
Developmental Bisphenol A Exposure Modulates Immune-Related Diseases

Abstract
:1. Introduction
2. Mechanisms of Immune Modulation and Disease Exacerbation Following Developmental BPA Exposure
2.1. Receptor Modification
2.2. Epigenetics
2.3. Microbiome
2.4. Cell Signaling Pathways
3. Immune System Alteration Following Developmental BPA Exposure
3.1. Innate Immune System
3.2. Adaptive Immune System
3.3. Cytokine/Chemokine, Antibody Production and Host Resistance
4. Diseases Related to Immune System Alteration Following Developmental BPA Exposure
4.1. Multiple Sclerosis
4.2. Type 1 Diabetes Mellitus
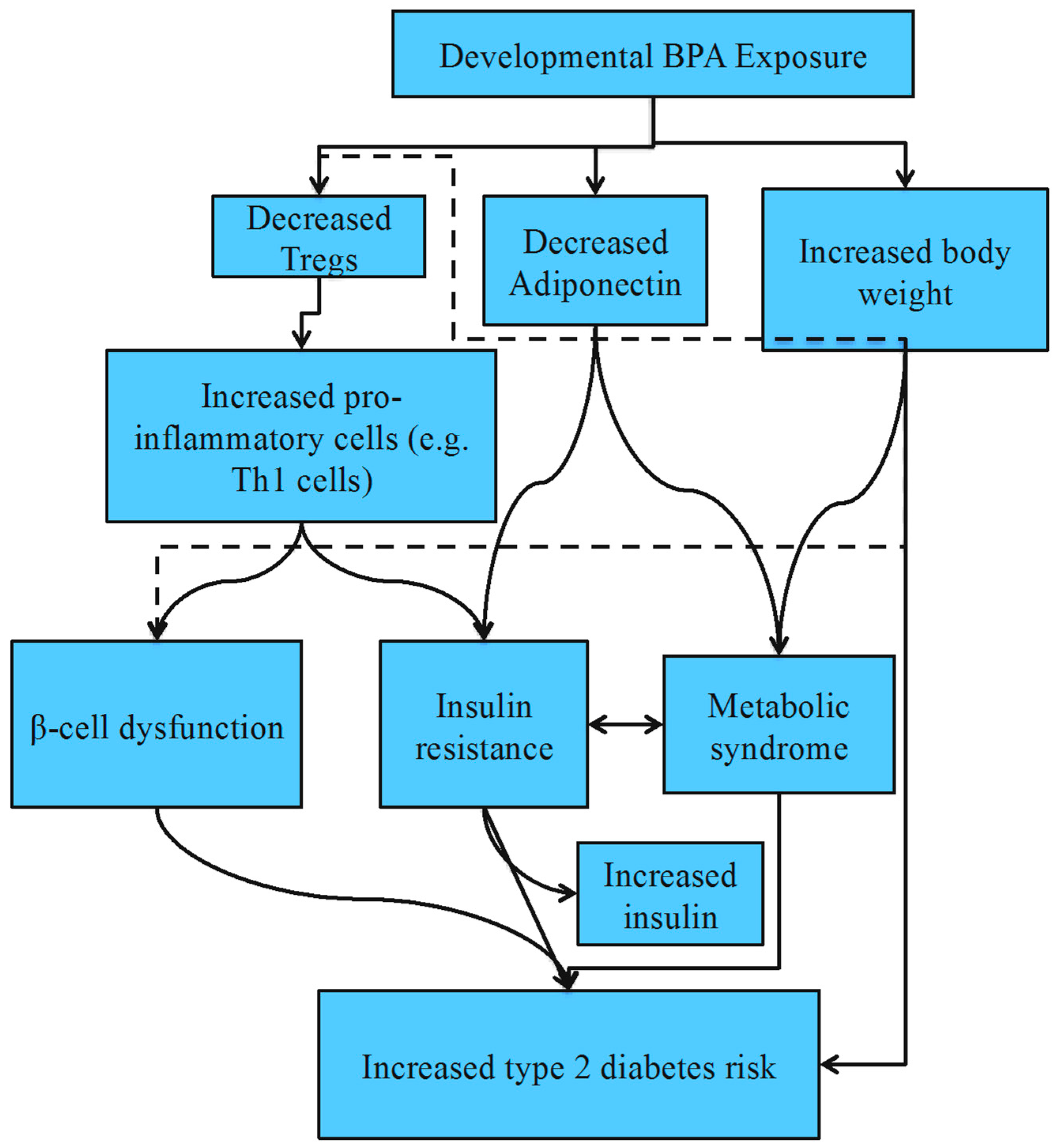
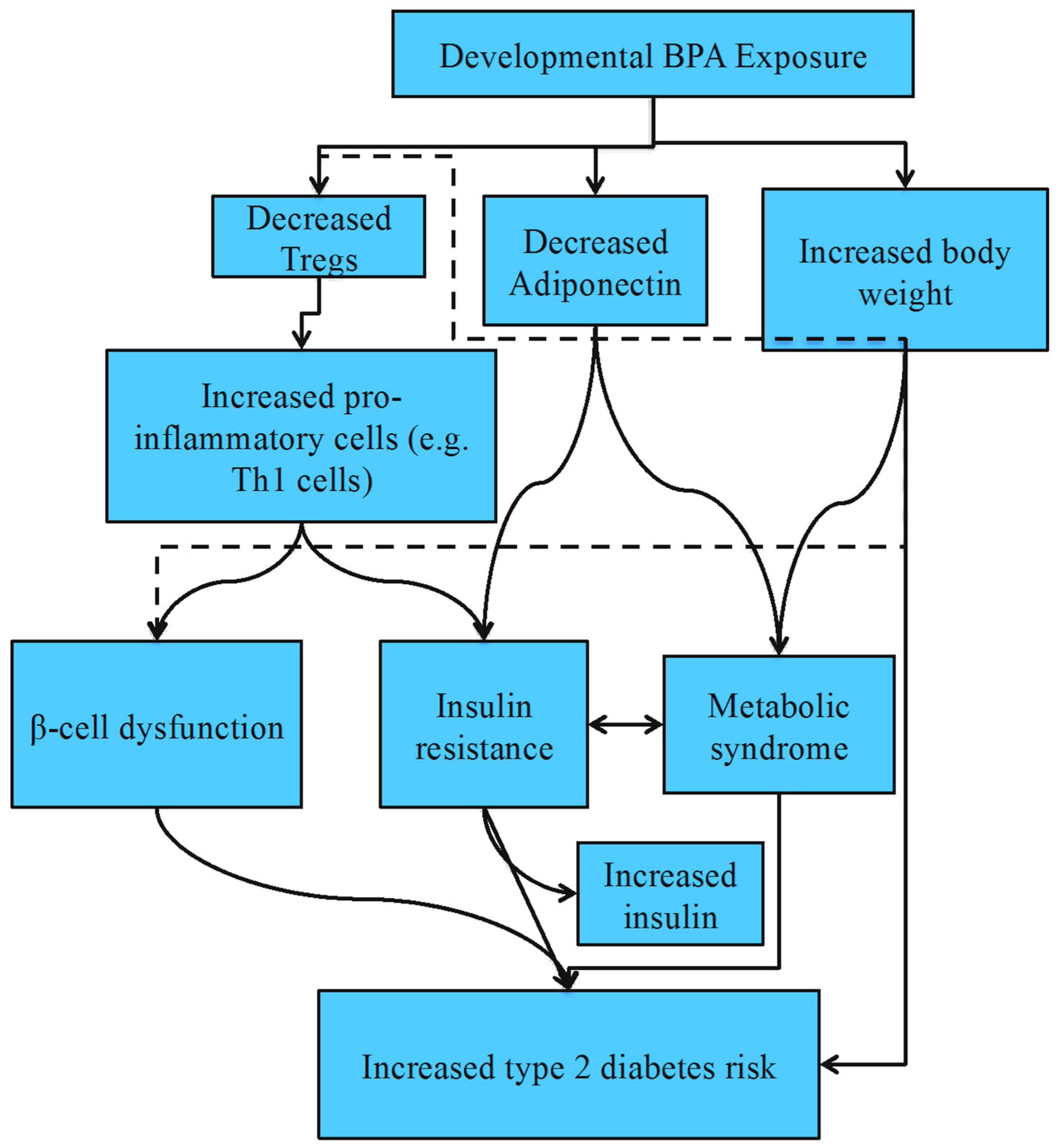
4.3. Type 2 Diabetes Mellitus
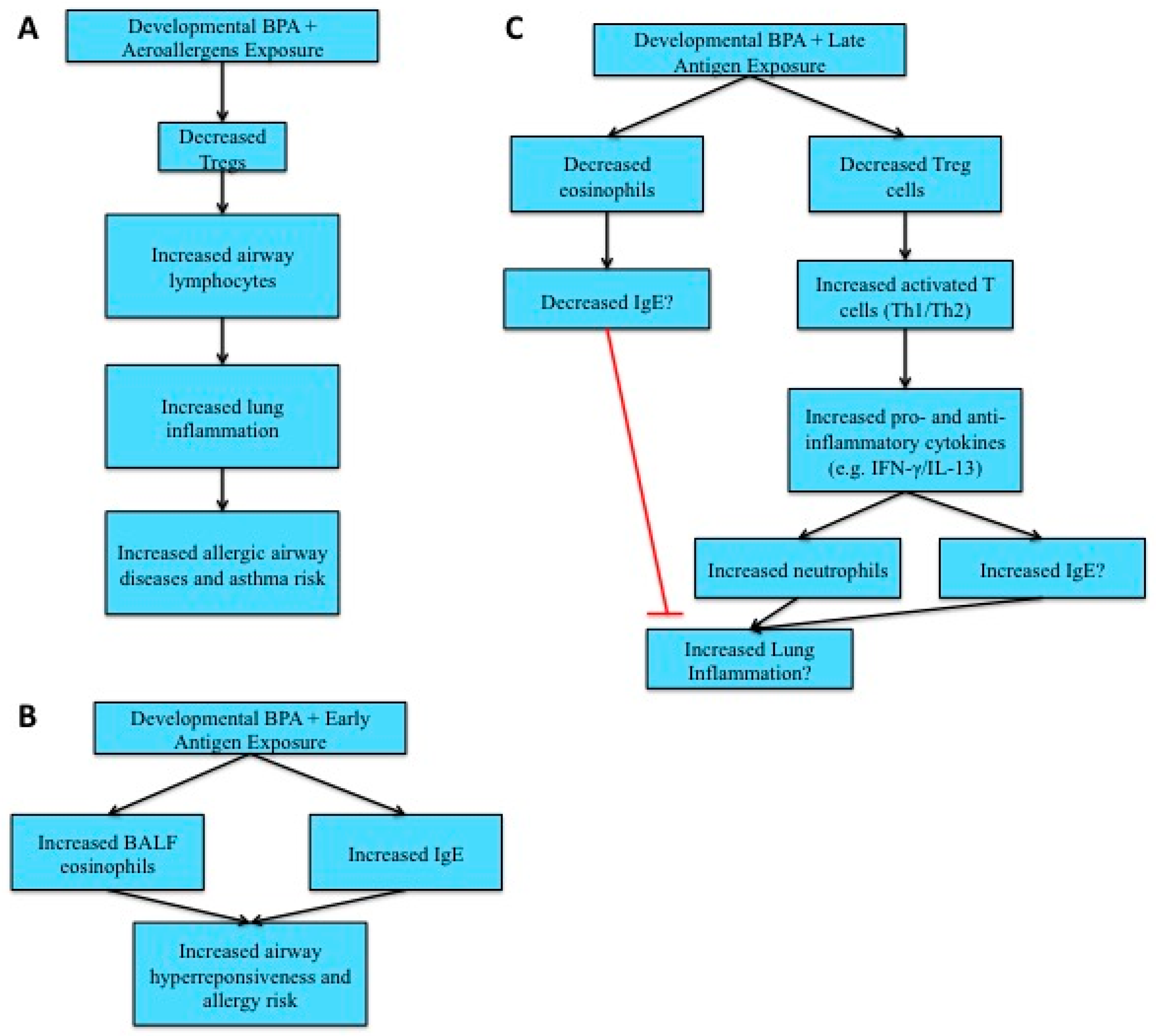
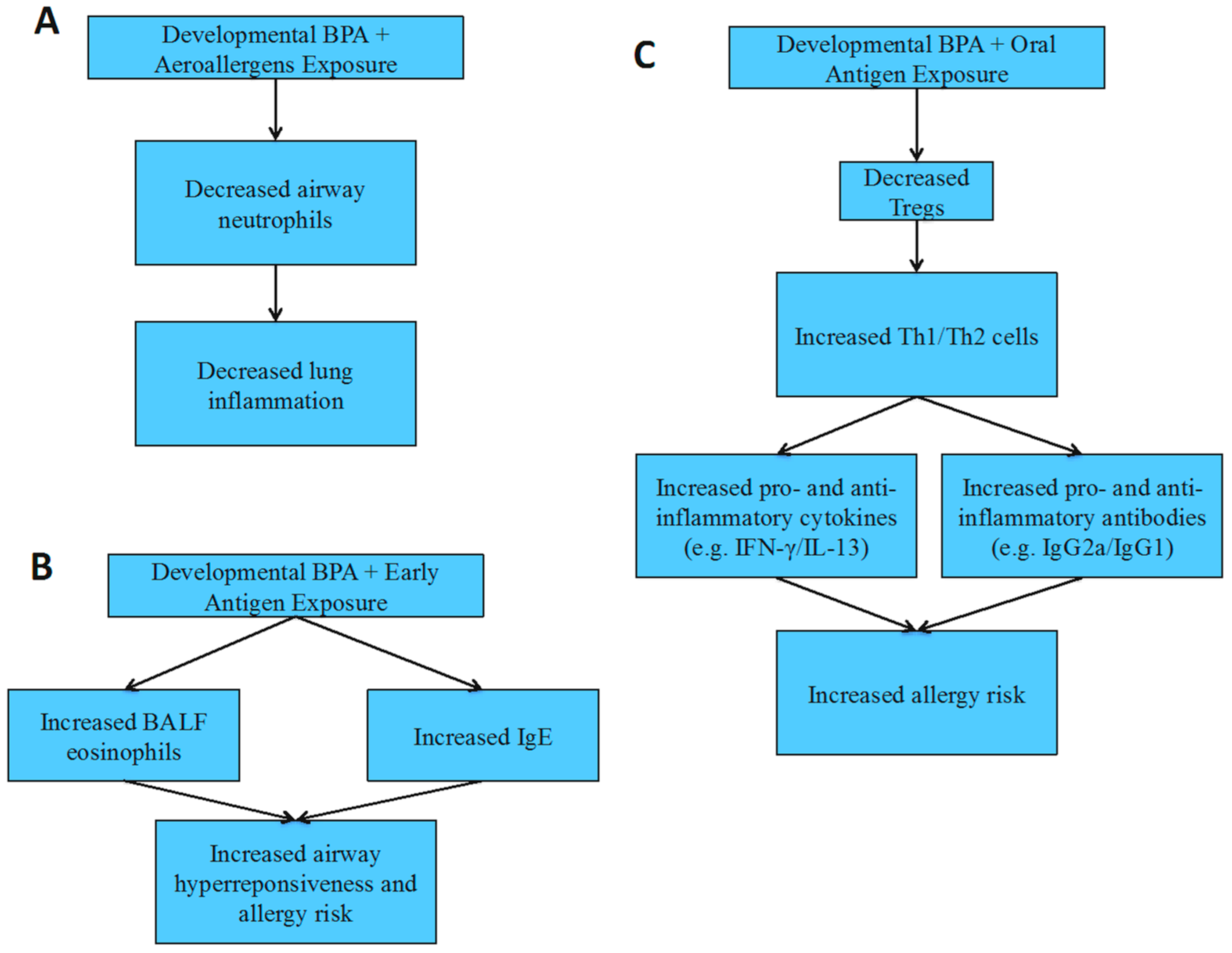
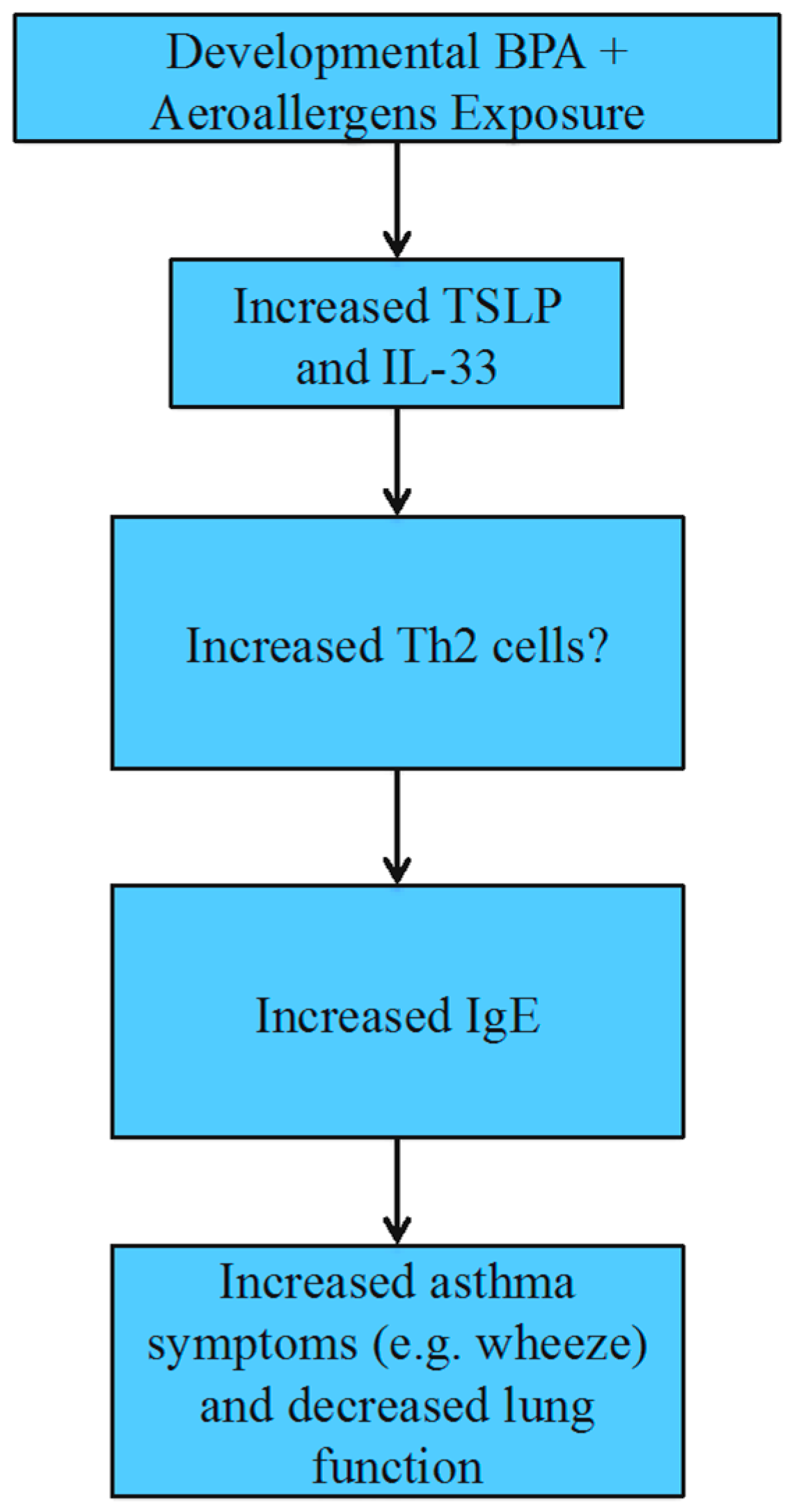
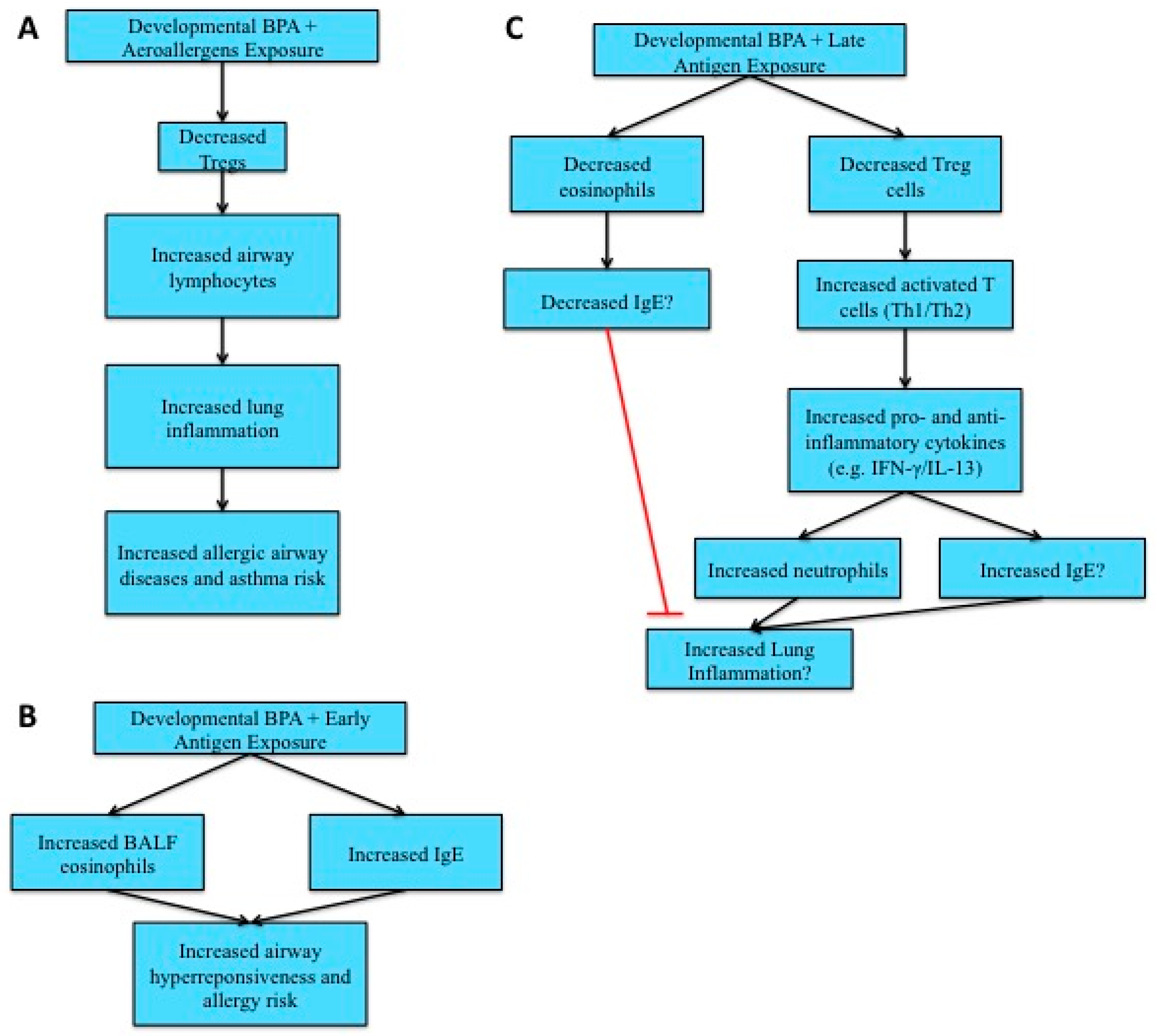
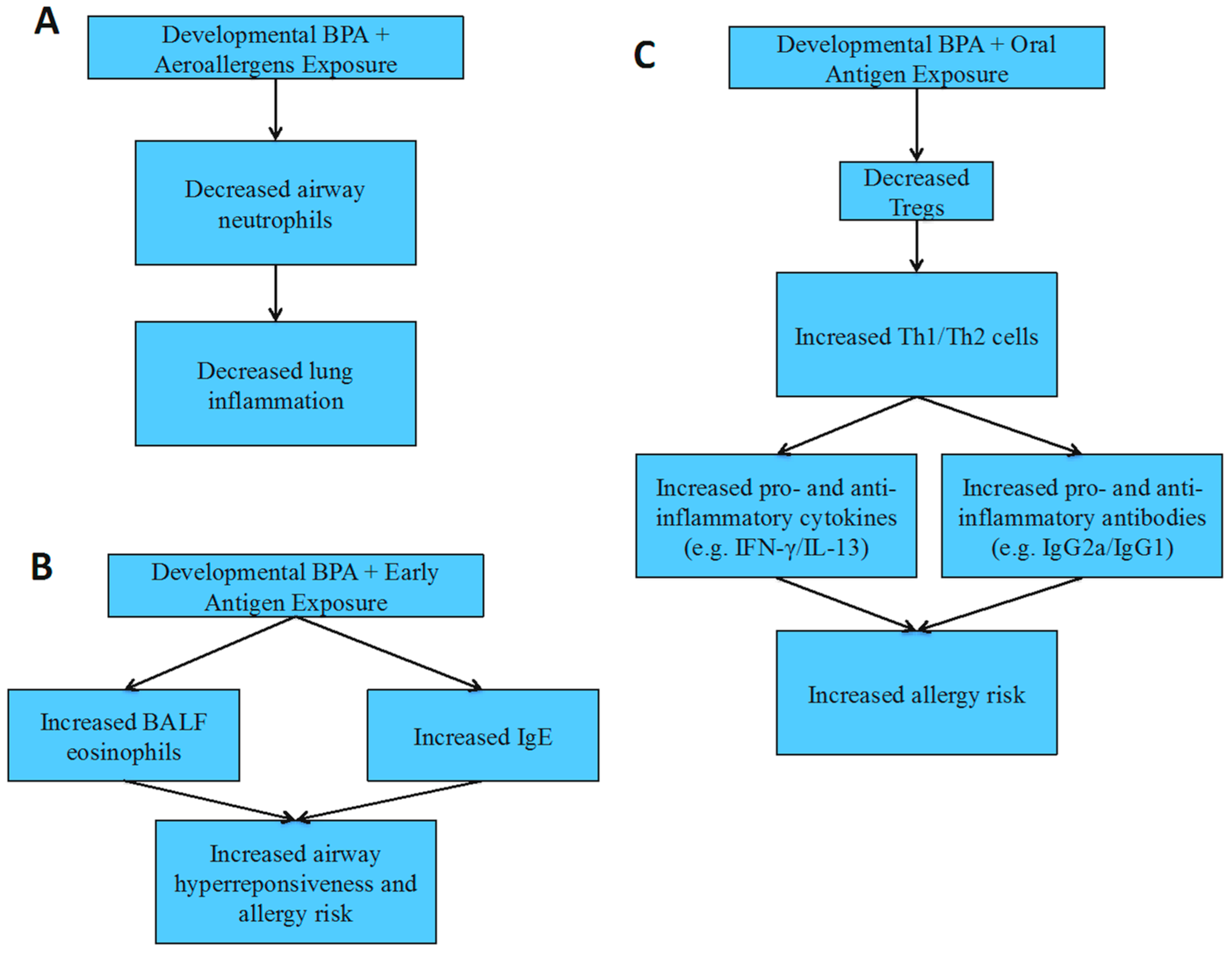
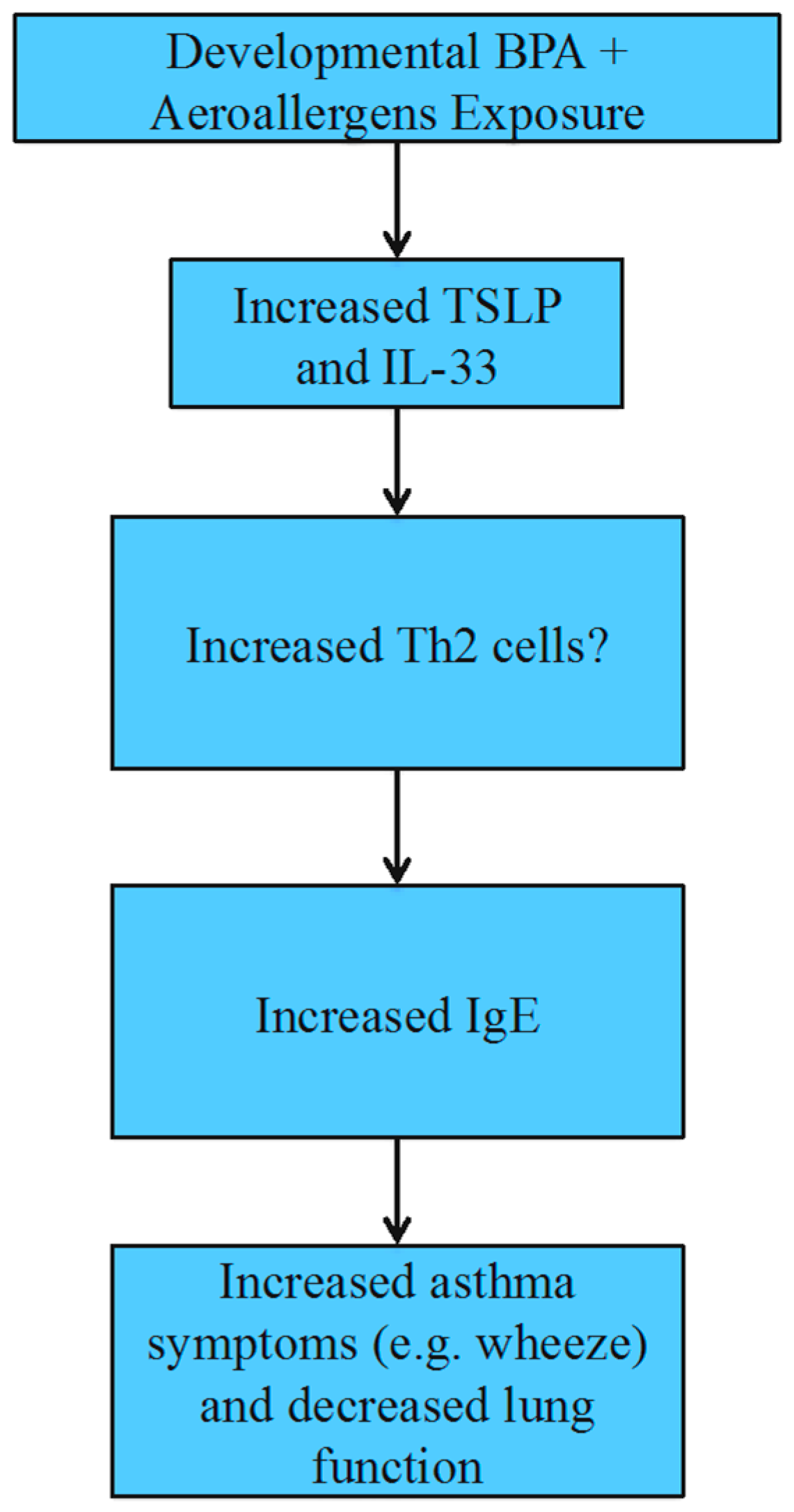
4.4. Allergies and Asthma
4.5. Mammary Cancer
5. Bisphenol S: An Alternative for BPA
6. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Belcher, S.M.; Gear, R.B.; Kendig, E.L. Bisphenol A alters autonomic tone and extracellular matrix structure and induces sex-specific effects on cardiovascular function in male and female CD-1 mice. Endocrinology 2015, 156, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Bushnik, T.; Haines, D.; Levallois, P.; Levesque, J.; Van Oostdam, J.; Viau, C. Lead and bisphenol A concentrations in the Canadian population. Health Rep. 2010, 21, 7–18. [Google Scholar] [PubMed]
- Corrales, J.; Kristofco, L.A.; Steele, W.B.; Yates, B.S.; Breed, C.S.; Williams, E.S.; Brooks, B.W. Global Assessment of Bisphenol A in the Environment: Review and analysis of its occurrence and bioaccumulation. Dose-Response Publ. Int. Hormesis Soc. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, R.W.; Welshons, W.V.; Swan, S.H. Bisphenol A data in NHANES suggest longer than expected half-life, substantial nonfood exposure, or both. Environ. Health Perspect. 2009, 117, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Deceuninck, Y.; Bichon, E.; Marchand, P.; Boquien, C.Y.; Legrand, A.; Boscher, C.; Antignac, J.P.; Le Bizec, B. Determination of bisphenol A and related substitutes/analogues in human breast milk using gas chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.H.; Yang, S.N.; Kuo, P.L.; Hung, C.H. Immunomodulatory effects of environmental endocrine disrupting chemicals. Kaohsiung J. Med. Sci. 2012, 28, S37–S42. