Commonalities between Copper Neurotoxicity and Alzheimer’s Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Alzheimer’s Disease
1.2. Copper
2. Methodology
3. Copper Pathways to the Brain
3.1. Role of Copper in the Human Body
3.2. Common Forms of Copper Intake
3.3. Alzheimer’s Disease and Copper Pathways
4. Beta-Amyloid and Copper Relationship
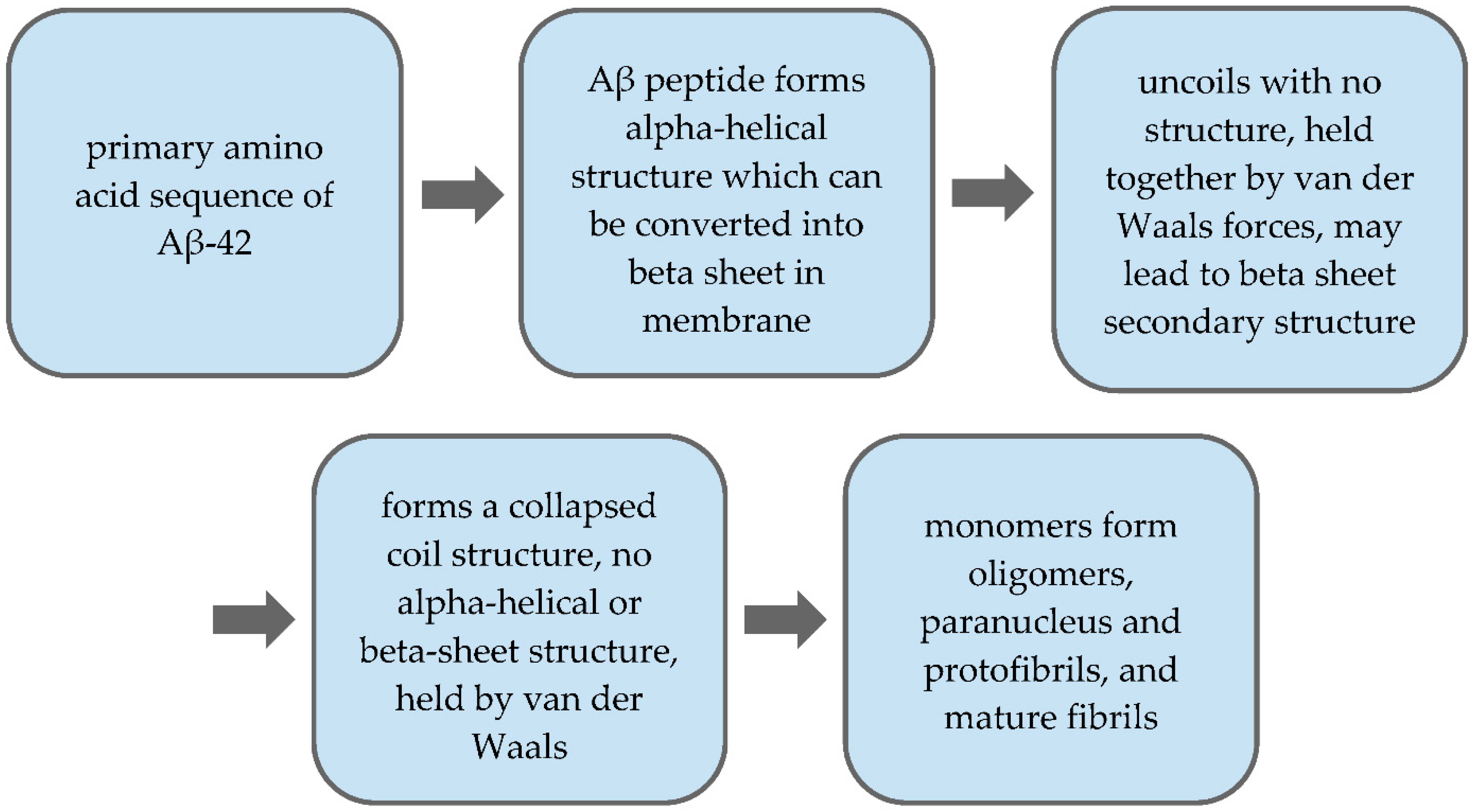
