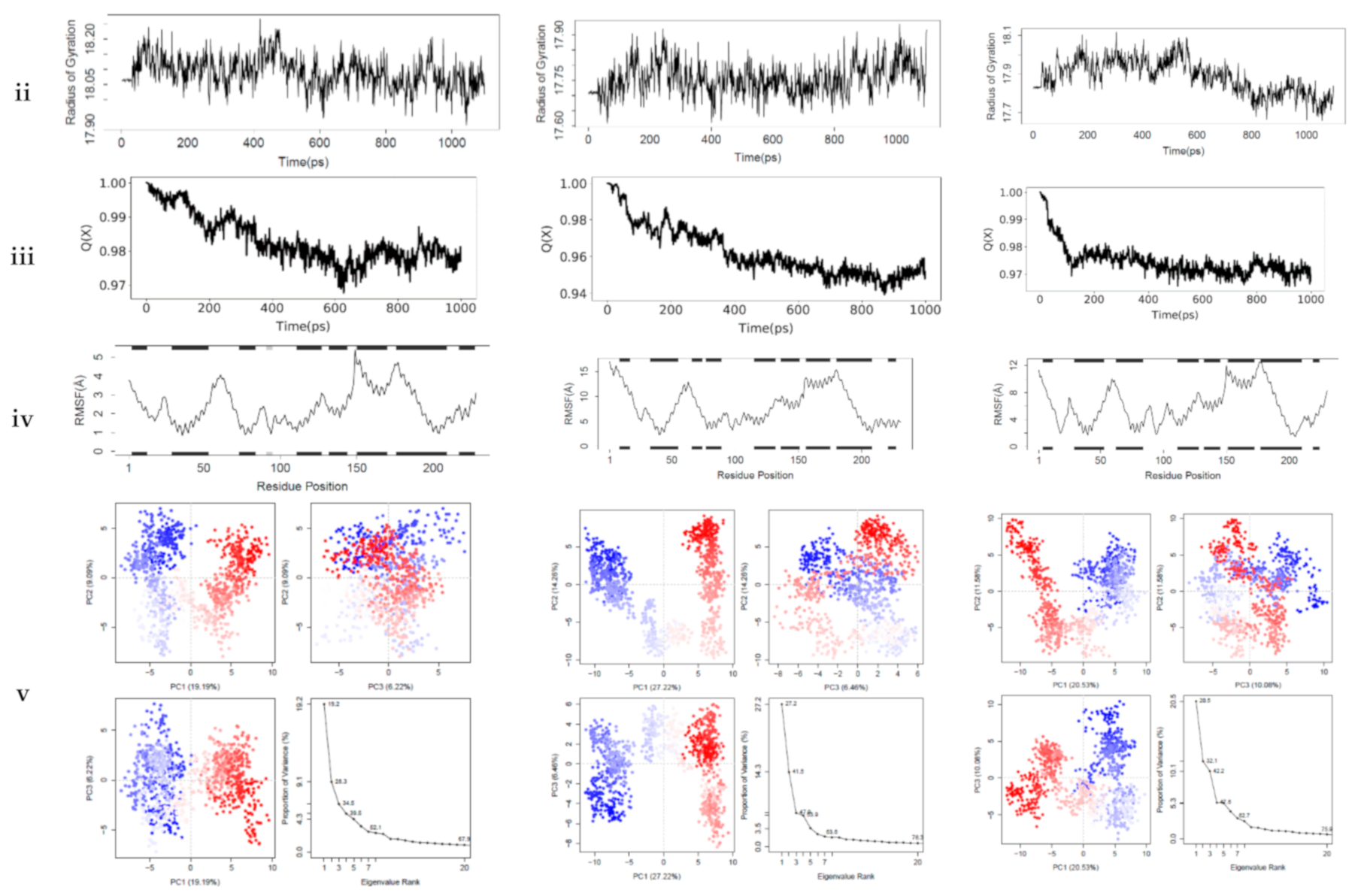
54]. The interactions between the top-ranking toxicants and their various docked complexes (AR-perfluorooctanoic acid, ER-α-diethylstilbestrol, and ER-β-diethylstilbestrol) were stable with <1.5 Å RMSD, while their radius of gyration (Rg) was within ±0.2 Å from the start point over the simulation timestep (as illustrated in
Figure 6i,ii).