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, S.; Manabe, N.; Nishizawa, H.; Morita, M.; Sugimoto, M.; Iwahori, M.; Miyamoto, H. Effects of oral exposure of bisphenol A on mRNA expression of nuclear receptors in murine placentae assessed by DNA microarray. J. Reprod. Dev. 2003, 49, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Morck, T.J.; Sorda, G.; Bechi, N.; Rasmussen, B.S.; Nielsen, J.B.; Ietta, F.; Rytting, E.; Mathiesen, L.; Paulesu, L.; Knudsen, L.E. Placental transport and in vitro effects of Bisphenol A. Reprod. Toxicol. (Elmsford, NY) 2010, 30, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.A.; Metz, L.; Yong, V.W. Review: Endocrine disrupting chemicals and immune responses: A focus on bisphenol-A and its potential mechanisms. Mol. Immunol. 2013, 53, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.L.; Tsai, M.H.; Lai, S.H.; Yao, T.C.; Hua, M.C.; Yeh, K.W.; Chiang, C.H.; Huang, S.Y.; Huang, J.L. Prenatal exposure to bisphenol-A is associated with Toll-like receptor-induced cytokine suppression in neonates. Pediatr. Res. 2016, 79, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.C.; Prince, L.R.; Sabroe, I. Translational mini-review series on Toll-like receptors: Networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin. Exp. Immunol. 2007, 147, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Giguere, V. Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim. Biophys. Acta 2011, 1812, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L. Estrogen, a double-edged sword: Modulation of TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr. Drug Targets Inflamm. Allergy 2004, 3, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Miao, S.; Gao, Z.; Kou, Z.; Xu, G.; Su, C.; Liu, N. Influence of bisphenol a on developing rat estrogen receptors and some cytokines in rats: A two-generational study. J. Toxicol. Environ. Health A 2008, 71, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shimizu, T.; Yu, H.P.; Hsieh, Y.C.; Choudhry, M.A.; Chaudry, I.H. Salutary effects of 17beta-estradiol on T-cell signaling and cytokine production after trauma-hemorrhage are mediated primarily via estrogen receptor-alpha. Am. J. Physiol. Cell Physiol. 2007, 292, C2103–C2111. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, S.; Tokunaga, T.; Liu, X.; Okada, H.; Matsushima, A.; Shimohigashi, Y. Endocrine disruptor bisphenol A strongly binds to human estrogen-related receptor γ (ERRγ) with high constitutive activity. Toxicol. Lett. 2006, 167, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Liu, X.; Sumiyoshi, M.; Matsushima, A.; Shimohigashi, M.; Shimohigashi, Y. Placenta expressing the greatest quantity of bisphenol A receptor ERR{gamma} among the human reproductive tissues: Predominant expression of type-1 ERRgamma isoform. J. Biochem. 2009, 146, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Ranhotra, H.S. The estrogen-related receptors: Orphans orchestrating myriad functions. J. Recept. Signal Trans. 2012, 32, 47–56. [Google Scholar] [CrossRef]
