Zinc Oxide Nanoparticles Induced Oxidative DNA Damage, Inflammation and Apoptosis in Rat’s Brain after Oral Exposure


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Kits
2.2. Particle Characterization
2.3. Study Design
2.4. Processing of Brain Tissue for Biochemical Studies
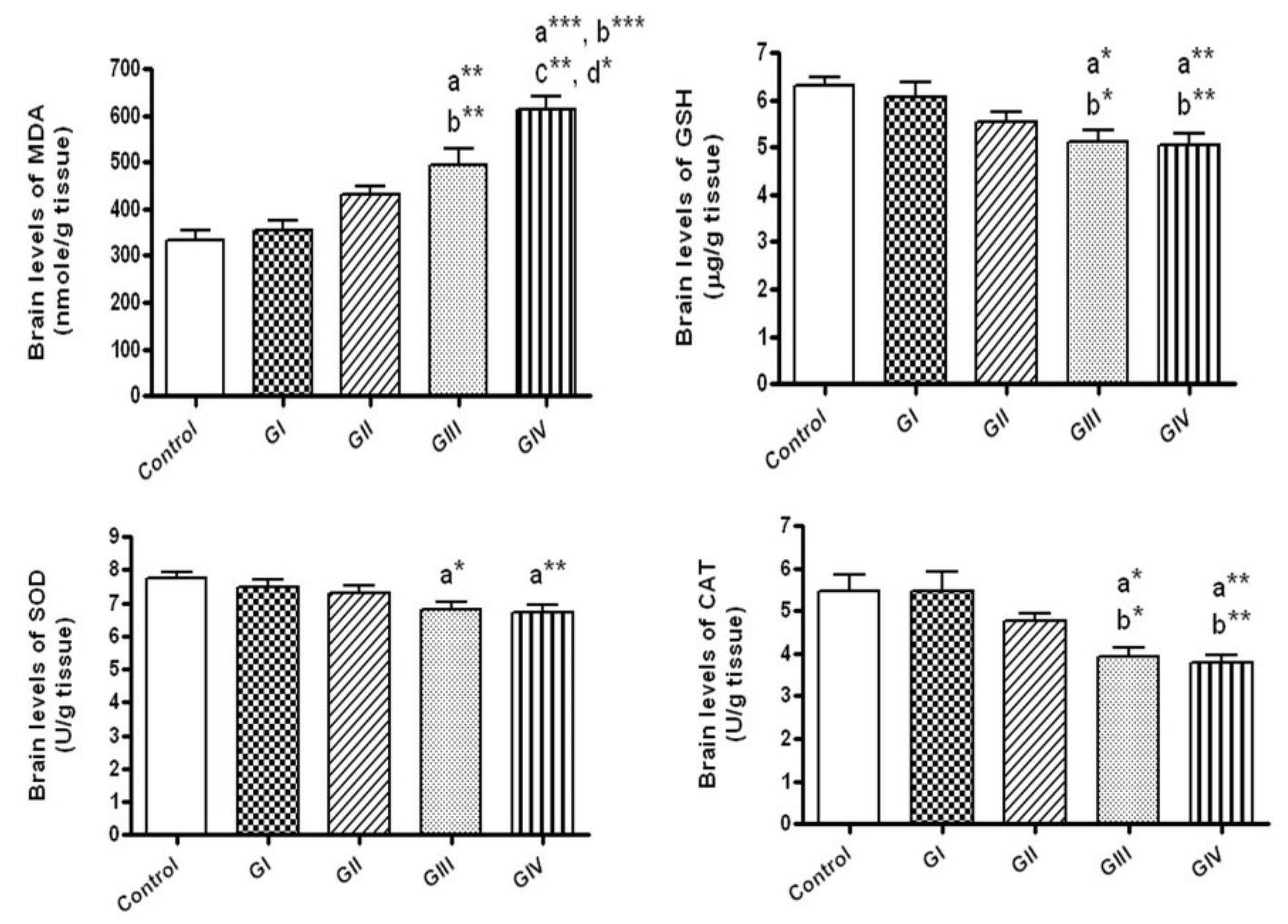
2.5. Assessment of Oxidative Stress Markers (Lipid Peroxidation and Antioxidants) in Brain Tissue
2.6. Measurement of Nitrite (an Index of Nitric Oxide)
2.7. Assay of HSP-70 in Brain Tissues
2.8. Assessment of Inflammatory Markers (TNF-α & IL-1β) in Brain Tissues
2.8.1. Measurement of IL-1β
2.8.2. Measurement of Tumor Necrosis Factor-α (TNF-α)
2.9. Assay of Apoptotic Markers (Fas & Caspase-3) in Brain Tissues
2.10. Assay of DNA Damage Markers
Measurement of DNA Fragmentation
2.11. Comet DNA Assay
2.12. Statistical Analysis
3. Results
3.1. Particle Characterization
3.2. Biochemical Measurements
3.3. Effect of ZnONPs on the Oxidative Stress Markers in Brain Tissue
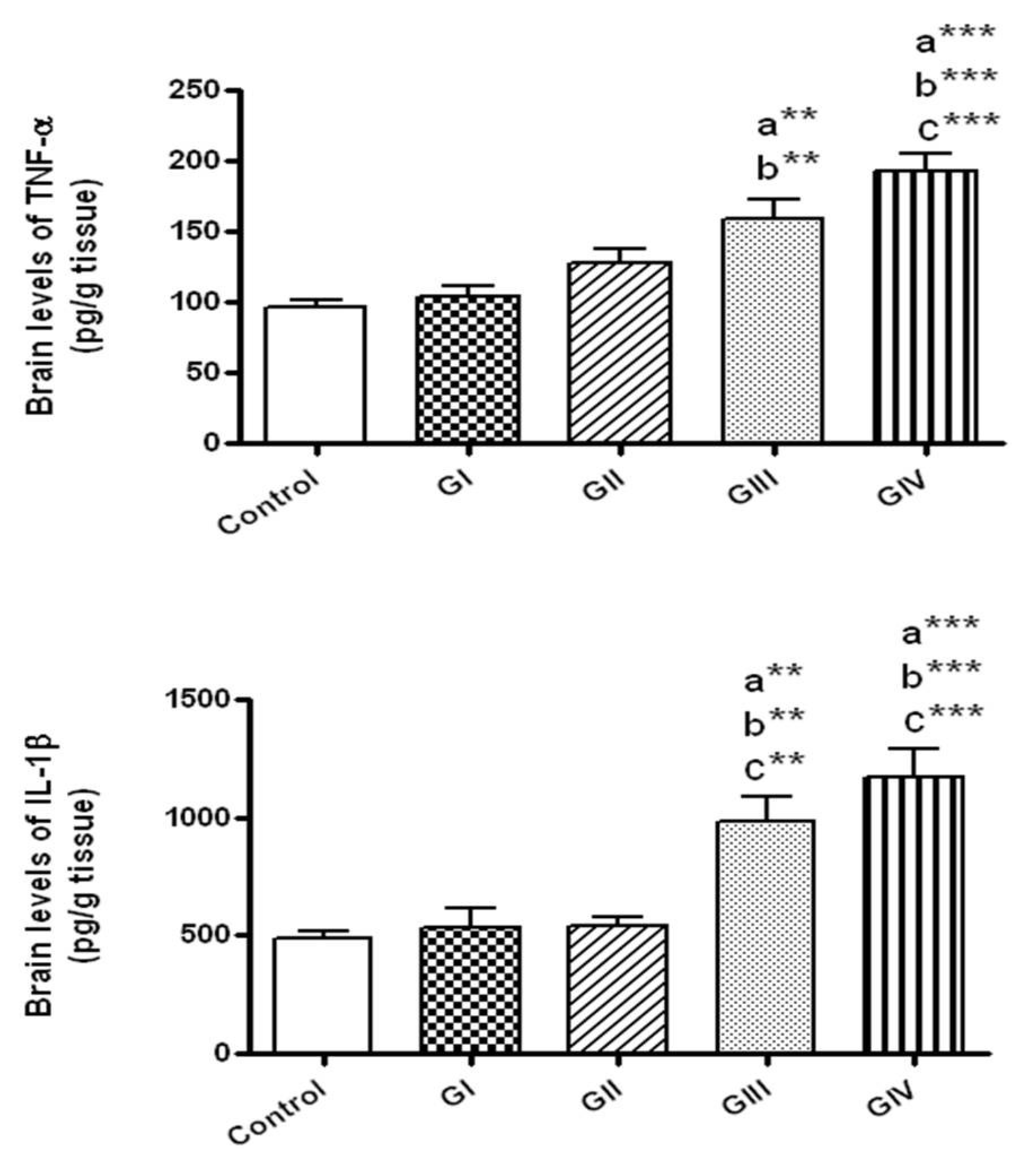
3.4. Effect of ZnONPs on the Inflammatory Markers in Brain Tissue
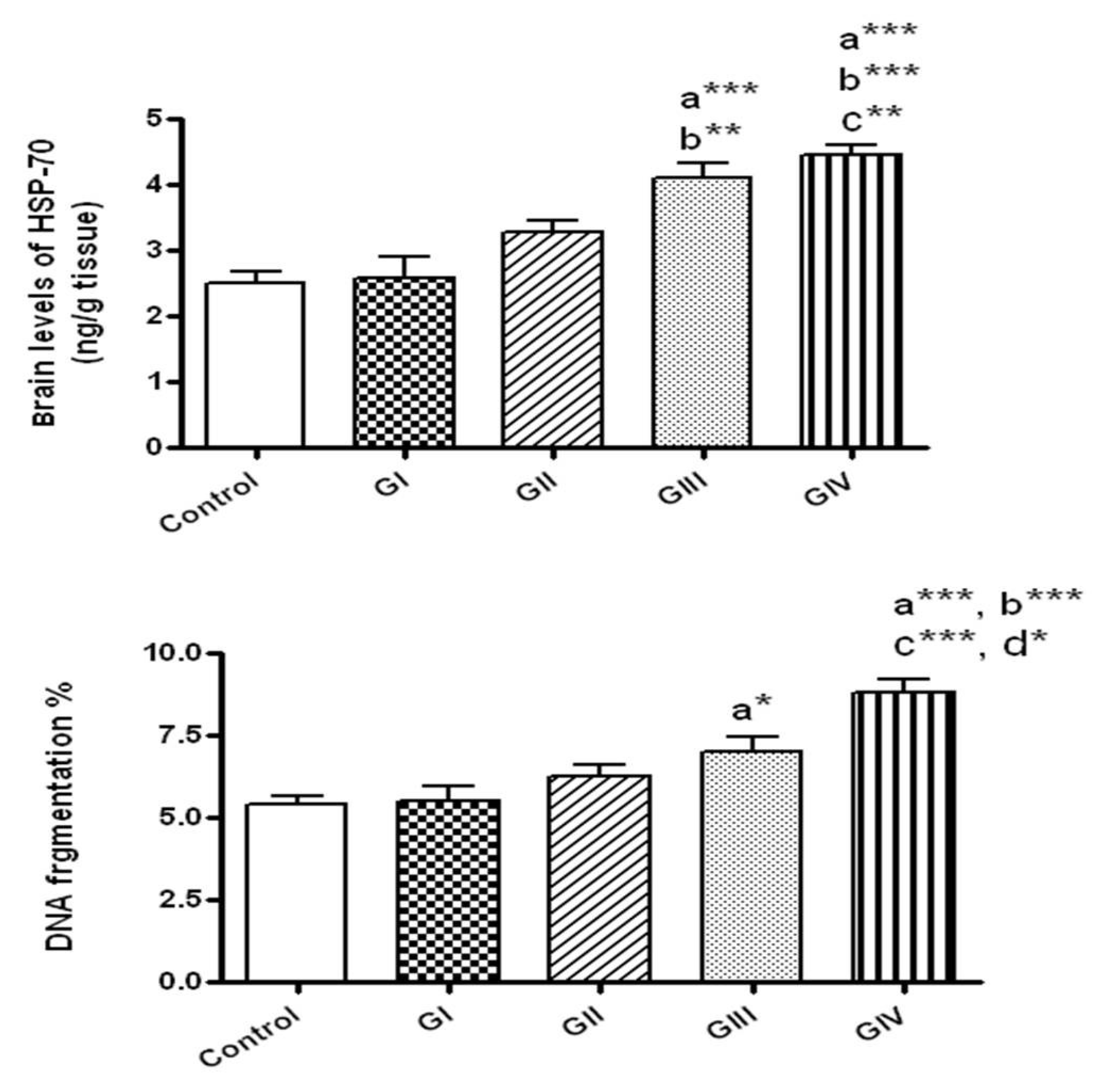
3.5. Effect of ZnONPs on HSP-70 in Brain Tissue
3.6. Effect of ZnONPs on DNA Fragmentation in Brain Tissue
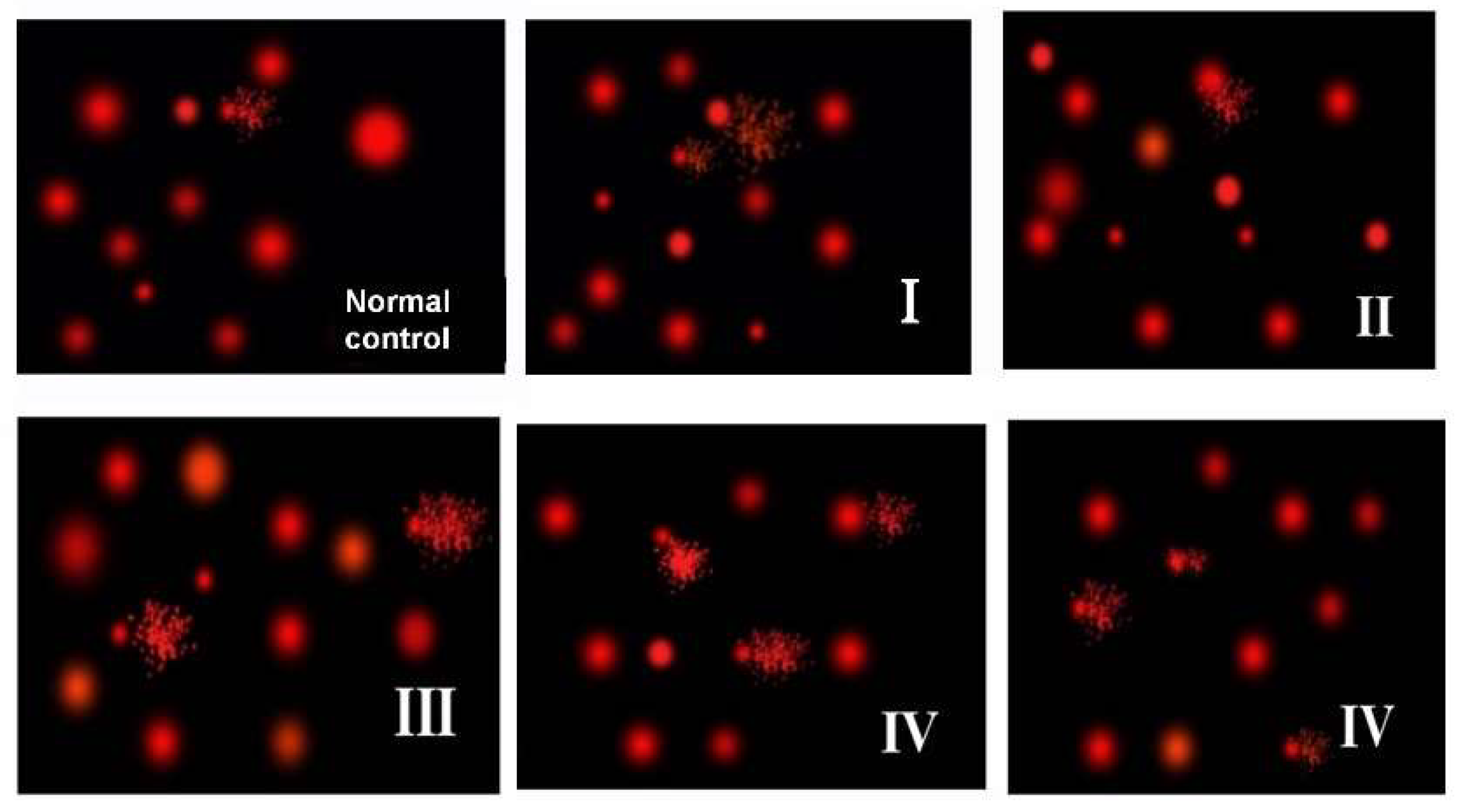
3.7. Effect of ZnONPs on DNA Comet Assay Indices in Brain Tissue
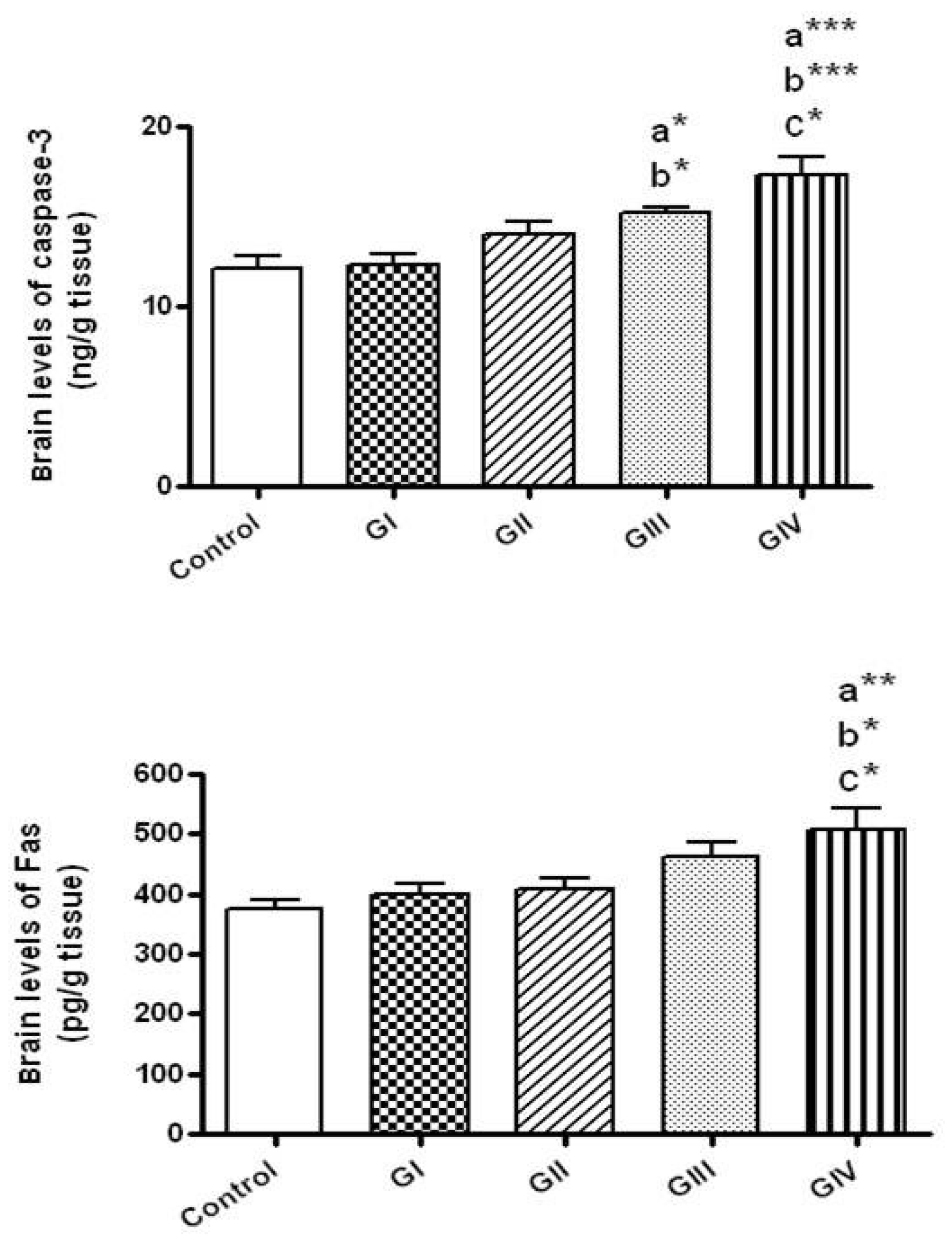
3.8. Effect of ZnONPs on Apoptotic Markers in Brain Tissue
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Silva, G.A. Neuroscience nanotechnology: Progress, opportunities and challenges. Nat. Rev. Neurosci. 2006, 7, 65–74. Available online: https://www.ncbi.nlm.nih.gov/pubmed/16371951 (accessed on 3 May 2017). [CrossRef] [PubMed]
