Nicotine Causes Nephrotoxicity through the Induction of NLRP6 Inflammasome and Alpha7 Nicotinic Acetylcholine Receptor

 , , ,
, , ,
Abstract

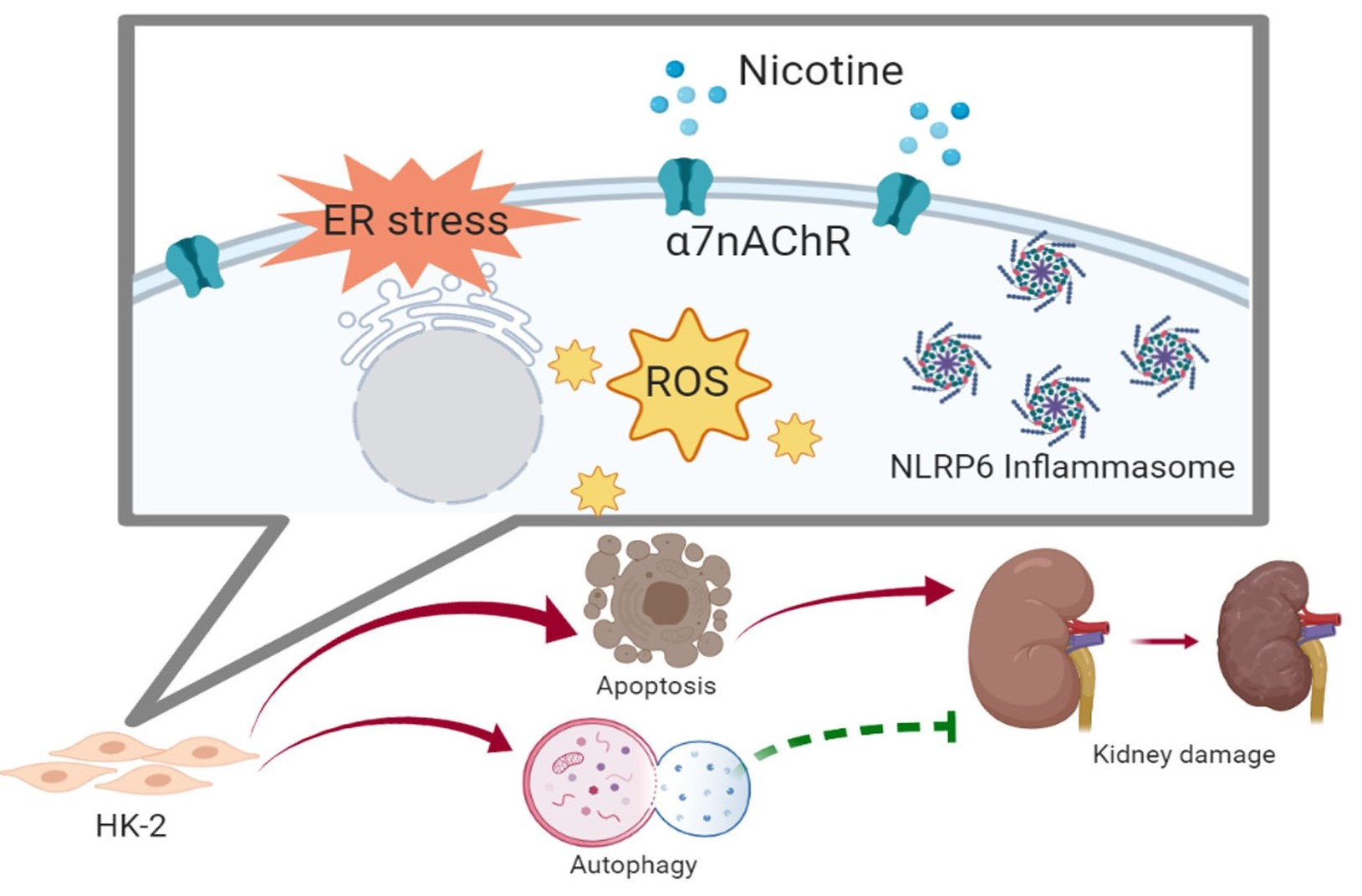{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Nicotine Treatment
2.2. SRB Cell Viability Assay
2.3. Measurement of Intracellular Reactive Oxygen Species (ROS) Level
2.4. Western Blotting
2.5. Detection of IL-1β by ELISA
2.6. Annexin V and Propidium Iodide (PI)
2.7. Immunofluorescence Assay
2.8. RNA Interference (RNAi)
2.9. Animal Studies
2.10. Histological and Immunohistochemical Analysis
2.11. Detection of Cystatin C by ELISA
2.12. Statistical Analysis
3. Results
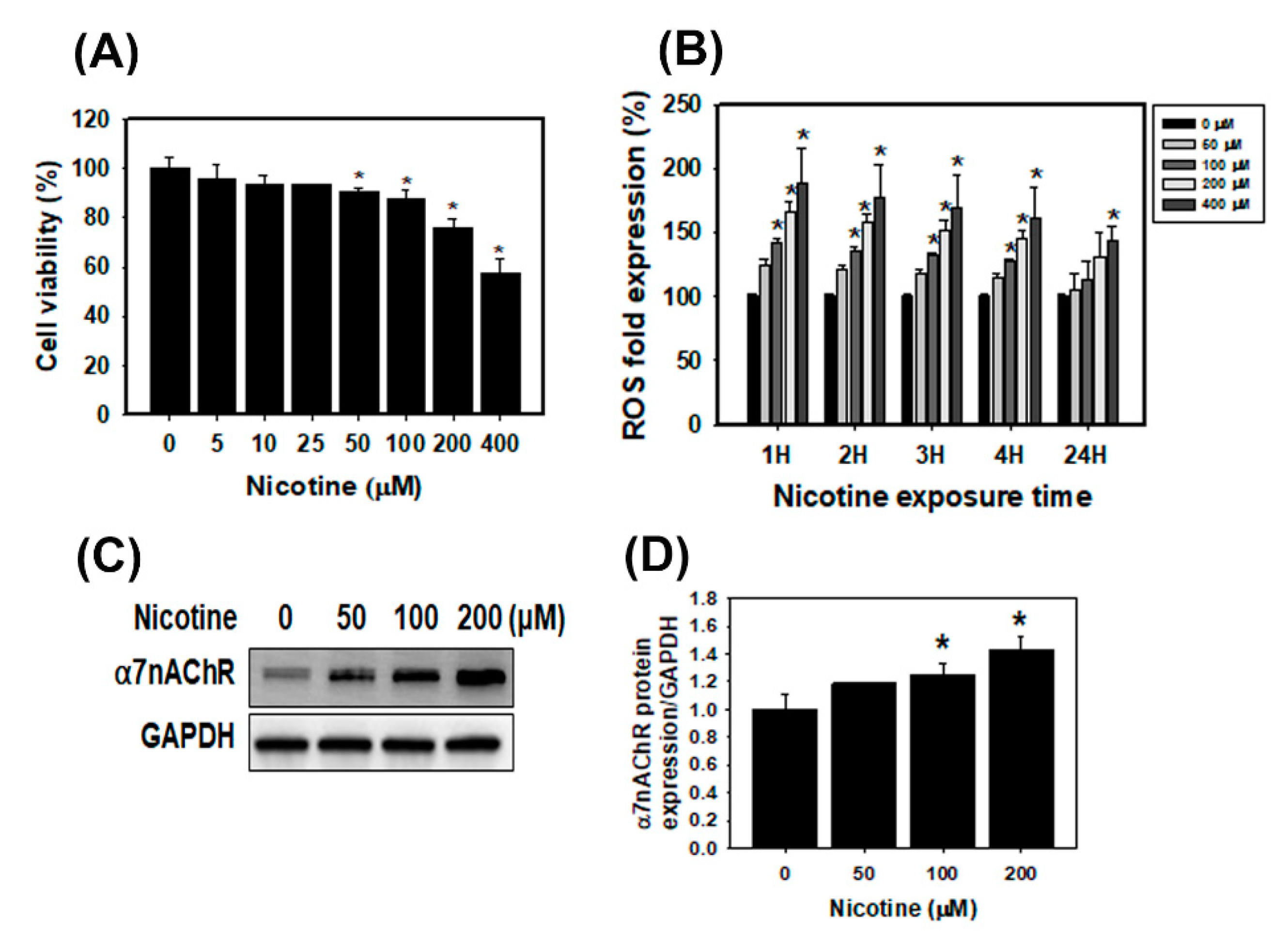
3.1. The Cell Viability, ROS Generation and α7nAChR Expression in Human Kidney Cells Treated with Nicotine
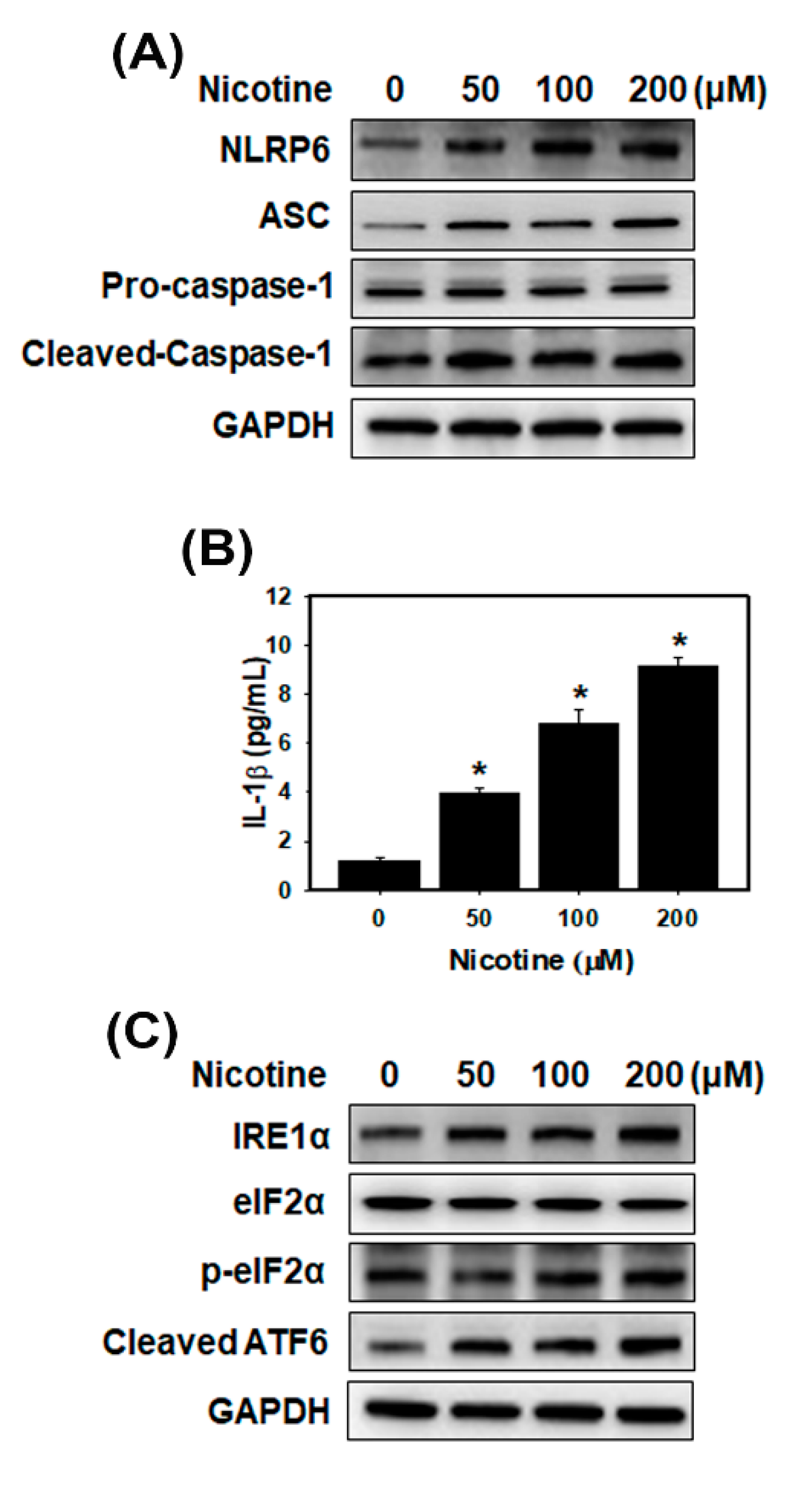
3.2. Nicotine Activates the NLRP6 Inflammasome and Induces ER Stress in Human Kidney Cells
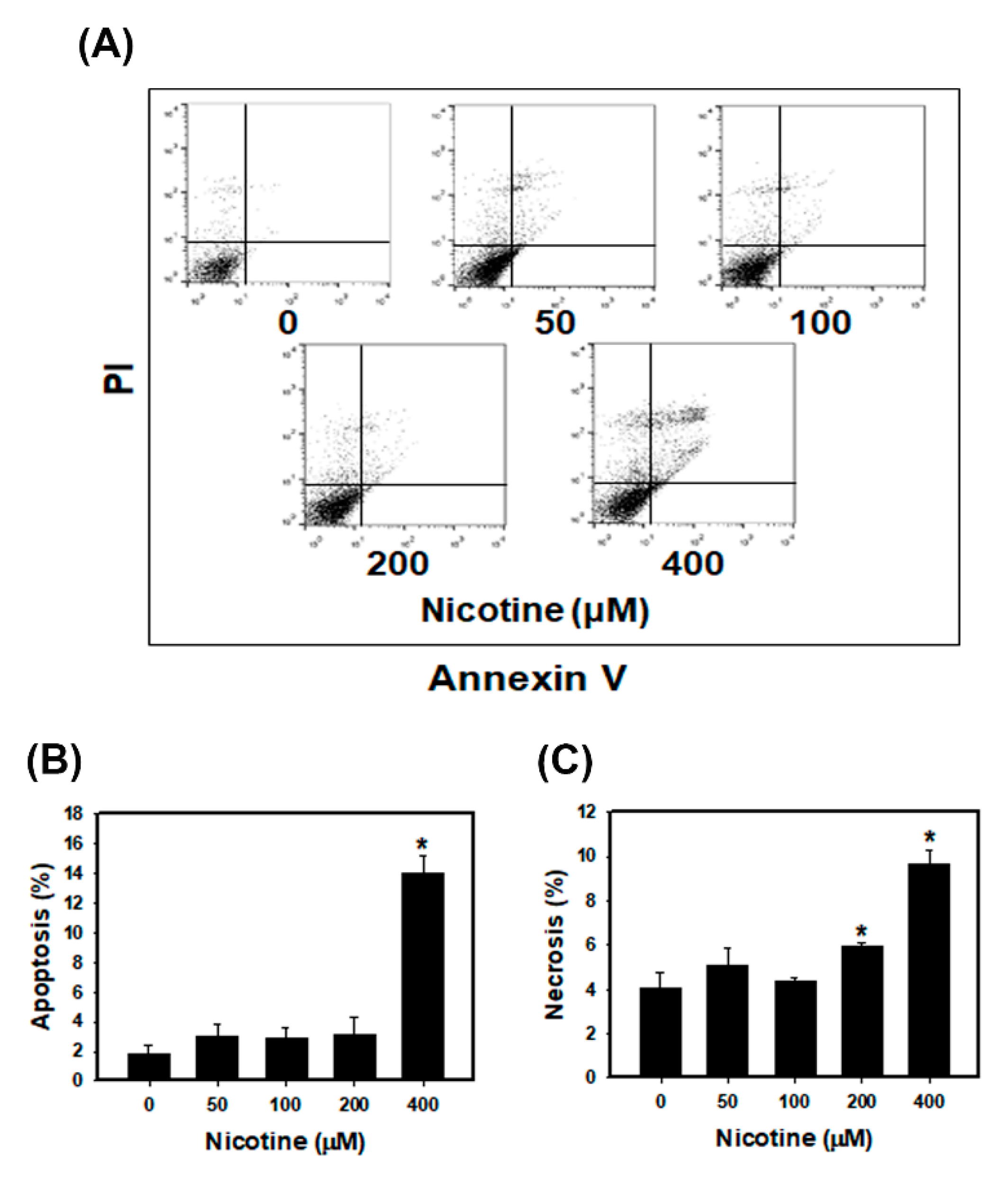
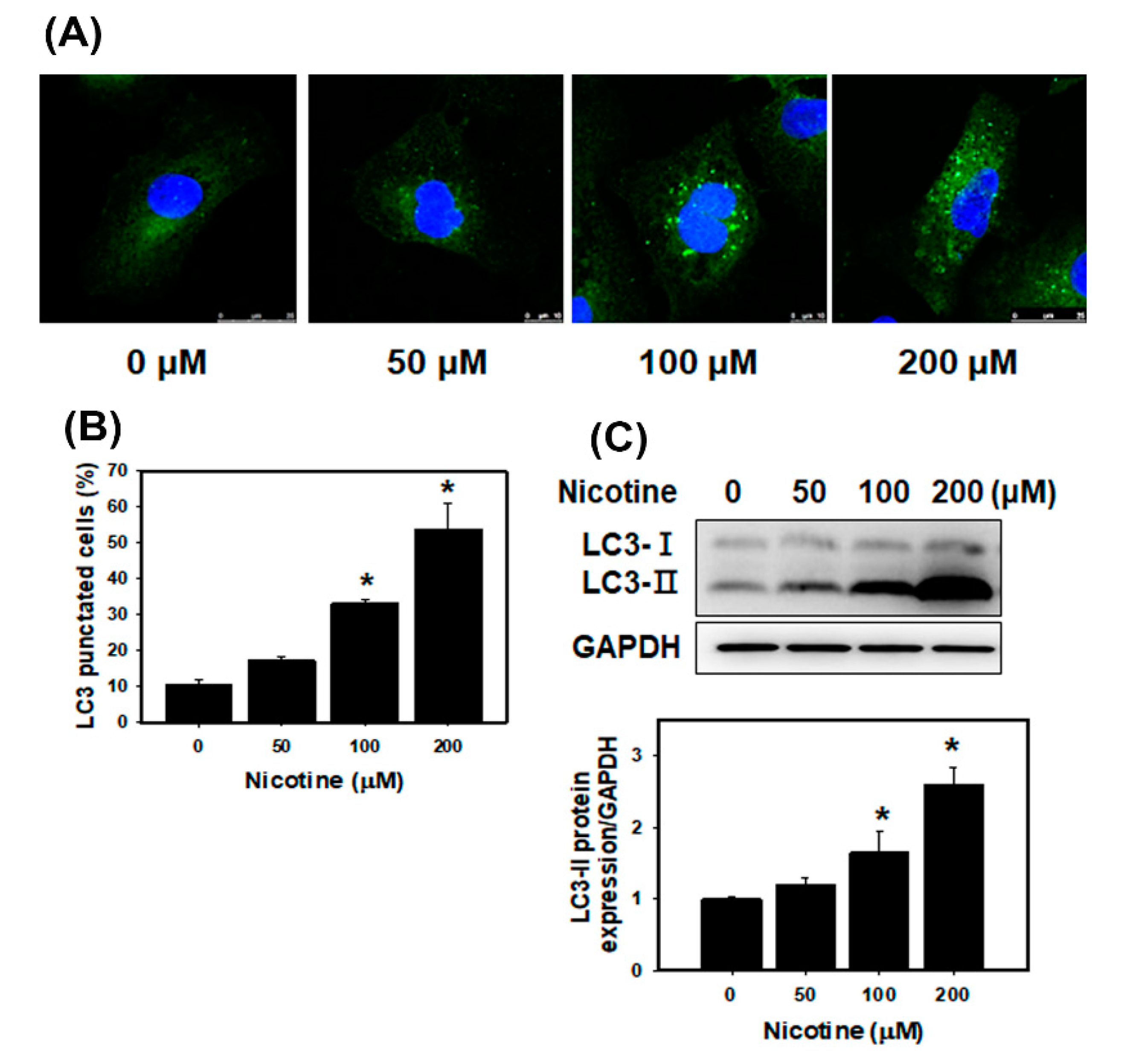
3.3. Nicotine Induces Mild Apoptosis and Necrosis but Triggers Significant Autophagy in Human Kidney Cells
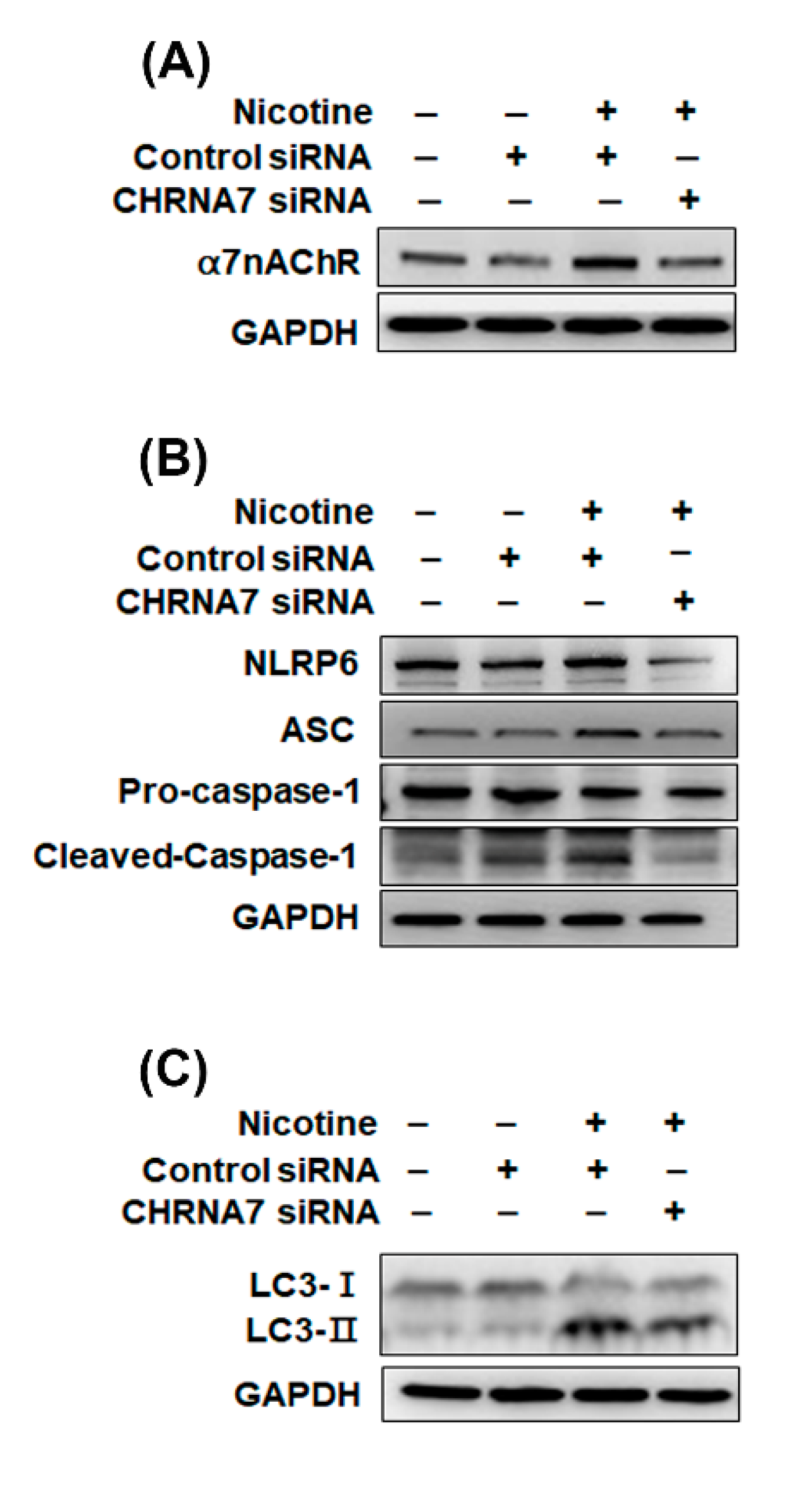
3.4. The Relationship between α7nAChR, NLRP6 Inflammasome and Autophagy in Kidney Cells That Were Treated with Nicotine
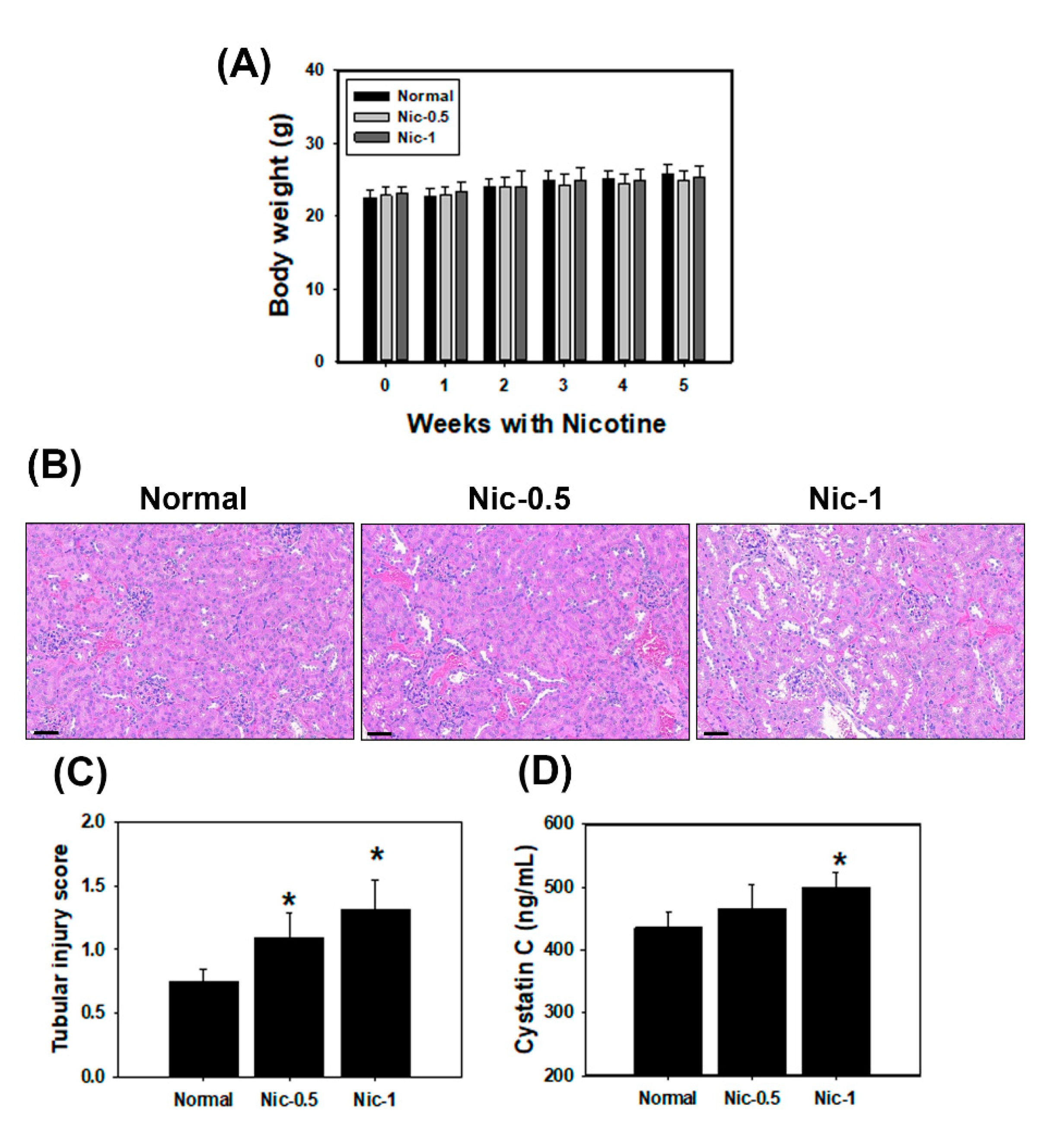