- Cipelli, R.; Harries, L.; Okuda, K.; Yoshihara, S.; Melzer, D.; Galloway, T. Bisphenol A modulates the metabolic regulator oestrogen-related receptor-α in T-cells. Reproduction 2014, 147, 419–426. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Nichols, A.G.; Inoue, M.; Kazmin, D.; Chang, C.-Y.; Dwyer, M.A.; Nelson, E.R.; Pollizzi, K.N.; Ilkayeva, O. Estrogen-related receptor-α is a metabolic regulator of effector T-cell activation and differentiation. Proc. Natl. Acad. Sci. USA 2011, 108, 18348–18353. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Gudsnuk, K.; Franks, B.; Madrid, J.; Miller, R.L.; Perera, F.P.; Champagne, F.A. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc. Natl. Acad. Sci. USA 2013, 110, 9956–9961. [Google Scholar] [CrossRef]
- Dhimolea, E.; Wadia, P.R.; Murray, T.J.; Settles, M.L.; Treitman, J.D.; Sonnenschein, C.; Shioda, T.; Soto, A.M. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ahmed, S.A. Epigenetic Regulation of Non-Lymphoid Cells by Bisphenol A, a Model Endocrine Disrupter: Potential Implications for Immunoregulation. Front. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Yin, H.; Wang, L.; Gershwin, M.E.; Lu, Q. The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J. Autoimmun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.J.; Patel, M.C.; Blanco, J.C.; Vogel, S.N. Epigenetic Mechanisms Governing Innate Inflammatory Responses. J. Interferon Cytokine Res. 2016, 36, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Casati, L.; Sendra, R.; Sibilia, V.; Celotti, F. Endocrine disrupters: The new players able to affect the epigenome. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.R.; Iqbal, K.; Tran, D.A.; Rivas, G.E.; Singh, P.; Pfeifer, G.P.; Szabo, P.E. Effects of endocrine disruptors on imprinted gene expression in the mouse embryo. Epigenetics 2011, 6, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Rozek, L.S.; Soliman, A.S.; Sartor, M.A.; Hablas, A.; Seifeldin, I.A.; Colacino, J.A.; Weinhouse, C.; Nahar, M.S.; Dolinoy, D.C. Bisphenol A-associated epigenomic changes in prepubescent girls: A cross-sectional study in Gharbiah, Egypt. Environ. Health 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Spahn, T.W.; Kucharzik, T. Modulating the intestinal immune system: The role of lymphotoxin and GALT organs. Gut 2004, 53, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Munyaka, P.M.; Khafipour, E.; Ghia, J.E. External influence of early childhood establishment of gut microbiota and subsequent health implications. Front. Pediatr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.K.; Hansen, C.H.; Krych, L.; Nielsen, D.S. Impact of the gut microbiota on rodent models of human disease. World J. Gastroenterol. 2014, 20, 17727–17736. [Google Scholar] [PubMed]