- De Matteis, V.; Rinaldi, R. Toxicity Assessment in the Nanoparticle Era. Adv. Exp. Med. Biol. 2018, 1048, 1–19. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29453529 (accessed on 6 May 2018). [CrossRef] [PubMed]
- Vandebriel, R.J.; De Jong, W.H. A review of mammalian toxicity of ZnO nanoparticles. Nanotechnol. Sci. Appl. 2012, 5, 61–71. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3781722/ (accessed on 11 November 2017). [CrossRef] [PubMed]
- Yah, C.S.; Simate, G.S.; Iyuke, S.E. Nanoparticles toxicity and their routes of exposures. Pak. J. Pharm. Sci. 2012, 25, 477–491. Available online: https://www.intechopen.com/books/recent-advances-in-novel-drug-carrier-systems/nanoparticles-toxicity-and-their-routes-of-exposures (accessed on 3 December 2017). [CrossRef] [PubMed]
- Singh, S.; Nalwa, H.S. Nanotechnology and health safety-toxicity and risk assessments of nanostructured materials on human health. J. Nanosci. Nanotechnol. 2007, 7, 3048–3070. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18019130 (accessed on 19 November 2017). [CrossRef] [PubMed]
- Sharma, H.S.; Sharma, A. Nanoparticles aggravate heat stress induced cognitive deficits, blood-brain barrier disruption, edema formation and brain pathology. Prog. Brain Res. 2007, 162, 245–273. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17645923 (accessed on 24 May 2017). [CrossRef] [PubMed]
- Sharma, H.S.; Sharma, A. Conference scene: Nanoneuroprotection and nanoneurotoxicity: Recent progress and future perspectives. Nanomedicine 2010, 5, 533–537. Available online: https://www.futuremedicine.com/doi/abs/10.2217/nnm.10.25 (accessed on 14 November 2017). [CrossRef] [PubMed]