3.5. Chronic Nicotine Exposure Results in Renal Injury
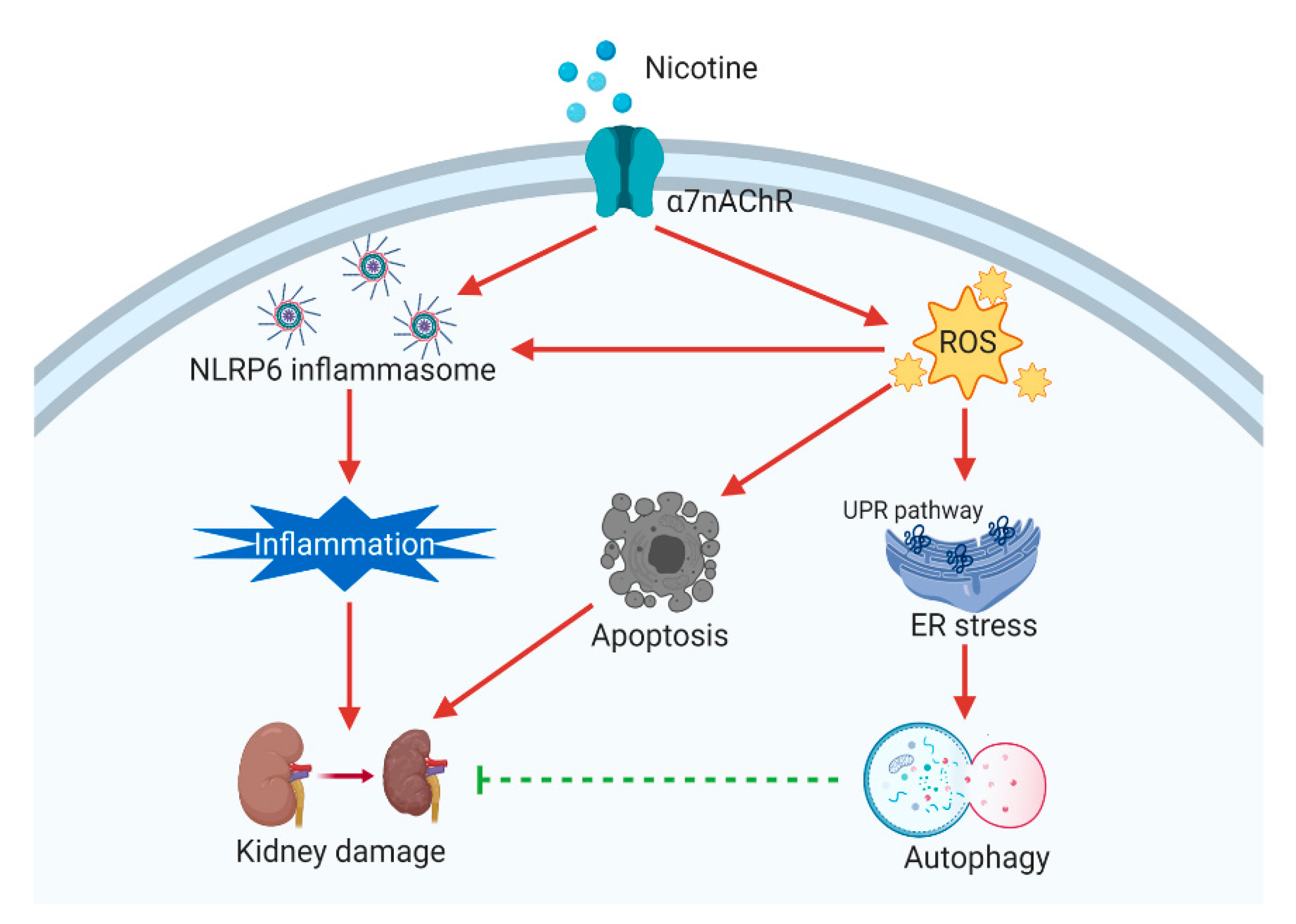
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chow, W.H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Guo, T.; Chen, Z.; Zhang, H.; Cai, S.; Yang, M.; Chen, P.; Guan, C.; Fang, X. Hypermethylation of mitochondrial transcription factor A induced by cigarette smoke is associated with chronic obstructive pulmonary disease. Exp. Lung Res. 2019, 45, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Jain, G.; Jaimes, E.A. Nicotine signaling and progression of chronic kidney disease in smokers. Biochem. Pharmacol. 2013, 86, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Pontieri, F.E.; Tanda, G.; Orzi, F.; Di Chiara, G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 1996, 382, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, R.; Albermann, K.; Lang, S. Nicotine and apoptosis. Apoptosis 2007, 12, 1927–1943. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, E.A.; Tian, R.X.; Joshi, M.S.; Raij, L. Nicotine augments glomerular injury in a rat model of acute nephritis. Am. J. Nephrol. 2009, 29, 319–326. [Google Scholar] [CrossRef]
- Kim, C.S.; Choi, J.S.; Joo, S.Y.; Bae, E.H.; Ma, S.K.; Lee, J.; Kim, S.W. Nicotine-Induced Apoptosis in Human Renal Proximal Tubular Epithelial Cells. PLoS ONE 2016, 11, e0152591. [Google Scholar] [CrossRef]