4.1. Beta-Amyloid Plaques
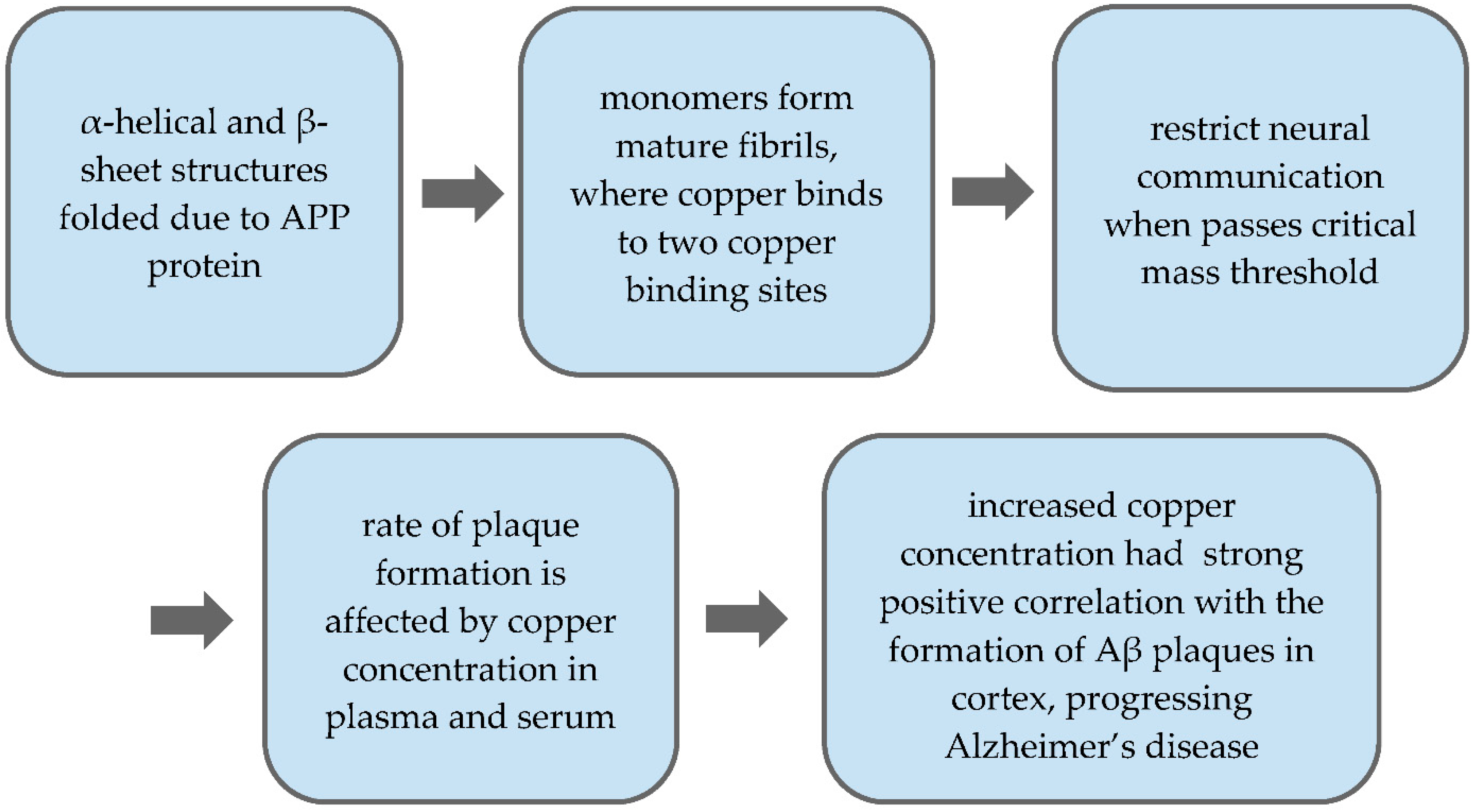
4.2. Copper Toxicity and Aβ Levels
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- National Institute on Aging What Happens to the Brain in Alzheimer’s Disease?|National Institute on Aging. Available online: https://www.nia.nih.gov/health/what-happens-brain-alzheimers-disease (accessed on 14 July 2020).