- Colvin, V.L. The potential environmental impact of engineered nanomaterials. Nat. Biotechnol. 2003, 21, 1166–1170. Available online: https://www.ncbi.nlm.nih.gov/pubmed/14520401 (accessed on 29 November 2017). [CrossRef] [PubMed]
- Memarzadeh, K.; Sharili, A.S.; Huang, J.; Rawlinson, S.C.; Allaker, R.P. Nanoparticulate zinc oxide as a coating material for orthopedic and dental implants. J. Biomed. Mater. Res. A 2015, 103, 981–989. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24862288 (accessed on 14 November 2017). [CrossRef] [PubMed]
- Osmond, M.J.; McCall, M.J. Zinc oxide nanoparticles in modern sunscreens: An analysis of potential exposure and hazard. Nanotoxicology 2010, 4, 15–41. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20795900 (accessed on 14 December 2017). [CrossRef] [PubMed]
- Jin, T.; Sun, D.; Su, J.Y.; Zhang, H.; Sue, H.J. Antimicrobial efficacy of zinc oxide quantum dots against Listeria monocytogenes, Salmonella enteritidis, and Escherichia coli. J. Food Sci. 2009, 74, M46–M52. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19200107 (accessed on 14 November 2017). [CrossRef] [PubMed]
- He, L.; Liu, Y.; Mustapha, A.; Lin, M. Antifungal activity of zinc oxide nanoparticles against Botrytis cinerea and Penicillium expansum. Microbiol. Res. 2011, 166, 207–215. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20630731 (accessed on 3 November 2017). [CrossRef] [PubMed]
- Rasmussen, J.W.; Martinez, E.; Louka, P.; Wingett, D.G. Zinc oxide nanoparticles for selective destruction of tumor cells and potential for drug delivery applications. Expert Opin. Drug Deliv. 2010, 7, 1063–1077. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20716019 (accessed on 3 January 2017). [CrossRef] [PubMed]
