
Neurotoxic Effects of Platinum Compounds: Studies in vivo on Intracellular Calcium Homeostasis in the Immature Central Nervous System


 ,
, 
Abstract
:1. Introduction
1.1. State of Art on Toxicity of Platinum Compounds and Aim
1.2. An Old Platinum Compound vs. a New Platinum Compound. CisPt vs. PtAcacDMS: Action Mechanisms and Cytotoxicity
1.3. Immature CNS Areas in the Postnatal Life of Mammals
2. Experimental Section
2.1. Animals and Schedule of the Experimental Plan
2.2. Immunocytochemistry in Light Microscopy
2.3. Immunocytochemistry in Fluorescence Microscopy
2.4. Determination of Cell Immunoreactivity Intensity
3. Results and Discussion
3.1. Platinum Compounds and Calcium Homeostasis
3.2. Histochemical Detection of Calcium Homeostasis and Differentiating Cells in the Immature Cerebellum and Hippocampus
3.2.1. Calbindin
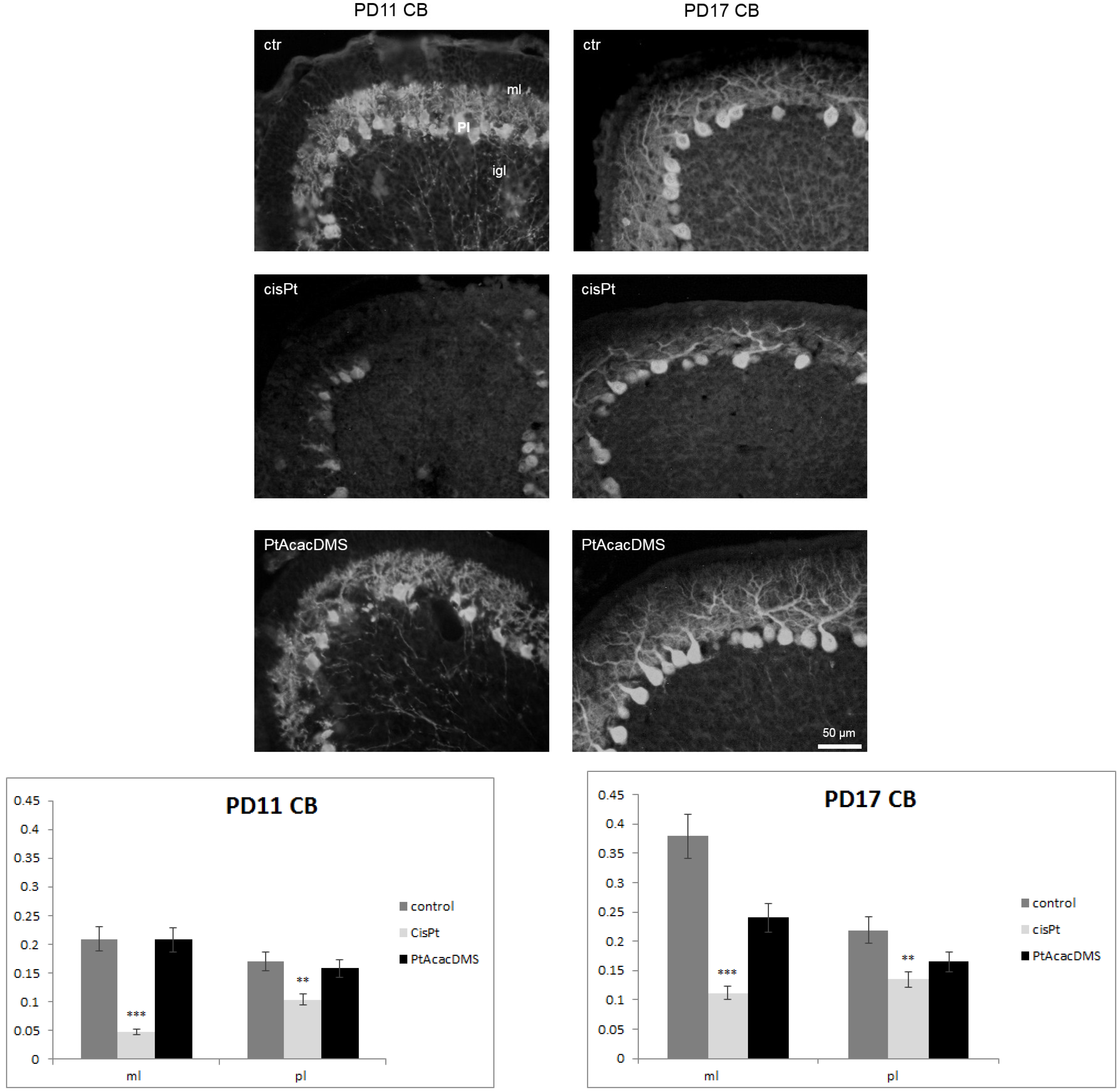
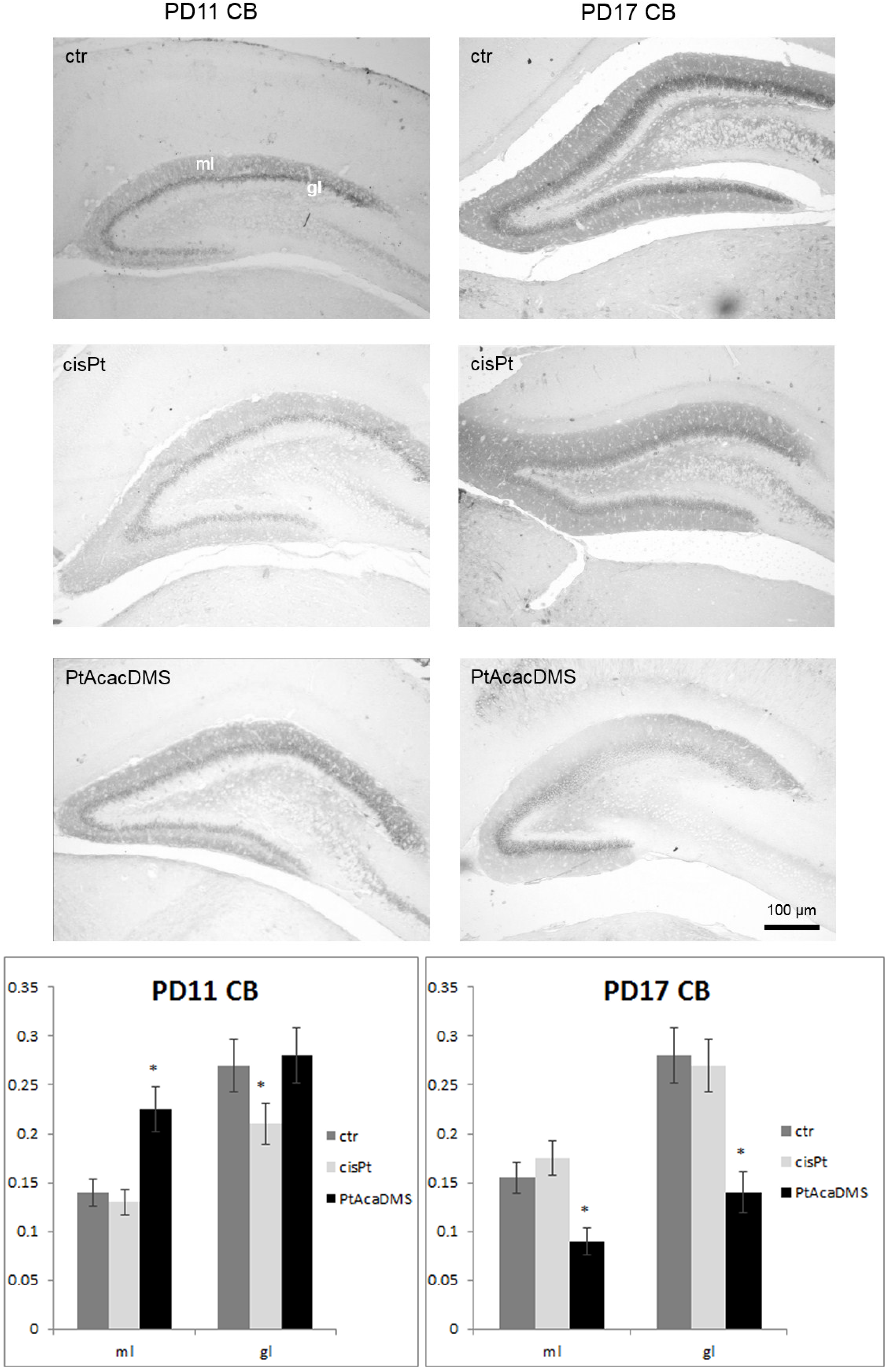
3.2.2. Plasma Membrane Calcium ATPase