- Derby, C.A. Trends in the Public Health Significance, Definitions of Disease, and Implications for Prevention of Alzheimer’s Disease. Curr. Epidemiol. Rep. 2020, 7, 68–76. [Google Scholar] [CrossRef]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer Disease in the United States (2010–2050) Estimated Using the 2010 Census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Friedman, E.M.; Shih, R.A.; Langa, K.M.; Hurd, M.D. US Prevalence And Predictors Of Informal Caregiving For Dementia. Health Aff. Proj. Hope 2015, 34, 1637–1641. [Google Scholar] [CrossRef]
- Jutkowitz, E.; Kane, R.L.; Gaugler, J.E.; MacLehose, R.F.; Dowd, B.; Kuntz, K.M. Societal and Family Lifetime Cost of Dementia: Implications for Policy. J. Am. Geriatr. Soc. 2017, 65, 2169–2175. [Google Scholar] [CrossRef]
- Ghazizadeh, H.; Yaghooti-Khorasani, M.; Darroudi, S.; Esmaily, H.; Sharifan, P.; Tayefi, M.; reza Seyedi, S.M.; Mohammadi-Bajgiran, M.; Ghaffarian-Zirak, R.; Tavallaie, S.; et al. Evaluation of the Association between the Healthy Eating Index and the Level of Serum and Dietary Intake of Copper and Zinc. Obes. Med. 2020, 19, 100277. [Google Scholar] [CrossRef]
- Freeborn, D.; Haldeman-Englert, C. Total Copper (Blood)—Health Encyclopedia—University of Rochester Medical Center. Available online: https://www.urmc.rochester.edu/encyclopedia/content.aspx?contenttypeid=167&contentid=total_copper_blood (accessed on 6 October 2020).
- Roberts, E.A.; Sarkar, B. Liver as a Key Organ in the Supply, Storage, and Excretion of Copper. Am. J. Clin. Nutr. 2008, 88, 851S–854S. [Google Scholar] [CrossRef]
- Royer, A.; Sharman, T. Copper Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Collins, J.F.; Klevay, L.M. Copper12. Adv. Nutr. 2011, 2, 520–522. [Google Scholar] [CrossRef]
- Pohanka, M. Copper and Copper Nanoparticles Toxicity and Their Impact on Basic Functions in the Body. Bratisl. Med. J. 2019, 120, 397–409. [Google Scholar] [CrossRef]
- De Bie, P.; Muller, P.; Wijmenga, C.; Klomp, L.W.J. Molecular Pathogenesis of Wilson and Menkes Disease: Correlation of Mutations with Molecular Defects and Disease Phenotypes. J. Med. Genet. 2007, 44, 673–688. [Google Scholar] [CrossRef]
- Ross, A.C.; Caballero, B.H.; Cousins, R.J.; Tucker, K.L.; Ziegler, T.R. Modern Nutrition in Health and Disease: Eleventh Edition; Wolters Kluwer Health Adis (ESP): Alphen aan den Rijn, The Netherlands, 2012; ISBN 978-1-60547-461-8. [Google Scholar]
- Ramos, D.; Mar, D.; Ishida, M.; Vargas, R.; Gaite, M.; Montgomery, A.; Linder, M.C. Mechanism of Copper Uptake from Blood Plasma Ceruloplasmin by Mammalian Cells. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Harvey, P.J.; Handley, H.K.; Taylor, M.P. Widespread Copper and Lead Contamination of Household Drinking Water, New South Wales, Australia. Environ. Res. 2016, 151, 275–285. [Google Scholar] [CrossRef]