- Couturier, S.; Bertrand, D.; Matter, J.M.; Hernandez, M.C.; Bertrand, S.; Millar, N.; Valera, S.; Barkas, T.; Ballivet, M. A neuronal nicotinic acetylcholine receptor subunit (alpha 7) is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron 1990, 5, 847–856. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha 7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Hoover, D.B. Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol. Ther. 2017, 179, 1–16. [Google Scholar] [CrossRef]
- Kiss, A.; Tratsiakovich, Y.; Mahdi, A.; Yang, J.; Gonon, A.T.; Podesser, B.K.; Pernow, J. Vagal nerve stimulation reduces infarct size via a mechanism involving the alpha-7 nicotinic acetylcholine receptor and downregulation of cardiac and vascular arginase. Acta Physiol. 2017, 221, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation—A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int. 2019, 96, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.A.; Kim, W.Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-beta and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 78. [Google Scholar] [CrossRef]
- Lin, Y.F.; Lee, Y.H.; Hsu, Y.H.; Chen, Y.J.; Lin, Y.F.; Cheng, F.Y.; Chiu, H.W. Resveratrol-loaded nanoparticles conjugated with kidney injury molecule-1 as a drug delivery system for potential use in chronic kidney disease. Nanomedicine 2017, 12, 2741–2756. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, B.; Zhu, J.; Wang, D.; Wang, Y. Nicotine-mediated autophagy of vascular smooth muscle cell accelerates atherosclerosis via nAChRs/ROS/NF-κB signaling pathway. Atherosclerosis 2019, 284, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, H.; Inagi, R. Stress Signal Network between Hypoxia and ER Stress in Chronic Kidney Disease. Front. Physiol. 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.A.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Wong, M.K.; Holloway, A.C.; Hardy, D.B. Nicotine Directly Induces Endoplasmic Reticulum Stress Response in Rat Placental Trophoblast Giant Cells. Toxicol. Sci. 2016, 151, 23–34. [Google Scholar] [CrossRef][Green Version]
- Seoane-Collazo, P.; Martínez de Morentin, P.B.; Fernø, J.; Diéguez, C.; Nogueiras, R.; López, M. Nicotine improves obesity and hepatic steatosis and ER stress in diet-induced obese male rats. Endocrinology 2014, 155, 1679–1689. [Google Scholar] [CrossRef]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Saremi, S.; Atyabi, F.; Akhlaghi, S.P.; Ostad, S.N.; Dinarvand, R. Thiolated chitosan nanoparticles for enhancing oral absorption of docetaxel: Preparation, in vitro and ex vivo evaluation. Int. J. Nanomed. 2011, 6, 119–128. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.