- Chuang, H.C.; Chuang, K.J.; Chen, J.K.; Hua, H.E.; Shen, Y.L.; Liao, W.N.; Lee, C.H.; Pan, C.H.; Chen, K.Y.; Lee, K.Y.; et al. Pulmonary pathobiology induced by zinc oxide nanoparticles in mice: A 24-hour and 28-day follow-up study. Toxicol. Appl. Pharmacol. 2017, 327, 13–22. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28433709 (accessed on 5 June 2017). [CrossRef] [PubMed]
- Chien, C.C.; Yan, Y.H.; Juan, H.T.; Cheng, T.J.; Liao, J.B.; Lee, H.P.; Wang, J.S. Sustained renal inflammation following 2 weeks of inhalation of occupationally relevant levels of zinc oxide nanoparticles in Sprague Dawley rats. J. Toxicol. Pathol. 2017, 30, 307–314. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5660952/ (accessed on 4 June 2017). [CrossRef] [PubMed]
- Hussein, M.M.; Ali, H.A.; Saadeldin, I.M.; Ahmed, M.M. Querectin Alleviates Zinc Oxide Nanoreprotoxicity in Male Albino Rats. J. Biochem. Mol. Toxicol. 2016, 30, 489–496. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27111109 (accessed on 16 July 2017). [CrossRef] [PubMed]
- Chuang, H.C.; Juan, H.T.; Chang, C.N.; Yan, Y.H.; Yuan, T.H.; Wang, J.S.; Chen, H.C.; Hwang, Y.H.; Lee, C.H.; Cheng, T.J. Cardiopulmonary toxicity of pulmonary exposure to occupationally relevant zinc oxide nanoparticles. Nanotoxicology 2014, 8, 593–604. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23738974 (accessed on 3 December 2017). [CrossRef] [PubMed]
- Nounou, H.; Attia, H.; Shalaby, M.; Arafa, M. Oral exposure to zinc oxide nanoparticles induced oxidative damage, inflammation and genotoxicity in rat’s lung. Life Sci. 2013, 10, 1969–1979. Available online: https://pdfs.semanticscholar.org/1733/b8b740354e6dd7b76fb0e061ea953adf5ca7.pdf (accessed on 3 February 2017).