- Cox, L.M.; Yamanishi, S.; Sohn, J.; Alekseyenko, A.V.; Leung, J.M.; Cho, I.; Kim, S.G.; Li, H.; Gao, Z.; Mahana, D.; et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014, 158, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Davis-Richardson, A.G.; Triplett, E.W. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia 2015, 58, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Jouault, A.; Gaultier, E.; Polizzi, A.; Buisson-Brenac, C.; Leveque, M.; Martin, P.G.; Theodorou, V.; Fioramonti, J.; Houdeau, E. Impact of oral bisphenol A at reference doses on intestinal barrier function and sex differences after perinatal exposure in rats. Proc. Natl. Acad. Sci. USA 2010, 107, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Yang, O.; Kim, H.L.; Weon, J.I.; Seo, Y.R. Endocrine-disrupting Chemicals: Review of Toxicological Mechanisms Using Molecular Pathway Analysis. J. Cancer Prev. 2015, 20, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.H.; Hlaing, M.M.; Zhang, X.; Yan, C.; Duan, Z.; Zhu, L.; Ung, C.Y.; Mathavan, S.; Ong, C.N.; Gong, Z. Toxicogenomic and phenotypic analyses of bisphenol-A early-life exposure toxicity in zebrafish. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Saili, K.S.; Tilton, S.C.; Waters, K.M.; Tanguay, R.L. Global gene expression analysis reveals pathway differences between teratogenic and non-teratogenic exposure concentrations of bisphenol A and 17beta-estradiol in embryonic zebrafish. Reprod. Toxicol. (Elmsford, NY) 2013, 38, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Y.; Wang, J.S.; Liu, J.; Liu, M.M.; Yang, H.B. The roles of Egr-2 in autoimmune diseases. Inflammation 2015, 38, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.; Sumitomo, S.; Morita, K.; Iwasaki, Y.; Inoue, M.; Nakachi, S.; Komai, T.; Shoda, H.; Miyazaki, J.; Fujio, K.; et al. TGF-beta3-expressing CD4+CD25(-)LAG3+ regulatory T cells control humoral immune responses. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Jiang, H.; Zheng, J.; Li, J.; Wei, Y.; Xu, G.; Li, H. MicroRNA-106b regulates pro-allergic properties of dendritic cells and Th2 polarisation by targeting early growth response-2 in vitro. Int. Immunopharmacol. 2015, 28, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Pal, S.K.; Reckamp, K.; Figlin, R.A.; Yu, H. STAT3: A target to enhance antitumor immune response. Curr. Top. Microbiol. Immunol. 2011, 344, 41–59. [Google Scholar] [PubMed]