- Babi, E.; Tariba, B.; Kovai, J.; Piznet, A.; Varnai, V.; Macan, J. Relevance of Serum Copper Elevation Induced by Oral Contraceptives: A Meta-Analysis—Contraception. Contraception 2012, 87, 790–800. [Google Scholar] [CrossRef]
- Jalali, K.; Nouairi, I.; Kallala, N.; M’Sehli, W.; Zribi, K.; Mhadhbi, H. Germination, Seedling Growth, and Antioxidant Activity in Four Legume (Fabaceae) Species under Copper Sulphate Fungcide Treatment. Pak. J. Bot. 2018, 50, 1599–1606. [Google Scholar]
- Snyder, J.S. Recalibrating the Relevance of Adult Neurogenesis. Trends Neurosci. 2019, 42, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Coupé, P.; Manjón, J.V.; Lanuza, E.; Catheline, G. Lifespan Changes of the Human Brain In Alzheimer’s Disease. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Graham, N.; Warner, J. Alzheimer’s Disease and Other Dementias; Family Doctor Publications Ltd.: Poole, UK, 2009; ISBN 978-1-903474-61-7. [Google Scholar]
- Brandes, R. The Current Neuroscientific Understanding of Alzheimer’s Disease. Purs. J. Undergrad. Res. Univ. Tenn. 2020, 10, 2. [Google Scholar]
- Li, Z.; Del-Aguila, J.L.; Dube, U.; Budde, J.; Martinez, R.; Black, K.; Xiao, Q.; Cairns, N.J.; Dougherty, J.D.; Lee, J.-M.; et al. Genetic Variants Associated with Alzheimer’s Disease Confer Different Cerebral Cortex Cell-Type Population Structure. Genome Med. 2018, 10, 43. [Google Scholar] [CrossRef]
- Hu, X.; Hu, Z.-L.; Li, Z.; Ruan, C.-S.; Qiu, W.-Y.; Pan, A.; Li, C.-Q.; Cai, Y.; Shen, L.; Chu, Y.; et al. Sortilin Fragments Deposit at Senile Plaques in Human Cerebrum. Front. Neuroanat. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Dobson, R.J.B.; Newhouse, S.J. A Meta-Analysis of Alzheimer’s Disease Brain Transcriptomic Data. J. Alzheimer’s Dis. 2019, 68, 1635–1656. [Google Scholar] [CrossRef] [PubMed]
- Yarris, L. Copper on the Brain. Available online: https://newscenter.lbl.gov/2013/05/24/copper-on-the-brain/ (accessed on 14 July 2020).
- Pal, A.; Badyal, R.K.; Vasishta, R.K.; Attri, S.V.; Thapa, B.R.; Prasad, R. Biochemical, Histological, and Memory Impairment Effects of Chronic Copper Toxicity: A Model for Non-Wilsonian Brain Copper Toxicosis in Wistar Rat. Biol. Trace Elem. Res. 2013, 153, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Younger, S.H.; Zhang, L.; Thompson-Peer, K.L.; Li, T.; Jan, L.Y.; Jan, Y.N. Glial Ensheathment of the Somatodendritic Compartment Regulates Sensory Neuron Structure and Activity. Proc. Natl. Acad. Sci. USA 2019, 116, 5126–5134. [Google Scholar] [CrossRef]
- Nazer, H.; Nazer, D.; Windle, M.; Cuffari, C.; Deodhar, J. Pediatric Fulminant Hepatic Failure: Background, Pathophysiology, Epidemiology. 2020. Available online: https://emedicine.medscape.com/article/929028-overview (accessed on 6 January 2021).