- Li, C.H.; Shen, C.C.; Cheng, Y.W.; Huang, S.H.; Wu, C.C.; Kao, C.C.; Liao, J.W.; Kang, J.J. Organ biodistribution, clearance, and genotoxicity of orally administered zinc oxide nanoparticles inmice. Nanotoxicology 2012, 6, 746–756. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21950449 (accessed on 12 January 2017). [CrossRef] [PubMed]
- Cho, W.S.; Duffin, R.; Howie, S.E.; Scotton, C.J.; Wallace, W.A.; Macnee, W.; Bradley, M.; Megson, I.L.; Donaldson, K. Progressive severe lung injury by zinc oxide nanoparticles; the role of Zn2+ dissolution inside lysosomes. Part. Fibre Toxicol. 2011, 8, 27. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21896169 (accessed on 21 March 2017). [CrossRef] [PubMed]
- Senapati, V.A.; Kumar, A.; Gupta, G.S.; Pandey, A.K.; Dhawan, A. ZnO nanoparticles induced inflammatory response and genotoxicity in human blood cells: A mechanistic approach. Food Chem. Toxicol. 2015, 85, 61–70. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21896169 (accessed on 13 November 2017). [CrossRef] [PubMed]
- Lee, S.H.; Pie, J.E.; Kim, Y.R.; Lee, H.R.; Son, S.W.; Kim, M.K. Effects of zinc oxide nanoparticles on gene expression profile in human keratinocytes. Mol. Cell. Toxicol. 2012, 8, 113–118. Available online: https://link.springer.com/content/pdf/10.1007/s13273-012-0014-8.pdf (accessed on 6 February 2017). [CrossRef]
- Gojova, A.; Guo, B.; Kota, R.S.; Rutledge, J.C.; Kennedy, I.M.; Barakat, A.I. Induction of inflammation in vascular endothelial cells by metal oxide nanoparticles: Effect of particle composition. Environ. Health Perspect. 2007, 115, 403–409. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1849911/ (accessed on 15 February 2017). [CrossRef] [PubMed]
- Roy, R.; Tripathi, A.; Das, M.; Dwivedi, P.D. Cytotoxicity and uptake of zinc oxide nanoparticles leading to enhanced inflammatory cytokines levels in murine macrophages: Comparison with bulk zinc oxide. J. Biomed. Nanotechnol. 2011, 7, 110–111. Available online: http://www.ingentaconnect.com/content/asp/jbn/2011/00000007/00000001/art00056 (accessed on 5 February 2017). [CrossRef] [PubMed]
- Hsiao, I.L.; Huang, Y.J. Effects of various physicochemical characteristics on the toxicities of ZnO and TiO nanoparticles toward human lung epithelial cells. Sci. Total Environ. 2011, 409, 1219–1228. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21255821 (accessed on 14 March 2017). [CrossRef] [PubMed]
- Hou, J.; Wu, Z.; Li, X.; Wei, B.; Li, S.; Wang, X. Toxic effects of different types of zinc oxide nanoparticles on algae, plants, invertebrates, vertebrates and microorganisms. Chemosphere 2018, 193, 852–860. Available online: https://www.sciencedirect.com/science/article/pii/S0045653517318544 (accessed on 6 May 2018). [CrossRef]
- Wei, L.; Wang, J.; Chen, A.; Liu, J.; Feng, X.; Shao, L. Involvement of PINK1/parkin-mediated mitophagy in ZnO nanoparticle-induced toxicity in BV-2 cells. Int. J. Nanomed. 2017, 12, 1891–1903. Available online: https://www.dovepress.com/involvement-of-pink1parkin-mediated-mitophagy-in-zno-nanoparticle-indu-peer-reviewed-article-IJN (accessed on 3 June 2017). [CrossRef] [PubMed]
- Sharma, A.K.; Singh, V.; Gera, R.; Purohit, M.P.; Ghosh, D. Zinc Oxide Nanoparticle Induces Microglial Death by NADPH-Oxidase-Independent Reactive Oxygen Species as well as Energy Depletion. Mol. Neurobiol. 2017, 54, 6273–6286. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27714634 (accessed on 14 May 2017). [CrossRef] [PubMed]
- Deng, X.; Luan, Q.; Chen, W.; Wang, Y.; Wu, M.; Zhang, H.; Jiao, Z. Nanosized zinc oxide particles induce neural stem cell apoptosis. Nanotechnology 2009, 20, 115101. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19420431 (accessed on 23 December 2017). [CrossRef] [PubMed]
- Zhao, J.; Xu, L.; Zhang, T.; Ren, G.; Yang, Z. Influences of nanoparticle zinc oxide on acutely isolated rat hippocampal CA3 pyramidal neurons. Neurotoxicology 2009, 30, 220–230. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19146874 (accessed on 10 February 2017). [CrossRef] [PubMed]
- Jeng, H.A.; Swanson, J. Toxicity of metal oxide nanoparticles in mammalian cells. J. Environ. Sci. Health Part A 2006, 41, 2699–2711. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17114101 (accessed on 3 March 2017). [CrossRef] [PubMed]
- Cho, W.S.; Kang, B.C.; Lee, J.K.; Jeong, J.; Che, J.H.; Seok, S.H. Comparative absorption, distribution, and excretion of titanium dioxide and zinc oxide nanoparticles after repeated oral administration. Part. Fibre Toxicol. 2013, 10, 9. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23531334 (accessed on 3 June 2017). [CrossRef] [PubMed]
- Shim, K.H.; Jeong, K.H.; Bae, S.O.; Kang, M.O.; Maeng, E.H.; Choi, C.S.; Kim, Y.R.; Hulme, J.; Lee, E.K.; Kim, M.K.; et al. Assessment of ZnO and SiO2 nanoparticle permeability through and toxicity to the blood-brain barrier using Evans blue and TEM. Int. J. Nanomed. 2014, 9, 225–233. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25565840 (accessed on 12 April2017). [CrossRef]
- Shim, K.H.; Hulme, J.; Maeng, E.H.; Kim, M.K.; An, S.S. Analysis of zinc oxide nanoparticles binding proteins in rat blood and brain homogenate. Int. J. Nanomed. 2014, 9, 217–224. Available online: https://www.dovepress.com/analysis-of-zinc-oxide-nanoparticles-binding-proteins-in-rat-blood-and-peer-reviewed-article-IJN (accessed on 4 July 2017). [CrossRef]