- Rebe, C.; Vegran, F.; Berger, H.; Ghiringhelli, F. STAT3 activation: A key factor in tumor immunoescape. JAK-STAT 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Queval, C.J.; Song, O.R.; Deboosere, N.; Delorme, V.; Debrie, A.S.; Iantomasi, R.; Veyron-Churlet, R.; Jouny, S.; Redhage, K.; Deloison, G.; et al. STAT3 Represses Nitric Oxide Synthesis in Human Macrophages upon Mycobacterium tuberculosis Infection. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, M.E.; Carow, B. SOCS3 and STAT3, major controllers of the outcome of infection with Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Yamamoto, T.; Yumioka, T.; Imoto, S.; Kojima, H.; Matsuda, T. Cross-talk between endocrine-disrupting chemicals and cytokine signaling through estrogen receptors. Biochem. Biophys. Res. Commun. 2004, 315, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fang, Y.; Shi, X.; Zhang, M.; Wang, X.; Tan, Y. Effect of bisphenol A on the EGFR-STAT3 pathway in MCF-7 breast cancer cells. Mol. Med. Rep. 2012, 5, 41–47. [Google Scholar] [PubMed]
- Liu, Y.; Mei, C.; Liu, H.; Wang, H.; Zeng, G.; Lin, J.; Xu, M. Modulation of cytokine expression in human macrophages by endocrine-disrupting chemical Bisphenol-A. Biochem. Biophys. Res. Commun. 2014, 451, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Couleau, N.; Falla, J.; Beillerot, A.; Battaglia, E.; D’innocenzo, M.; Plançon, S.; Laval-Gilly, P.; Bennasroune, A. Effects of endocrine disruptor compounds, alone or in combination, on human macrophage-like THP-1 cell response. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Jeong, H.G. Down-regulation of inducible nitric oxide synthase and tumor necrosis factor-alpha expression by bisphenol A via nuclear factor-kappaB inactivation in macrophages. Cancer Lett. 2003, 196, 69–76. [Google Scholar] [PubMed]
- Xu, H.; Yang, M.; Qiu, W.; Pan, C.; Wu, M. The impact of endocrine-disrupting chemicals on oxidative stress and innate immune response in zebrafish embryos. Environ. Toxicol. Chem./SETAC 2013, 32, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Bodin, J.; Bølling, A.K.; Wendt, A.; Eliasson, L.; Becher, R.; Kuper, F.; Løvik, M.; Nygaard, U.C. Exposure to bisphenol A, but not phthalates, increases spontaneous diabetes type 1 development in NOD mice. Toxicol. Rep. 2015, 2, 99–110. [Google Scholar] [CrossRef]
- Zeliger, H.I. Exposure to lipophilic chemicals as a cause of neurological impairments, neurodevelopmental disorders and neurodegenerative diseases. Interdiscip. Toxicol. 2013, 6, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Sugita-Konishi, Y.; Shimura, S.; Nishikawa, T.; Sunaga, F.; Naito, H.; Suzuki, Y. Effect of Bisphenol A on non-specific immunodefenses against non-pathogenic Escherichia coli. Toxicol. Lett. 2003, 136, 217–227. [Google Scholar] [CrossRef]
- Pyo, M.Y.; Kim, H.J.; Back, S.K.; Yang, M. Downregulation of peritoneal macrophage activity in mice exposed to bisphenol A during pregnancy and lactation. Arch. Pharm. Res. 2007, 30, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M. Diabetes Mystery: Why Are Type 1 Cases Surging. Available online: http://www.scientificamerican.com/article/a-diabetes-cliffhanger/ (accessed on 14 July 2016).