The obtained RMSD and Rg values would suggest the complexes formed were equilibrated and had low oscillation with a folded polypeptide structure that was relatively stable. It can also be hypothesised that there is no shift in ligand position as contrarily reported in a study by Ali [
55], suggesting protein–ligand complex compactness. A steady decline in the fraction of native contact was observed from 1.0 to 0.97 (AR-Perfluorooctanoic acid and ER-β-Diethylstilbestrol) and 0.94 Q[X] (ER-α-Diethylstilbestrol) with RMSF fluctuations being dominant majorly at the loop region (as illustrated in
Figure 6iii,iv). The larger fluctuations observed in the loops are normal as they are the most flexible protein structure regions. From the docking analysis, they are not involved in binding the toxicants; hence, they are surface loops, energetically favouring the decline in the fraction of native contact. These RMSF values of the helical structures suggest a little variation of the ligands from the initial binding pose after the MD simulation [
56].
In AR-perfluorooctanoic acid PCA scree plot, PC1, PC2, and PC3 accounted for 19.2, 9.1, and 6.2% of the variance, respectively; subsequently, the individual PC’s contribution dropped below 4.3%. From the PCA scree plot for ER-α-diethylstilbestrol, PC1 accounts for more than one-quarter of the overall variance, strongly dominating the overall variance. When computed with the next 2 PC’s, they made up 41.9% of the variance. After the 4th PC, individual component contributions were below 3.5%. PC1 accounted for more than one-fifth of the overall variance, with PC2 and PC3 contributing a little over 10%, strongly dominating the overall variance. A drop below 5% was subsequently observed for other individual component contributions as depicted in ER-β-diethylstilbestrol PCA scree plot (as illustrated in
Figure 6v). PC1, PC2, and PC3 retained most of the variance while providing a useful description of the original distribution’s atomic fluctuations. The large initial intensive motions rapidly declined to more localised fluctuations. The first 3 PC’s were used from the cluster analysis to analyse conformational transitions of the simulated systems by projecting their