- Xiaoli, F.; Junrong, W.; Xuan, L.; Yanli, Z.; Limin, W.; Jia, L.; Longquan, S. Prenatal exposure to nanosized zinc oxide in rats: Neurotoxicity and postnatal impaired learning and memory ability. Nanomedicine 2017, 12, 777–795. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28322126 (accessed on 5 January 2017). [CrossRef] [PubMed]
- Downes, N.; Mullins, P. The development of myelin in the brain of the juvenile rat. Toxicol. Pathol. 2013, 42, 913–922. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24129760 (accessed on 6 May 2018). [CrossRef] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. Available online: https://www.ncbi.nlm.nih.gov/pubmed/36810 (accessed on 16 March 2017). [CrossRef]
- Moron, M.S.; Depierre, J.; Mannervik, B. Levels of glutathione, glutathione reductase and glutathione–S-transferase activities in rat lung and liver. Biochem. Biophys. Acta 1979, 582, 67–78. Available online: https://www.sciencedirect.com/science/article/pii/0304416579902897?via%3Dihub (accessed on 18 March 2017). [CrossRef]
- Wang, Y.; Oberley, L.W.; Murhhammer, D.W. Evidence of oxidative stress following the viral infection of two Lepidopteran insect cell lines. Free Rad. Biol. Med. 2001, 31, 1448–1455. Available online: https://www.ncbi.nlm.nih.gov/pubmed/11728817 (accessed on 17 November 2017). [CrossRef]
- Kakkar, P.; Das, B.; Viswanathan, P.N. A modified spectrophotometric assay of superoxide dismutase. Indian J. Biochem. Biophys. 1984, 21, 2130–2132. Available online: https://www.ncbi.nlm.nih.gov/pubmed/6490072 (accessed on 18 March 2017). [CrossRef]
- Green, L.; Wagner, D.; Glogowski, J.; Skipper, P.; Wishnok, J.; Tannenbaum, S. Analysis of nitrate, nitrite and [15N] nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7181105 (accessed on 23 June 2017). [CrossRef]
- Macejak, D.; Rayfield, M.; Luftig, R. Isolation and characterization of human HSP70 expressed in Escherichia coli. Arch. Biochem. Biophys. 1990, 280, 53–60. Available online: https://www.sciencedirect.com/science/article/pii/0003986190905173?via%3Dihub (accessed on 2 December 2017). [CrossRef]
- March, C.J.; Mosley, B.; Larsen, A.; Cerretti, D.P.; Braedt, G.; Price, V.; Gillis, S.; Henney, C.S.; Kronheim, S.R.; Grabstein, K. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985, 315, 641–647. Available online: https://www.nature.com/articles/315641a0 (accessed on 12 November 2017). [CrossRef] [PubMed]
- Engelmann, H.; Novick, D.; Wallach, D. Two tumor necrosis factor-binding proteins purified from human urine. Evidence for immunological cross-reactivity with cell surface tumor necrosis factor receptors. J. Biol. Chem. 1990, 265, 1531–1563. Available online: http://www.jbc.org/content/265/3/1531.full.pdf?sid=9167c08f-d4c9-4a88-b38c-5bc8bdaf8e45 (accessed on 25 January 2017). [PubMed]
- Suda, T.; Takahashi, T.; Golstein, P.; Nagata, S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 1993, 75, 1169–1178. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7505205 (accessed on 15 July 2017). [CrossRef]
- Liu, W.; Wang, G.; Yakovlev, A.G. Identification and Functional Analysis of the Rat Caspase-3 Gene Promoter. J. Biol. Chem. 2002, 277, 8273–8278. Available online: http://www.jbc.org/content/277/10/8273.full (accessed on 3 January 2017). [CrossRef] [PubMed]
- Burton, K.A. Study of the Conditions and Mechanism of the Diphenylamine Reaction for the Colorimetric Estimation of Deoxyribonucleic Acid. Biochem. J. 1956, 62, 315–323. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1215910/ (accessed on 15 June 2017). [CrossRef] [PubMed]
- Suenobu, N.; Shichiri, M.; Iwashina, M.; Marumo, F.; Hirata, Y. Natriuretic Peptides and Nitric Oxide Induce Endothelial Apoptosis via a cGMP–Dependent Mechanism. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 140–146. Available online: http://atvb.ahajournals.org/content/19/1/140 (accessed on 18 July 2017). [CrossRef] [PubMed]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. Available online: https://iths.pure.elsevier.com/en/publications/a-simple-technique-for-quantitation-of-low-levels-of-dna-damage-i (accessed on 20 December 2017). [CrossRef]
- Ansar, S.; Abudawood, M.; Hamed, S.S.; Aleem, M.M. Exposure to Zinc Oxide Nanoparticles Induces Neurotoxicity and Proinflammatory Response: Amelioration by Hesperidin. Biol. Trace Elem. Res. 2017, 175, 360–366. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27300038 (accessed on 1 April 2017). [CrossRef] [PubMed]
- Chen, A.; Feng, X.; Sun, T.; Zhang, Y.; An, S.; Shao, L. Evaluation of the effect of time on the distribution of zinc oxide nanoparticles in tissues of rats and mice: A systematic review. ET Nanobiotechnol. 2016, 10, 97–106. Available online: https://ieeexplore.ieee.org/document/7484966/ (accessed on 3 November 2017). [CrossRef] [PubMed]
- Ruas, C.B.G.; Dos Santos Carvalho, C.; De Araújo, H.S.S.; Espíndola, E.L.G.; Fernandes, M.N. Oxidative stress biomarkers of exposure in the blood of cichlid species from a metal-contaminated river. Ecotoxicol. Environ. 2008, 71, 86–93. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17936357 (accessed on 10 June 2017). [CrossRef] [PubMed]