- Quesada, I.; Tudurí, E.; Ripoll, C.; Nadal, Á. Physiology of the pancreatic α-cell and glucagon secretion: Role in glucose homeostasis and diabetes. J. Endocrinol. 2008, 199, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Thornley, T.B.; Agarwal, K.A.; Kyriazis, P.; Ma, L.; Chipashvili, V.; Aker, J.E.; Korniotis, S.; Csizmadia, E.; Strom, T.B.; Koulmanda, M. Contrasting Roles of Islet Resident Immunoregulatory Macrophages and Dendritic Cells in Experimental Autoimmune Type 1 Diabetes. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Yamaki, K.; Li, X.; Sai, T.; Yanagisawa, R.; Takano, H.; Taneda, S.; Hayashi, H.; Mori, Y. Prenatal exposure to bisphenol A up-regulates immune responses, including T helper 1 and T helper 2 responses, in mice. Immunology 2004, 112, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Dornmair, K.; Goebels, N.; Weltzien, H.U.; Wekerle, H.; Hohlfeld, R. T-cell-mediated autoimmunity: Novel techniques to characterize autoreactive T-cell receptors. Am. J. Pathol. 2003, 163, 1215–1226. [Google Scholar] [CrossRef]
- Dejaco, C.; Duftner, C.; Grubeck-Loebenstein, B.; Schirmer, M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology 2006, 117, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P.; Chen, Z. Autoimmune effector memory T cells: The bad and the good. Immunol. Res. 2013, 57, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Luo, D.; She, R.; Liu, T.; Ding, Y.; Yue, Z.; Xia, K. Effects of bisphenol A on the development of central immune organs of specific-pathogen-free chick embryos. Toxicol. Indus. Health 2014, 30, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zhang, J.; Chen, W. Thymic output: Influence factors and molecular mechanism. Cell. Mol. Immunol. 2006, 3, 341–350. [Google Scholar] [PubMed]
- Savino, W. The thymus is a common target organ in infectious diseases. PLoS Pathog. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Stolp, J.; Marino, E.; Batten, M.; Sierro, F.; Cox, S.L.; Grey, S.T.; Silveira, P.A. Intrinsic molecular factors cause aberrant expansion of the splenic marginal zone B cell population in nonobese diabetic mice. J. Immunol. (Baltim., MD, 1950) 2013, 191, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Holladay, S.D.; Xiao, S.; Diao, H.; Barber, J.; Nagy, T.; Ye, X.; Gogal, R.M., Jr. Perinatal bisphenol A exposure in C57B6/129svj male mice: Potential altered cytokine/chemokine production in adulthood. Int. J. Environ. Res. Public Health 2010, 7, 2845–2852. [Google Scholar] [CrossRef] [PubMed]
- Ashley-Martin, J.; Dodds, L.; Levy, A.R.; Platt, R.W.; Marshall, J.S.; Arbuckle, T.E. Prenatal exposure to phthalates, bisphenol A and perfluoroalkyl substances and cord blood levels of IgE, TSLP and IL-33. Environ. Res. 2015, 140, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R., Jr.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.A. The politics of plastics: The making and unmaking of bisphenol a “safety”. Am. J. Public Health 2009, 99, S559–S566. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Takamoto, M.; Sugane, K. Exposure to Bisphenol A prenatally or in adulthood promotes T(H)2 cytokine production associated with reduction of CD4CD25 regulatory T cells. Environ. Health Perspect 2008, 116, 514–519. [Google Scholar] [PubMed]
- Menard, S.; Guzylack-Piriou, L.; Lencina, C.; Leveque, M.; Naturel, M.; Sekkal, S.; Harkat, C.; Gaultier, E.; Olier, M.; Garcia-Villar, R.; et al. Perinatal exposure to a low dose of bisphenol A impaired systemic cellular immune response and predisposes young rats to intestinal parasitic infection. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Bauer, S.M.; Lawrence, B.P. Developmental exposure to bisphenol A modulates innate but not adaptive immune responses to influenza A virus infection. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Negri-Cesi, P. Bisphenol A Interaction With Brain Development and Functions. Dose-Response Publ. Int. Hormesis Soc. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wojtowicz, A.K. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol. Rep. PR 2013, 65, 1632–1639. [Google Scholar] [CrossRef]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Krementsov, D.N.; Katchy, A.; Case, L.K.; Carr, F.E.; Davis, B.; Williams, C.; Teuscher, C. Studies in experimental autoimmune encephalomyelitis do not support developmental bisphenol a exposure as an environmental factor in increasing multiple sclerosis risk. Toxicol. Sci. 2013, 135, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Luhder, F. Interleukin-17—extended features of a key player in multiple sclerosis. Am. J. Pathol. 2008, 172, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeyer-Langford, C.; Rodrigues, A.; Kochan, K.J.; Haney, R.; Rassu, F.; Steelman, A.J.; Young, C.; Riggs, P.; Storts, R.; Meagher, M.W.; et al. Consequences of perinatal bisphenol A exposure in a mouse model of multiple sclerosis. Autoimmunity 2014, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Bodin, J.; Bolling, A.K.; Samuelsen, M.; Becher, R.; Lovik, M.; Nygaard, U.C. Long-term bisphenol A exposure accelerates insulitis development in diabetes-prone NOD mice. Immunopharmacol. Immunotoxicol. 2013, 35, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA 2008, 300, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Bodin, J.; Bølling, A.K.; Becher, R.; Kuper, F.; Lovik, M.; Nygaard, U.C. Transmaternal bisphenol A exposure accelerates diabetes type 1 development in NOD mice. Toxicol. Sci. 2014, 137, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.J.; Benoist, C.; Mathis, D. The immune system’s involvement in obesity-driven type 2 diabetes. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 436–442. [Google Scholar]
- Ashley-Martin, J.; Dodds, L.; Arbuckle, T.E.; Ettinger, A.S.; Shapiro, G.D.; Fisher, M.; Morisset, A.-S.; Taback, S.; Bouchard, M.F.; Monnier, P. A birth cohort study to investigate the association between prenatal phthalate and bisphenol A exposures and fetal markers of metabolic dysfunction. Environ. Health 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- García-Arevalo, M.; Alonso-Magdalena, P.; Dos Santos, J.R.; Quesada, I.; Carneiro, E.M.; Nadal, A. Exposure to bisphenol-A during pregnancy partially mimics the effects of a high-fat diet altering glucose homeostasis and gene expression in adult male mice. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xia, W.; Wang, D.; Wan, Y.; Xu, B.; Chen, X.; Li, Y.; Xu, S. Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia 2013, 56, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Lin, Y.; Li, Y.; Ying, C.; Chen, J.; Song, L.; Zhou, Z.; Lv, Z.; Xia, W.; Chen, X. Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology 2011, 152, 3049–3061. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Vieira, E.; Soriano, S.; Menes, L.; Burks, D.; Quesada, I.; Nadal, A. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ. Health Perspect. 2010, 118. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Haller, A.M.; Sorrell, J.E.; Woods, S.C.; Jandacek, R.J.; Seeley, R.J. Perinatal exposure to bisphenol-a and the development of metabolic syndrome in CD-1 mice. Endocrinology 2010, 151, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Teppala, S. Relationship between urinary bisphenol A levels and diabetes mellitus. J. Clin. Endocrinol. Metab. 2011, 96, 3822–3826. [Google Scholar] [CrossRef] [PubMed]