trajectories on a 2D subspace. The continuous colour scale, which changed from blue to white and then red, indicates that the conformational behavior of the three simulated systems exhibited no observable difference between the blue and red conformations as projected along the direction of PC2–PC3, while a difference along the direction of PC1–PC2 and PC1–PC3 between the 2 colour conformations were observed (as illustrated in
Figure 6v). The continuous colour scale changed from blue to white and then red, indicating intermittent transitions between these conformations [
57]. Electrostatic interactions contributed little, while Van der Waals interactions were the most contributors to the total free energy and binding free energy in all 3 complexes. The total free energy was counteracted by the entropy generated (as illustrated in
Table 7). The difference in the proportion of variance observed in the principal component analysis (PCA) was evident in the predicted binding energy by MM-PB/GBSA (as illustrated in
Table 7). Studies reported a positive correlation between MM-PB/GBSA predicted binding energy and experimental results [
58,
59]. The calculated MM-PB/GBSA binding energy corroborates the docking analysis that was earlier reported, suggesting these toxicants can interact with these receptors and elicit their toxic effects. The increased free binding energy observed in ER-β-diethylstilbestrol may be due to lower Van der Waals energy contribution [
60].
3.5. Gene Expression Network Modified by Toxicants
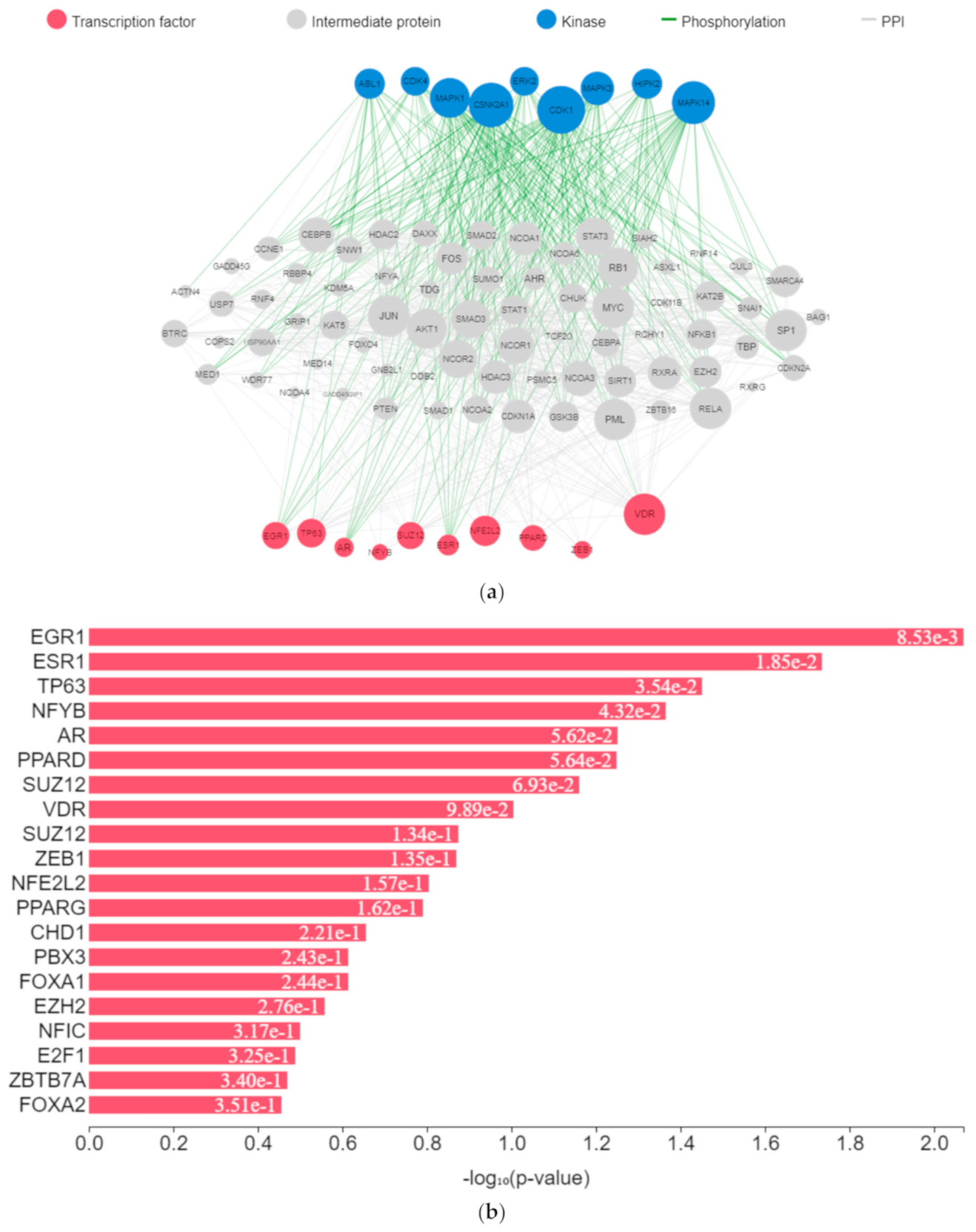
The expression of genes, proteins, and transcription factors upregulated by toxicants upon sex hormone receptor binding from the gene network analysis is depicted in
Figure 7a.
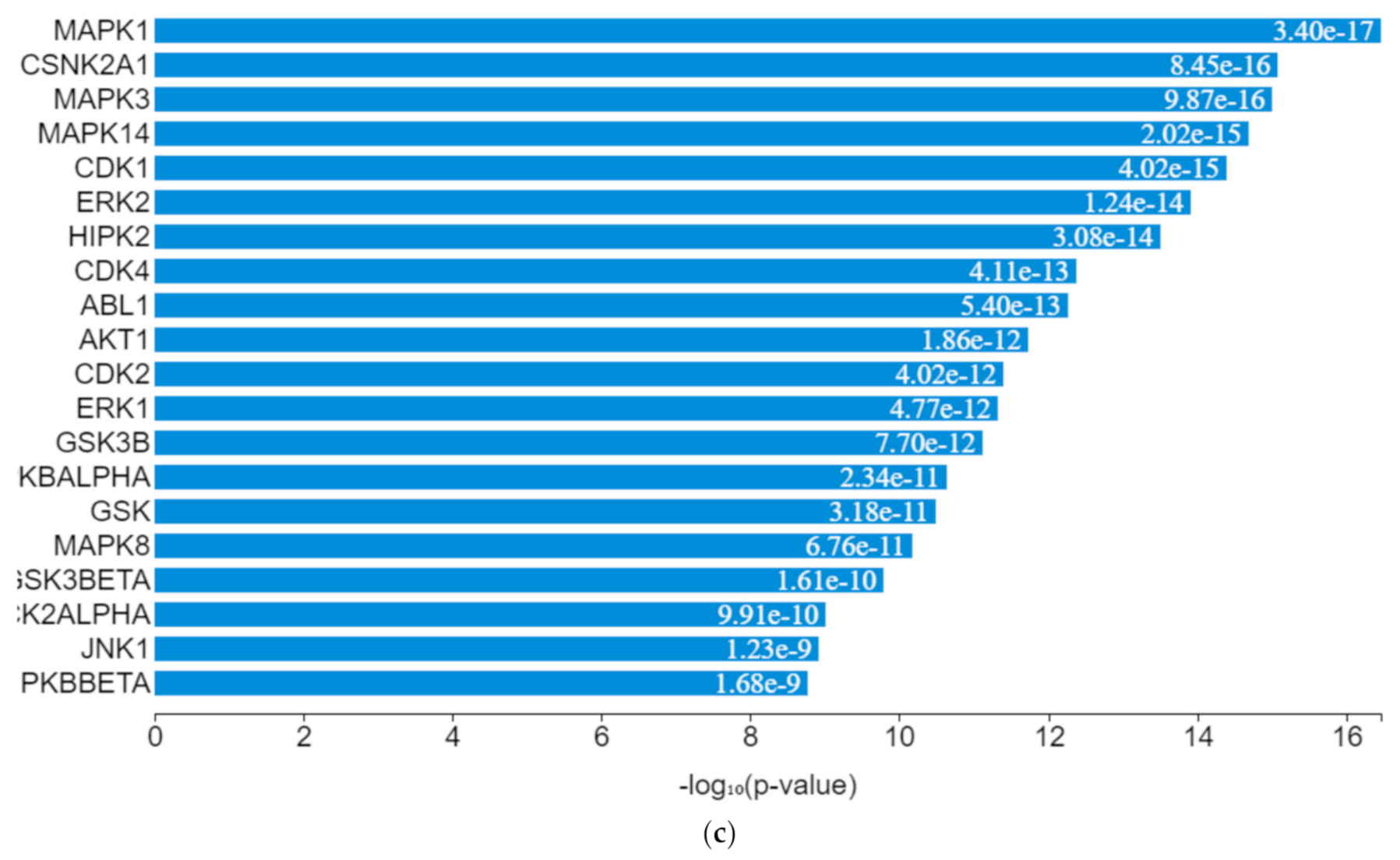
EGR1 (early growth response protein 1), ESR1 (estrogen receptor 1), VDR (vitamin D receptor), SUZ12 (polycomb protein), PPARD (peroxisome proliferator-activated receptors delta), NFE2L2 (nuclear factor erythroid 2-related factor 2), ZEB1 (zinc finger e-box binding homeobox 1), NFYB (nuclear transcription factor y subunit beta), and TP63 (tumour protein P63) were the transcription factors predicted to be induced by the toxicants upon