-Z.; Xu, Z.-Q.; Chen, X.-W.; Wei, W.; Liu, C. Activating α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of β-arrestin-1. CNS Neurosci. Ther. 2017, 23, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Dharnidharka, V.R.; Kwon, C.; Stevens, G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: A meta-analysis. Am. J. Kidney Dis. 2002, 40, 221–226. [Google Scholar] [CrossRef]
- Vaidya, V.S.; Ferguson, M.A.; Bonventre, J.V. Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 463–493. [Google Scholar] [CrossRef]
- Orth, S.R.; Hallan, S.I. Smoking: A risk factor for progression of chronic kidney disease and for cardiovascular morbidity and mortality in renal patients--absence of evidence or evidence of absence? Clin. J. Am. Soc. Nephrol. 2008, 3, 226–236. [Google Scholar] [CrossRef]
- Ishani, A.; Grandits, G.A.; Grimm, R.H.; Svendsen, K.H.; Collins, A.J.; Prineas, R.J.; Neaton, J.D. Association of single measurements of dipstick proteinuria, estimated glomerular filtration rate, and hematocrit with 25-year incidence of end-stage renal disease in the multiple risk factor intervention trial. J. Am. Soc. Nephrol. 2006, 17, 1444–1452. [Google Scholar] [CrossRef]
- Huang, M.F.; Lin, W.L.; Ma, Y.C. A study of reactive oxygen species in mainstream of cigarette. Indoor Air 2005, 15, 135–140. [Google Scholar] [CrossRef]