- Kaya, H.; Akbulut, M. Effects of waterborne lead exposure in Mozambiquetilapia: Oxidative stress, osmoregulatory responses, and tissue accumulation. J. Aquat. Anim. Health 2015, 27, 77–87. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25951052 (accessed on 22 May 2018). [CrossRef] [PubMed]
- Afifi, M.; Saddick, S.; Abu Zinada, O.A. Toxicity of silver nanoparticles on the brain of Oreochromis niloticus and Tilapia zillii. Saudi J. Biol. Sci. 2016, 23, 754–760. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27872573 (accessed on 9 May 2018). [CrossRef] [PubMed]
- Davies, M.G.; Fulton, G.J.; Hagen, P.O. Clinical biology of nitric oxide. Br. J. Surg. 1995, 82, 1598–1610. Available online: https://onlinelibrary.wiley.com/doi/pdf/10.1002/bjs.1800821206 (accessed on 11 February 2017). [CrossRef] [PubMed]
- Sharma, V.; Singh, P.; Pandey, A.K.; Dhawan, A. Induction of oxidative stress, DNA damage and apoptosis in mouse liver after sub-acute oral exposure to zinc oxide nanoparticles. Mutat. Res. 2012, 745, 84–91. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22198329 (accessed on 30 January 2017). [CrossRef] [PubMed]
- Xia, T.; Kovochich, M.; Liong, M.; Mädler, L.; Gilbert, B.; Shi, H.; Yeh, J.I.; Zink, J.I.; Nel, A.E. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano 2008, 2, 2121–2134. Available online: https://pubs.acs.org/doi/abs/10.1021/nn800511k (accessed on 28 May 2017). [CrossRef] [PubMed]
- Foster, K.A.; Galeffi, F.; Gerich, F.J.; Turner, D.A.; Muller, M. Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog. Neurobiol. 2006, 79, 136–171. Available online: https://www.sciencedirect.com/science/article/pii/S0301008206000694 (accessed on 3 May 2017). [CrossRef] [PubMed]
- Kvietys, P.R.; Granger, D.N. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic. Biol. Med. 2012, 52, 556–592. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22154653 (accessed on 4 December 2017). [CrossRef] [PubMed]
- Park, E.J.; Park, K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol. Lett. 2009, 184, 18–25. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19022359 (accessed on 5 February 2017). [CrossRef] [PubMed]
- Tsou, T.C.; Yeh, S.C.; Tsai, F.Y.; Lin, H.J.; Cheng, T.J.; Chao, H.R.; Tai, L.A. Zinc oxide particles induce inflammatory responses in vascular endothelial cells via NF-kappaB signaling. J. Hazard. Mater. 2010, 183, 182–188. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20674161 (accessed on 11 July2017). [CrossRef] [PubMed]
- Kumar, A.; Dhawan, A. Genotoxic and carcinogenic potential of engineered nanoparticles: An update. Arch. Toxicol. 2013, 87, 1883–1900. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24068037 (accessed on 3 August 2017). [CrossRef] [PubMed]
- Sharma, V.; Shukla, R.K.; Saxena, N.; Parmar, D.; Das, M.; Dhawan, A. DNA damaging potential of zinc oxide nanoparticles in human epidermal cells. Toxicol. Lett. 2009, 185, 211–218. Available online: https://www.sciencedirect.com/science/article/pii/S037842740900023X (accessed on 3 January 2017). [CrossRef] [PubMed]
- Sharma, V.; Anderson, D.; Dhawan, A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis 2012, 17, 852–870. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22395444 (accessed on 14 September 2017). [CrossRef] [PubMed]
- Totsuka, Y.; Ishino, K.; Kato, T.; Goto, S.; Tada, Y.; Nakae, D.; Watanabe, M.; Wakabayashi, K. Magnetite nanoparticles induce genotoxicity in the lungs of mice via inflammatory response. Nanomaterials 2014, 4, 175–188. Available online: https://www.mdpi.com/2079-4991/4/1/175 (accessed on 15 September 2017). [CrossRef] [PubMed]
- Harangi, M.; Seres, I.; Varga, Z.; Emri, G.; Szilvássy, Z.; Paragh, G. Atorvastatin effect on high-density lipoprotein-associated paraoxonase activity and oxidative DNA damage. Eur J. Clin Pharmacol. 2004, 60, 685–691. Available online: https://www.ncbi.nlm.nih.gov/pubmed/15490140 (accessed on 23 October 2017). [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. Available online: https://www.ncbi.nlm.nih.gov/pubmed/17562483 (accessed on 22 May 2018). [CrossRef] [PubMed]
- Yoo, K.C.; Yoon, C.H.; Kwon, D.; Hyun, K.H.; Woo, S.J.; Kim, R.K.; Lim, E.J.; Suh, Y.; Kim, M.J.; Yoon, T.H.; et al. Titanium dioxide induces apoptotic cell death through reactive oxygen species-mediated Fas upregulation and Bax activation. Int. J. Nanomed. 2012, 7, 1203–1214. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3298386/ (accessed on 13 August 2017). [CrossRef]
- Sruthi, S.; Mohanan, P.V. Investigation on cellular interactions of astrocytes with zinc oxide nanoparticles using rat C6 cell lines. Colloid Surf. B-Biointerfaces 2015, 133, 1–11. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26057374 (accessed on 12 October 2017). [CrossRef] [PubMed]
- Wahab, R.; Kaushik, N.K.; Verma, A.K.; Mishra, A.; Hwang, I.H.; Yang, Y.B. Fabrication and growth mechanism of ZnO nanostructures and their cytotoxic effect on human brain tumor U87, cervical cancer HeLa, and normal HEK cells. J. Biol. Inorg. Chem. 2011, 16, 431–442. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21140179 (accessed on 12 April 2017). [CrossRef] [PubMed]
- Wang, J.; Deng, X.; Zhang, F.; Chen, D.; Ding, W. ZnO nanoparticle-induced oxidative stress triggers apoptosis by activating JNK signaling pathway in cultured primary astrocytes. Nanoscale Res. Lett. 2014, 9, 117. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24624962 (accessed on 30 March 2017). [CrossRef] [PubMed]