protein–protein interaction. They interacted with mitogen-activated protein kinase (MAPK), casein kinase (CSNK), cyclin-dependent kinase (CDK), extracellular receptor kinase (ERK), homeodomain-interacting protein kinase (HIPK), tyrosine-protein kinase (ABL), and some intermediate proteins, namely PTEN, JUN, AKT1, NfKB1, MYC, RB1, SP1, SMAD3 and others as depicted in the interaction nodes. Some of these transcription factors, kinases, and intermediate proteins were previously identified as key genes and pathways implicated in T2DM prognosis and pathogenesis and potential pharmacological targets substantiating the result [
61,
62]. EGR1 was identified upon analysis as the most enriched transcription factor expressed by the toxicants followed by ESR1 and TP63, while MAPK1 was the most enriched kinase expressed by the toxicants followed by casein kinase 2 alpha 1 (CSNK2A1) and MAPK3 based on the hypergeometric
p-value.
Upregulation of EGR1 in diabetes-related diseases was noted in numerous studies. It enhances extracellular matrix production and proliferation of mesangial cells by interacting with TGF-β1 and upregulation of downstream genes [
63,
64]. Glucose transporter type 1 (GLUT1) is a transcriptional target of TP63. Upon activation, there is an elevated expression of GLUT1, which leads to GLUT1-mediated glucose influx and generation of nicotinamide adenine dinucleotide phosphate (NADPH) from the pentose phosphate pathway (PPP) [
65]. Upregulation of CSNK2A1 gene expression and protein concentration is associated with T2DM and its associated pathologies [
66]. Upregulation of important intermediary proteins such as PTEN, AKT1, NfKB1, SP1, and SMAD3 was identified as a common occurrence in T2DM and its vascular complications [
67,
68,
69]. Various experimental studies showed that binding of environmental toxicants to sex hormone receptors led to increased AKT and CREB phosphorylation and increased PI3K and NfKβ activity and MAPK and ERK activation while inhibiting PPAR, corroborating the results [
70,
71,
72,
73,
74,
75,
76,
77]. These molecular activities lead to insulin action impairment and intermediary metabolism alteration [
70,
78,
79].


 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}