- Nehring, S.M.; Tadi, P.; Tenny, S. Cerebral Edema. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Pal, A.; Prasad, R. Regional Distribution of Copper, Zinc and Iron in Brain of Wistar Rat Model for Non-Wilsonian Brain Copper Toxicosis. Indian J. Clin. Biochem. 2016, 31, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of Astrocyte-Mediated Cerebral Edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Bourgade, K.; Khalil, A.; Zerif, E.; Larbi, A.; Hirokawa, K.; Pawelec, G.; Bocti, C.; Lacombe, G.; et al. Can an Infection Hypothesis Explain the Beta Amyloid Hypothesis of Alzheimer’s Disease? Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef]
- Kardos, J.; Héja, L.; Simon, Á.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper Signalling: Causes and Consequences. Cell Commun. Signal. 2018, 16. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper Toxicity in Alzheimer’s Disease: Cognitive Loss from Ingestion of Inorganic Copper. J. Trace Elem. Med. Biol. 2012, 26, 89–92. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A. The Role of Amyloid-Beta in the Regulation of Memory. Biochem. Pharmacol. 2014, 88, 479–485. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological Relevance and Mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid Cascade Hypothesis: Pathogenesis and Therapeutic Strategies in Alzheimer’s Disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Hillen, H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Overview of Neuron Structure and Function. In Molecular Cell Biology, 4th ed.; W.H. Freeman and Company: New York, NY, USA, 2000. [Google Scholar]
- Rudenko, G. Neurexins—Versatile Molecular Platforms in the Synaptic Cleft. Curr. Opin. Struct. Biol. 2019, 54, 112–121. [Google Scholar] [CrossRef]
- Tricaud, N. Myelinating Schwann Cell Polarity and Mechanically-Driven Myelin Sheath Elongation. Available online: https://www.frontiersin.org/articles/10.3389/fncel.2017.00414/full (accessed on 10 October 2020).
- Mukherjee, A.; Soto, C. Prion-Like Protein Aggregates and Type 2 Diabetes. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Greenwald, J.; Riek, R. Biology of Amyloid: Structure, Function, and Regulation. Structure 2010, 18, 1244–1260. [Google Scholar] [CrossRef]
- Han, X.-J.; Hu, Y.-Y.; Yang, Z.-J.; Jiang, L.-P.; Shi, S.-L.; Li, Y.-R.; Guo, M.-Y.; Wu, H.-L.; Wan, Y.-Y. Amyloid β-42 Induces Neuronal Apoptosis by Targeting Mitochondria. Mol. Med. Rep. 2017, 16, 4521–4528. [Google Scholar] [CrossRef]
- Bendlin, B.B.; Zetterberg, H. Screening with a High-Precision Blood-Based Assay for Alzheimer Disease. Neurology 2019, 93, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2017, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Talwar, P.; Grover, S.; Sinha, J.; Chandna, P.; Agarwal, R.; Kushwaha, S.; Kukreti, R. Multifactorial Analysis of a Biomarker Pool for Alzheimer Disease Risk in a North Indian Population. Dement. Geriatr. Cogn. Disord. 2017, 44, 25–34. [Google Scholar] [CrossRef]
- González, C.; Martín, T.; Cacho, J.; Breñas, M.T.; Arroyo, T.; García-Berrocal, B.; Navajo, J.A.; González-Buitrago, J.M. Serum Zinc, Copper, Insulin and Lipids in Alzheimer’s Disease Epsilon 4 Apolipoprotein E Allele Carriers. Eur. J. Clin. Investig. 1999, 29, 637–642. [Google Scholar] [CrossRef]
- Kitazawa, M.; Hsu, H.-W.; Medeiros, R. Copper Exposure Perturbs Brain Inflammatory Responses and Impairs Clearance of Amyloid-Beta. Toxicol. Sci. 2016, 152, 194–204. [Google Scholar] [CrossRef]
- Segal, M. Dendritic Spines: Morphological Building Blocks of Memory. Neurobiol. Learn. Mem. 2017, 138, 3–9. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, R.; Aschner, M. Commonalities between Copper Neurotoxicity and Alzheimer’s Disease. Toxics 2021, 9, 4. https://doi.org/10.3390/toxics9010004
Patel R, Aschner M. Commonalities between Copper Neurotoxicity and Alzheimer’s Disease. Toxics. 2021; 9(1):4. https://doi.org/10.3390/toxics9010004
Chicago/Turabian StylePatel, Roshni, and Michael Aschner. 2021. "Commonalities between Copper Neurotoxicity and Alzheimer’s Disease" Toxics 9, no. 1: 4. https://doi.org/10.3390/toxics9010004
APA StylePatel, R., & Aschner, M. (2021). Commonalities between Copper Neurotoxicity and Alzheimer’s Disease. Toxics, 9(1), 4. https://doi.org/10.3390/toxics9010004