- Akhtar, M.J.; Ahamed, M.; Kumar, S.; Khan, M.M.; Ahmad, J. Zinc oxide nanoparticles selectively induce apoptosis in human cancer cells through reactive oxygen species. Int. J. Nanomed. 2012, 7, 845–857. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22393286 (accessed on 17 August 2017). [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssiere, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. Available online: https://app.dimensions.ai/details/publication/pub.1001857722 (accessed on 4 November 2017). [CrossRef]
- Wu, P.P.; Liu, K.C.; Huang, W.W. Triptolide induces apoptosis in human adrenal cancer NCI-H295 cells through a mitochondrial dependent pathway. Oncol. Rep. 2011, 25, 551–557. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21152873 (accessed on 3 December 2017). [CrossRef] [PubMed]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18485876 (accessed on 11 June 2017). [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. Available online: https://www.sciencedirect.com/science/article/pii/S009286740300521X (accessed on 3 August 2017). [CrossRef]
- De Maio, A. Heat shock proteins: Facts, thoughts, and dreams. Shock 1999, 11, 1–12. Available online: https://insights.ovid.com/crossref?an=00024382-199901000-00001 (accessed on 30 August 2017). [CrossRef] [PubMed]
- Simar, D.; Jacques, A.; Caillaud, C. Heat shock proteins induction reduces stress kinases activation, potentially improving insulin signalling in monocytes from obese subjects. Cell Stress Chaperones 2012, 17, 615–621. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3535161/ (accessed on 3 February 2017). [CrossRef] [PubMed]
- Kelly, S.; Yenari, M.A. Neuroprotection: Heat shock proteins. Curr. Med. Res. Opin. 2002, 18, s55–s60. Available online: https://www.tandfonline.com/doi/abs/10.1185/030079902125000732 (accessed on 4 October 2017). [CrossRef] [PubMed]
- Yenari, M.A. Heat shock proteins and neuroprotection. Adv. Exp. Med. Biol. 2002, 513, 281–299. Available online: http://www.eurekaselect.com/90008/article/heat-shock-proteins-and-neuroprotection (accessed on 20 December 2017). [CrossRef] [PubMed]
- Ying, G.Y.; Jing, C.H.; Li, J.R.; Wu, C.; Yan, F.; Chen, J.Y.; Wang, L.; Dixon, B.J.; Chen, G. Neuroprotective Effects of Valproic Acid on Blood-Brain Barrier Disruption and Apoptosis-Related Early Brain Injury in Rats Subjected to Subarachnoid Hemorrhage Are Modulated by Heat Shock Protein 70/Matrix Metalloproteinases and Heat Shock Protein 70/AKT Pathways. Neurosurgery 2016, 79, 286–295. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27244466 (accessed on 22 December 2017). [CrossRef] [PubMed]
- Miyata, Y.; Rauch, J.N.; Jinwal, U.K.; Thompson, A.D.; Srinivasan, S.; Dickey, C.A.; Gestwicki, J.E. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem. Biol. 2012, 19, 1391–1399. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23177194 (accessed on 16 May 2017). [CrossRef] [PubMed]
- Afolayan, A.J.; Teng, R.J.; Eis, A.; Rana, U.; Broniowska, K.A.; Corbett, J.A.; Pritchard, K.; Konduri, G.G. Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L351–L360. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3920226/ (accessed on 4 June 2017). [CrossRef] [PubMed]
- Carocci, A.; Rovito, N.; Sinicropi, M.S.; Genchi, G. Mercury toxicity and neurodegenerative effects. Rev. Environ. Contam. Toxicol. 2014, 229, 1–18. Available online: https://link.springer.com/chapter/10.1007%2F978-3-319-03777-6_1 (accessed on 11 May 2018). [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Dosage | Duration |
|---|---|---|
| Group I | 40 mg/kg ZnONPs | 24 h |
| Group II | 100 mg/kg ZnONPs | 24 h |
| Group III | 40 mg/kg ZnONPs, daily | 7 days |
| Group IV | 100 mg/kg ZnONPs, daily | 7 days |
| Control A | Normal Saline | 24 h |
| Control B | Normal Saline, daily | 7 days |
| Normal Control | G I (40 mg/kg/24 h) | G II (100 mg/kg/24 h) | G III (40 mg/kg/day for 7 days) | G IV (100 mg/kg/day for 7 days) | |
|---|---|---|---|---|---|
| Tailed DNA % | 4 ± 0.3 | 4.37 ± 0.29 | 5.03 ± 0.3 | 4.85 ± 0.28 | 5.6 ± 0.21 a**b* |
| Tail length (µm) | 2.5 ± 0.17 | 2.7 ± 0.18 | 2.9 ± 0.146 | 3.3 a* ± 0.157 | 4.18 ± 0.18 a***b***c*** d* |
| Tail intensity (%) | 2.12 ± 0.15 | 2.22 ± 0.28 | 2.19 ± 0.21 | 2.54 ± 0.216 | 3.38 ± 0.12 a**b**c**d* |
| Tail moment (Unit) | 4.66 ± 0.25 | 5.55 ± 0.293 | 5.07 ± 0.31 | 5.82 a* ± 0.26 | 7.53 ± 0.263 a***b***c**d** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attia, H.; Nounou, H.; Shalaby, M. Zinc Oxide Nanoparticles Induced Oxidative DNA Damage, Inflammation and Apoptosis in Rat’s Brain after Oral Exposure. Toxics 2018, 6, 29. https://doi.org/10.3390/toxics6020029
Attia H, Nounou H, Shalaby M. Zinc Oxide Nanoparticles Induced Oxidative DNA Damage, Inflammation and Apoptosis in Rat’s Brain after Oral Exposure. Toxics. 2018; 6(2):29. https://doi.org/10.3390/toxics6020029
Chicago/Turabian StyleAttia, Hala, Howaida Nounou, and Manal Shalaby. 2018. "Zinc Oxide Nanoparticles Induced Oxidative DNA Damage, Inflammation and Apoptosis in Rat’s Brain after Oral Exposure" Toxics 6, no. 2: 29. https://doi.org/10.3390/toxics6020029
APA StyleAttia, H., Nounou, H., & Shalaby, M. (2018). Zinc Oxide Nanoparticles Induced Oxidative DNA Damage, Inflammation and Apoptosis in Rat’s Brain after Oral Exposure. Toxics, 6(2), 29. https://doi.org/10.3390/toxics6020029