- Andra, S.S.; Kalyvas, H.; Andrianou, X.D.; Charisiadis, P.; Christophi, C.A.; Makris, K.C. Preliminary evidence of the association between monochlorinated bisphenol A exposure and type II diabetes mellitus: A pilot study. J. Environ. Sci. Health A Toxic/Hazard. Subst. Environ. Eng. 2015, 50, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Park, H. Association between urinary concentrations of bisphenol A and type 2 diabetes in Korean adults: A population-based cross-sectional study. Int. J. Hyg. Environ. Health 2013, 216, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.; Bi, Y.; Wang, T.; Xu, M.; Xu, Y.; Huang, Y.; Li, M.; Li, X.; Wang, W.; Chen, Y.; et al. Relationship of urinary bisphenol A concentration to risk for prevalent type 2 diabetes in Chinese adults: A cross-sectional analysis. Ann. Intern. Med. 2011, 155, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Wang, W.; Xu, M.; Wang, T.; Lu, J.; Xu, Y.; Dai, M.; Chen, Y.; Zhang, D.; Sun, W.; et al. Diabetes Genetic Risk Score Modifies Effect of Bisphenol A Exposure on Deterioration in Glucose Metabolism. J. Clin. Endocrinol. Metab. 2016, 101, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Kendziorski, J.A.; Kendig, E.L.; Gear, R.B.; Belcher, S.M. Strain specific induction of pyometra and differences in immune responsiveness in mice exposed to 17alpha-ethinyl estradiol or the endocrine disrupting chemical bisphenol A. Reprod. Toxicol. (Elmsford, NY) 2012, 34, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Midoro-Horiuti, T.; Tiwari, R.; Watson, C.S.; Goldblum, R.M. Maternal bisphenol a exposure promotes the development of experimental asthma in mouse pups. Environ. Health Perspect. 2010, 118. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Akdis, C.A.; Finkelman, F.D.; Rothenberg, M.E. Advances and highlights in mechanisms of allergic disease in 2015. J. Allergy Clin. Immunol. 2016, 137, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, Y.; Yamada, A.; Tokuriki, S.; Yasutomi, M.; Omata, N.; Mayumi, M. Transmaternal exposure to bisphenol a modulates the development of oral tolerance. Pediatr. Res. 2007, 62, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.; Miller, R. The Impact of Bisphenol A and Phthalates on Allergy, Asthma, and Immune Function: A Review of Latest Findings. Curr. Environ. Health Rep. 2015, 2, 379–387. [Google Scholar] [CrossRef]
- Bauer, S.M.; Roy, A.; Emo, J.; Chapman, T.J.; Georas, S.N.; Lawrence, B.P. The effects of maternal exposure to bisphenol A on allergic lung inflammation into adulthood. Toxicol. Sci. 2012, 130, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Goldblum, R.M.; Midoro-Horiuti, T. Fetal exposure to bisphenol A as a risk factor for the development of childhood asthma: An animal model study. Environ. Health 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Bergin, I.L.; Dolinoy, D.C.; Zaslona, Z.; Little, R.J.; Tao, Y.; Peters-Golden, M.; Mancuso, P. Perinatal bisphenol A exposure beginning before gestation enhances allergen sensitization, but not pulmonary inflammation, in adult mice. J. Dev. Orig. Health Dis. 2014, 5, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Petzold, S.; Averbeck, M.; Simon, J.C.; Lehmann, I.; Polte, T. Lifetime-dependent effects of bisphenol A on asthma development in an experimental mouse model. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Menard, S.; Guzylack-Piriou, L.; Leveque, M.; Braniste, V.; Lencina, C.; Naturel, M.; Moussa, L.; Sekkal, S.; Harkat, C.; Gaultier, E. Food intolerance at adulthood after perinatal exposure to the endocrine disruptor bisphenol A. FASEB J. 2014, 28, 4893–4900. [Google Scholar] [CrossRef] [PubMed]
- Spanier, A.J.; Kahn, R.S.; Kunselman, A.R.; Hornung, R.; Xu, Y.; Calafat, A.M.; Lanphear, B.P. Prenatal exposure to bisphenol A and child wheeze from birth to 3 years of age. Environ. Health Perspect 2012, 120, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Donohue, K.M.; Miller, R.L.; Perzanowski, M.S.; Just, A.C.; Hoepner, L.A.; Arunajadai, S.; Canfield, S.; Resnick, D.; Calafat, A.M.; Perera, F.P.; et al. Prenatal and postnatal bisphenol A exposure and asthma development among inner-city children. J. Allergy Clin. Immunol. 2013, 131, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Gascon, M.; Casas, M.; Morales, E.; Valvi, D.; Ballesteros-Gómez, A.; Luque, N.; Rubio, S.; Monfort, N.; Ventura, R.; Martínez, D. Prenatal exposure to bisphenol A and phthalates and childhood respiratory tract infections and allergy. J. Allergy Clin. Immunol. 2015, 135, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Spanier, A.J.; Kahn, R.S.; Kunselman, A.R.; Schaefer, E.W.; Hornung, R.; Xu, Y.; Calafat, A.M.; Lanphear, B.P. Bisphenol a exposure and the development of wheeze and lung function in children through age 5 years. JAMA Pediatr. 2014, 168, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Standish, L.J.; Sweet, E.S.; Novack, J.; Wenner, C.A.; Bridge, C.; Nelson, A.; Martzen, M.; Torkelson, C. Breast cancer and the immune system. J. Soc. Integr. Oncol. 2008, 6, 158–168. [Google Scholar] [PubMed]
- Soto, A.M.; Brisken, C.; Schaeberle, C.; Sonnenschein, C. Does cancer start in the womb? Altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J. Mammary Gland Biol. Neoplasia 2013, 18, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, C.; Anderson, O.S.; Bergin, I.L.; Vandenbergh, D.J.; Gyekis, J.P.; Dingman, M.A.; Yang, J.; Dolinoy, D.C. Dose-dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ. Health Perspect. 2014, 122. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.A.; Ho, S.-M.; Hunt, P.A.; Knudsen, K.E.; Soto, A.M.; Prins, G.S. An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod. Toxicol. 2007, 24, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Mamillapalli, R.; Goetz, L.G.; Jorgenson, E.; Ilagan, Y.; Taylor, H.S. Bisphenol A (BPA) exposure in utero leads to immunoregulatory cytokine dysregulation in the mouse mammary gland: A potential mechanism programming breast cancer risk. Hormones Cancer 2016, 7, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Paulose, T.; Speroni, L.; Sonnenschein, C.; Soto, A.M. Estrogens in the wrong place at the wrong time: Fetal BPA exposure and mammary cancer. Reprod. Toxicol. 2015, 54, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Betancourt, A.M.; Mobley, J.A.; Russo, J.; Lamartiniere, C.A. Proteomic analysis in mammary glands of rat offspring exposed in utero to bisphenol A. J. Proteom. 2010, 73, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Moral, R.; Wang, R.; Russo, I.H.; Lamartiniere, C.A.; Pereira, J.; Russo, J. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J. Endocrinol. 2008, 196, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Brady, N.J.; Chuntova, P.; Schwertfeger, K.L. Macrophages: Regulators of the Inflammatory Microenvironment during Mammary Gland Development and Breast Cancer. Mediat. Inflamm. 2016, 2016, 4549676. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Liu, F.; Alomirah, H.; Loi, V.D.; Mohd, M.A.; Moon, H.-B.; Nakata, H.; Kannan, K. Bisphenol S in urine from the United States and seven Asian countries: Occurrence and human exposures. Environ. Sci. Technol. 2012, 46, 6860–6866. [Google Scholar] [CrossRef] [PubMed]
- Kinch, C.D.; Ibhazehiebo, K.; Jeong, J.-H.; Habibi, H.R.; Kurrasch, D.M. Low-dose exposure to bisphenol A and replacement bisphenol S induces precocious hypothalamic neurogenesis in embryonic zebrafish. Proc. Natl. Acad. Sci. USA 2015, 112, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.; Sánchez, P.; Torres, J.M.; Ortega, E. Bisphenol A, bisphenol F and bisphenol S affect differently 5α-reductase expression and dopamine—Serotonin systems in the prefrontal cortex of juvenile female rats. Environ. Res. 2015, 142, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Fic, A.; Mlakar, S.J.; Juvan, P.; Mlakar, V.; Marc, J.; Dolenc, M.S.; Broberg, K.; Mašič, L.P. Genome-wide gene expression profiling of low-dose, long-term exposure of human osteosarcoma cells to bisphenol A and its analogs bisphenols AF and S. Toxicol. In Vitro 2015, 29, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Multiple Sclerosis Disease Models | Animal Model | Exposure Windows | BPA Dose | Routes of Administration | Diet | Effects | Reference |