- Saldivar, L.; Luna, M.; Reyes, E.; Soto, R.; Fortoul, T.I. Cadmium determination in Mexican-produced tobacco. Environ. Res. 1991, 55, 91–96. [Google Scholar] [CrossRef]
- Fowles, J.; Dybing, E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob. Control 2003, 12, 424–430. [Google Scholar] [CrossRef]
- Guo, J.; Chu, M.; Abbeyquaye, T.; Chen, C.-Y. Persistent nicotine treatment potentiates amplification of the dihydrofolate reductase gene in rat lung epithelial cells as a consequence of Ras activation. J. Biol. Chem. 2005, 280, 30422–30431. [Google Scholar] [CrossRef]
- Jaimes, E.A.; Tian, R.-X.; Raij, L. Nicotine: The link between cigarette smoking and the progression of renal injury? Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H76–H82. [Google Scholar] [CrossRef]
- Ziyadeh, F.N.; Hoffman, B.B.; Han, D.C.; Iglesias-De La Cruz, M.C.; Hong, S.W.; Isono, M.; Chen, S.; McGowan, T.A.; Sharma, K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 8015–8020. [Google Scholar] [CrossRef]
- Eddy, A.A. Molecular basis of renal fibrosis. Pediatric Nephrol. 2000, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Corradi, J.; Bouzat, C. Understanding the Bases of Function and Modulation of α7 Nicotinic Receptors: Implications for Drug Discovery. Mol. Pharmacol. 2016, 90, 288–299. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.K.; Nicholson, C.J.; Holloway, A.C.; Hardy, D.B. Maternal nicotine exposure leads to impaired disulfide bond formation and augmented endoplasmic reticulum stress in the rat placenta. PLoS ONE 2015, 10, e0122295. [Google Scholar] [CrossRef]
- Lu, B.; Kwan, K.; Levine, Y.A.; Olofsson, P.S.; Yang, H.; Li, J.; Joshi, S.; Wang, H.; Andersson, U.; Chavan, S.S.; et al. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 2014, 20, 350–358. [Google Scholar] [CrossRef]
- Richter, K.; Sagawe, S.; Hecker, A.; Küllmar, M.; Askevold, I.; Damm, J.; Heldmann, S.; Pöhlmann, M.; Ruhrmann, S.; Sander, M.; et al. C-Reactive Protein Stimulates Nicotinic Acetylcholine Receptors to Control ATP-Mediated Monocytic Inflammasome Activation. Front. Immunol. 2018, 9, 1604. [Google Scholar] [CrossRef]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid. Med. Cell. Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef]
- Kaushal, G.P. Autophagy protects proximal tubular cells from injury and apoptosis. Kidney Int. 2012, 82, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Bi, P.; Luo, D.; Guan, Y.; Zeng, W.; Xiang, H.; Mi, Q.; Yang, G.; Li, X.; Yang, B. Nicotine-induced autophagy via AMPK/mTOR pathway exerts protective effect in colitis mouse model. Chem. Biol. Interact. 2020, 317, 108943. [Google Scholar] [CrossRef]