|---|---|---|---|---|---|---|---|
| Autoimmune Encephalomyelitis (EAE) | Male and Female Mice (C57BL/6J and SJL/JCrHsd) | Gestation and Lactation | 10 μg/mL | 1% ethanol in drinking water | AIN-93G (casein-based phytoestrogen-free) | No Effect on EAE or INF-γ; decreased IL-17 in females | [77] |
| Theiler’s Murine Encephalomyelitis Virus (TMEV) | Male and Female Mice (SJL) | Gestation and Lactation | 10 μg/kg BW | Charcoal-stripped corn oil via gavage | Not Specified | Earlier onset of disease; increased inflammation; decreased antibodies to virus | [80] |
| Diabetes Disease Model | Animal Model | Exposure Windows | BPA Dose | Routes of Administration | Diet | Effects | Reference |
|---|---|---|---|---|---|---|---|
| Type 1 Diabetes | Female NOD/ShiLtJ Mice Offspring | Gestation and Lactation | 0.1, 1 or 10 mg/L | Deionized autoclaved drinking water | 2919X (minimal phytoestrogen content) | Increased insulitis, diabetes, Treg cells and apoptosis of β-cells, α-cells and macrophages in highest dose only | [83] |
| Type 1 Diabetes | Female NOD/ShiLtJ Mice Offspring | From Gestation to End of Study | 1 mg/L | Deionized autoclaved drinking water | 2919X (minimal phytoestrogen content) | Increased insulitis, diabetes and apoptosis pancreatic cells macrophages; decreased phagocytic macrophages, IL-10, IL-4 and TNF-α | [52] |
| Type 2 Diabetes | Male and Female Human Infants | 1st Trimester | ≤0.34 to >1.7 μg/L (measured, not dosed) | Measured exposure from environment, etc. | Not Specified | Lower adiponectins in male cord blood | [85] |
| Type 2 Diabetes | Male Wistar Rat Offspring | Gestation and Lactation | 50 μg/kg | Gavage; dissolved in corn oil | Not Specified | Increased insulin and insulin resistance; reduced glycogen | [87] |
| Type 2 Diabetes | Male and Female Wistar Rat Offspring | Gestation and Lactation | 50, 250 or 1250 μg/kg | Gavage; in corn oil | Standard or high-fat diet | Low dose only: increased body weight and insulin; altered β-cell function; high-fat diet and male had a greater effect | [88] |
| Type 2 Diabetes | Male and Female OF-1 Mice Offspring | Prenatal (GD9-16) | 10 or 100 μg/kg | S.C. injection; in tocopherol-stripped corn oil | Soy/alfalfa-free | Low dose, males only: increased insulin, insulin sensitivity and glucose intolerance; altered β-cell function | [89] |
| Type 2 Diabetes | Male and Female CD-1 Mice Offspring | Gestation and Lactation | About 0.25 μg/kg | In food | Phytoestrogen-free until weaning then LFD and half mice after 9 weeks old high-fat diet | No effect on glucose tolerance | [90] |
| Type 2 Diabetes | Male OF-1 Mice Offspring | Prenatal (GD9-16) | 10 μg/kg | S.C. injection; in tocopherol-stripped corn oil | Soy/alfalfa-free | No effect for insulin sensitivity; glucose intolerance; increased NEFA | [86] |
| OVA Sensitization | Animal Model/Sex | Exposure Windows | BPA Dose | Routes of Administration | Diet | Effects | Reference |
|---|---|---|---|---|---|---|---|
| Airway Sensitization | Male and Female C57BL/6 Offspring | GD6-PND21 | 0.5, 5, 50 or 500 μg/kg | Peanut oil via gavage | AIN76-semi-PD1RR chow (phytoestrogen-free) | Increased airway lymphocytes and lung inflammation in females; decreased airway neutrophils and lung inflammation in males; no effect on IgE, T cell subpopulations or BALF cytokines | [101] |
| “Suboptimal” Peritoneal Sensitization | BALB/c offspring | Gestation and Lactation | 5 or 10 μg/mL | Drinking water | Phytoestrogen-free | Increased AHR, BALF eosinophils and IgE; no effect for IgG1 | [97] |
| BALB/c offspring | Prenatal, perinatal or postnatal | 5 μg/mL | Drinking water | Phytoestrogen-free | Increased AHR and BALF eosinophils from prenatal and perinatal; no effect for postnatal only exposure | [102] | |
| Peritoneal Sensitization | Female C57BL/6 Offspring | GD6-PND21 | 0.5, 5, 50 or 500 μg/kg | Peanut oil via gavage | AIN76-semi-PD1RR chow (phytoestrogen-free) | Decreased airway eosinophils and IgE; no effect on AHR | [101] |
| Male and Female BALB/c Offspring | Gestation and Lactation | 50 ng, 50 μg or 50 mg/kg diet | In food | AIN-93G (phytoestrogen-free) | Increased IgE, IL-13 and INF-γ; decreased BALF leukocytes, eosinophils, IL-17 and CysLTs; decreased macrophages, PMN and lung inflammation in males; in females only: decreased BALF IL-4, IL-13 and TNF-α, increased lung RANTES and no effect on lung inflammation | [103] | |
| BALB/cByJ Offspring | One week after mating period until birth or PND21 | 5 μg/mL | Drinking water | C1000 (phytoestrogen-free) | Prenatal: no effect on AHR or airway inflammation; perinatal: increased lung inflammation, IgE and IL-13 | [104] | |
| Female Wistar rats offspring | GD15-PND21 | 0.5, 5 or 50 μg/kg | 4% ethanol in corn oil via oral | Rodent Diet 2018 (<20 pmol estrogen content) | Increased IgG, activated T cells, splenocyte proliferation, INF-γ, neutrophils and IL-10 (colon); no effect for IgE, Treg cells or IL-10 (spleen); decreased TGF-β (colon) | [105] | |
| Gavage Sensitization | Male heterozygous offspring of OVA-TCR-Tg crossed with BALB/c | Gestation and Lactation | 0, 0.1 or 1 ppm BPA | In Food | Not Specified | Increased IL-13, INFγ, anti-OVA IgG1 and anti-OVA IgG2a; no change in IL-4; decreased OVA-specific T cells and Treg response to OVA | [99] |
| Sex/Age | Time of BPA Measurement | BPA Measured | BPA Levels Assessed From | Effects | Reference |
|---|---|---|---|---|---|
| Male and Female Infants | 1st Trimester | 0.8 μg/L | Median urine concentration | Non-monotonic increase of TSLP, IL-33 and IgE in cord blood | [68] |
| Male and Female Children | 16 weeks gestation, 26 weeks gestation and birth | 2.4 μg BPA/g creatinine | Median urine concentration | Increased wheeze risk of 6 months old, but not 3 years | [106] |
| Male and Female Children | 3rd trimester, 3, 5 and 7 years old | 1.8 ng/mL (3rd trimester), 3.8 ng/mL (3 years), 3.1 ng/mL (5 years), 2.7 ng/mL (7 years) | Median urine concentration | Higher prenatal BPA levels inversely correlated with wheeze at 5 years and bronchodilator response; postnatal exposure increased wheeze, airway inflammation and aeroallergen sensitization at 7 years | [107] |
| Male and Female Children | 12 and 32 weeks gestation | 2.4 μg BPA/g creatinine | Median urine concentration | Increased wheeze, respiratory tract infection and bronchitis risk from 6 months–7 years old; no change in atopy/IgE levels | [108] |
| Male and Female Children | 16 weeks gestation, 26 weeks gestation and birth | 2.4 μg BPA/g creatinine | Median urine concentration | Decreased lung function at 4 years, but not 5 years; 16 week BPA only: increased wheeze and persistent wheeze risk | [109] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Huang, G.; Guo, T.L. Developmental Bisphenol A Exposure Modulates Immune-Related Diseases. Toxics 2016, 4, 23. https://doi.org/10.3390/toxics4040023
Xu J, Huang G, Guo TL. Developmental Bisphenol A Exposure Modulates Immune-Related Diseases. Toxics. 2016; 4(4):23. https://doi.org/10.3390/toxics4040023
Chicago/Turabian StyleXu, Joella, Guannan Huang, and Tai L. Guo. 2016. "Developmental Bisphenol A Exposure Modulates Immune-Related Diseases" Toxics 4, no. 4: 23. https://doi.org/10.3390/toxics4040023