- Xing, R.; Cheng, X.; Qi, Y.; Tian, X.; Yan, C.; Liu, D.; Han, Y. Low-dose nicotine promotes autophagy of cardiomyocytes by upregulating HO-1 expression. Biochem. Biophys. Res. Commun. 2020, 522, 1015–1021. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, C.-M.; Lee, Y.-H.; Chiu, I.-J.; Chiu, Y.-J.; Sung, L.-C.; Hsu, Y.-H.; Chiu, H.-W. Nicotine Causes Nephrotoxicity through the Induction of NLRP6 Inflammasome and Alpha7 Nicotinic Acetylcholine Receptor. Toxics 2020, 8, 92. https://doi.org/10.3390/toxics8040092
Zheng C-M, Lee Y-H, Chiu I-J, Chiu Y-J, Sung L-C, Hsu Y-H, Chiu H-W. Nicotine Causes Nephrotoxicity through the Induction of NLRP6 Inflammasome and Alpha7 Nicotinic Acetylcholine Receptor. Toxics. 2020; 8(4):92. https://doi.org/10.3390/toxics8040092
Chicago/Turabian StyleZheng, Cai-Mei, Yu-Hsuan Lee, I-Jen Chiu, Yu-Jhe Chiu, Li-Chin Sung, Yung-Ho Hsu, and Hui-Wen Chiu. 2020. "Nicotine Causes Nephrotoxicity through the Induction of NLRP6 Inflammasome and Alpha7 Nicotinic Acetylcholine Receptor" Toxics 8, no. 4: 92. https://doi.org/10.3390/toxics8040092
APA StyleZheng, C.-M., Lee, Y.-H., Chiu, I.-J., Chiu, Y.-J., Sung, L.-C., Hsu, Y.-H., & Chiu, H.-W. (2020). Nicotine Causes Nephrotoxicity through the Induction of NLRP6 Inflammasome and Alpha7 Nicotinic Acetylcholine Receptor. Toxics, 8(4), 92. https://doi.org/10.3390/toxics8040092