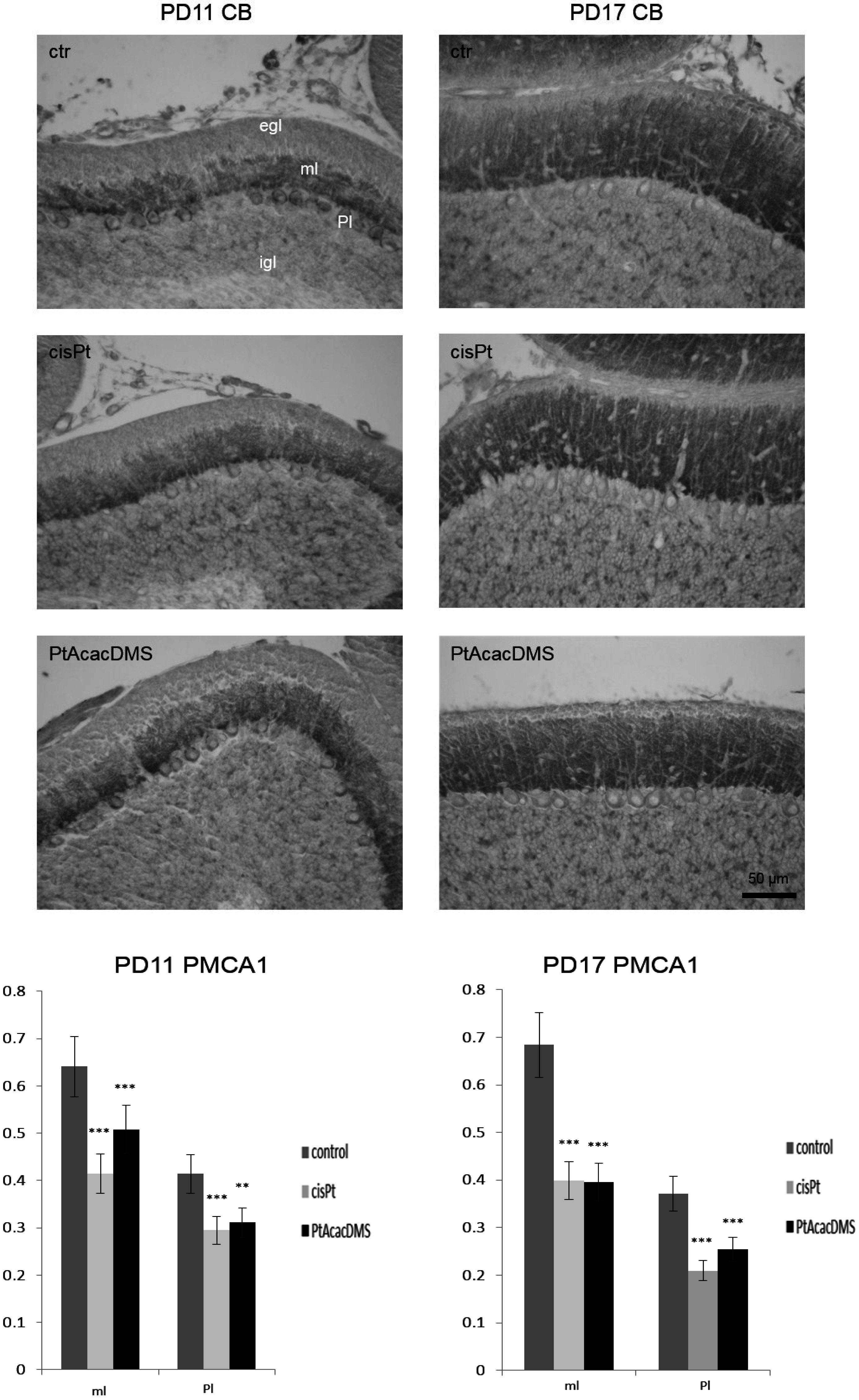
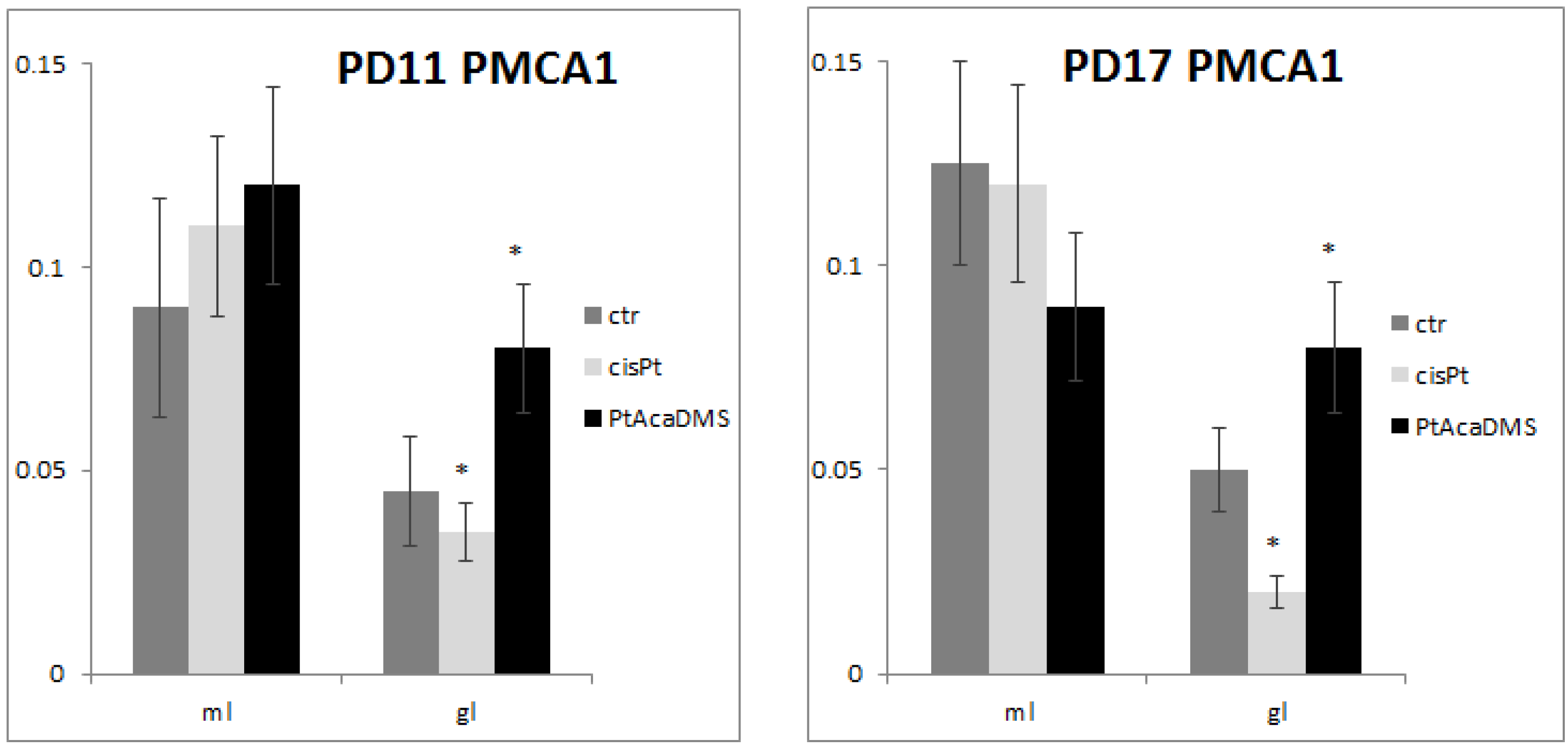
3.3. Platinum Compounds and Calcium Homeostasis in the Immature Neuroarchitecture

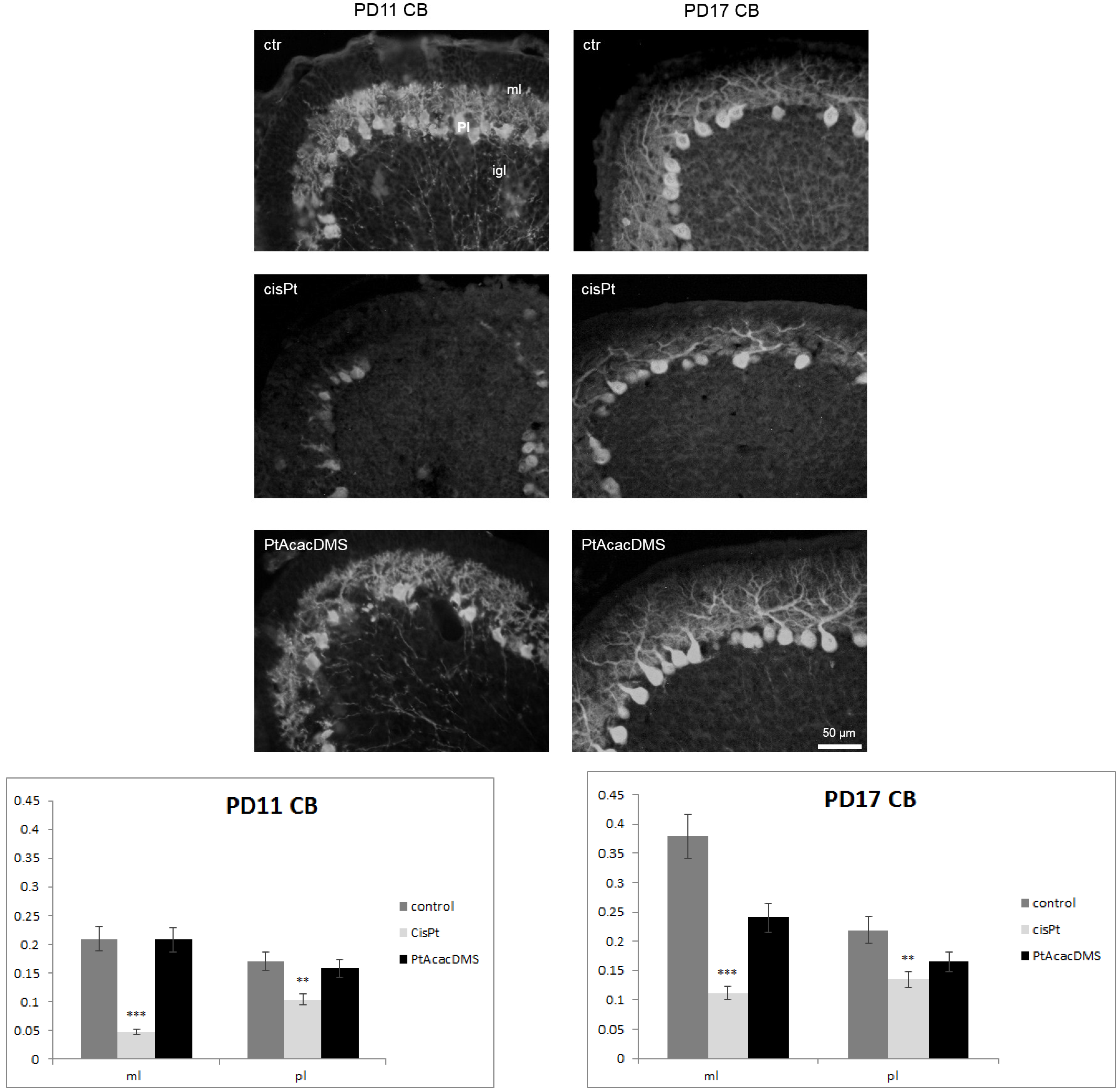{kind=link}
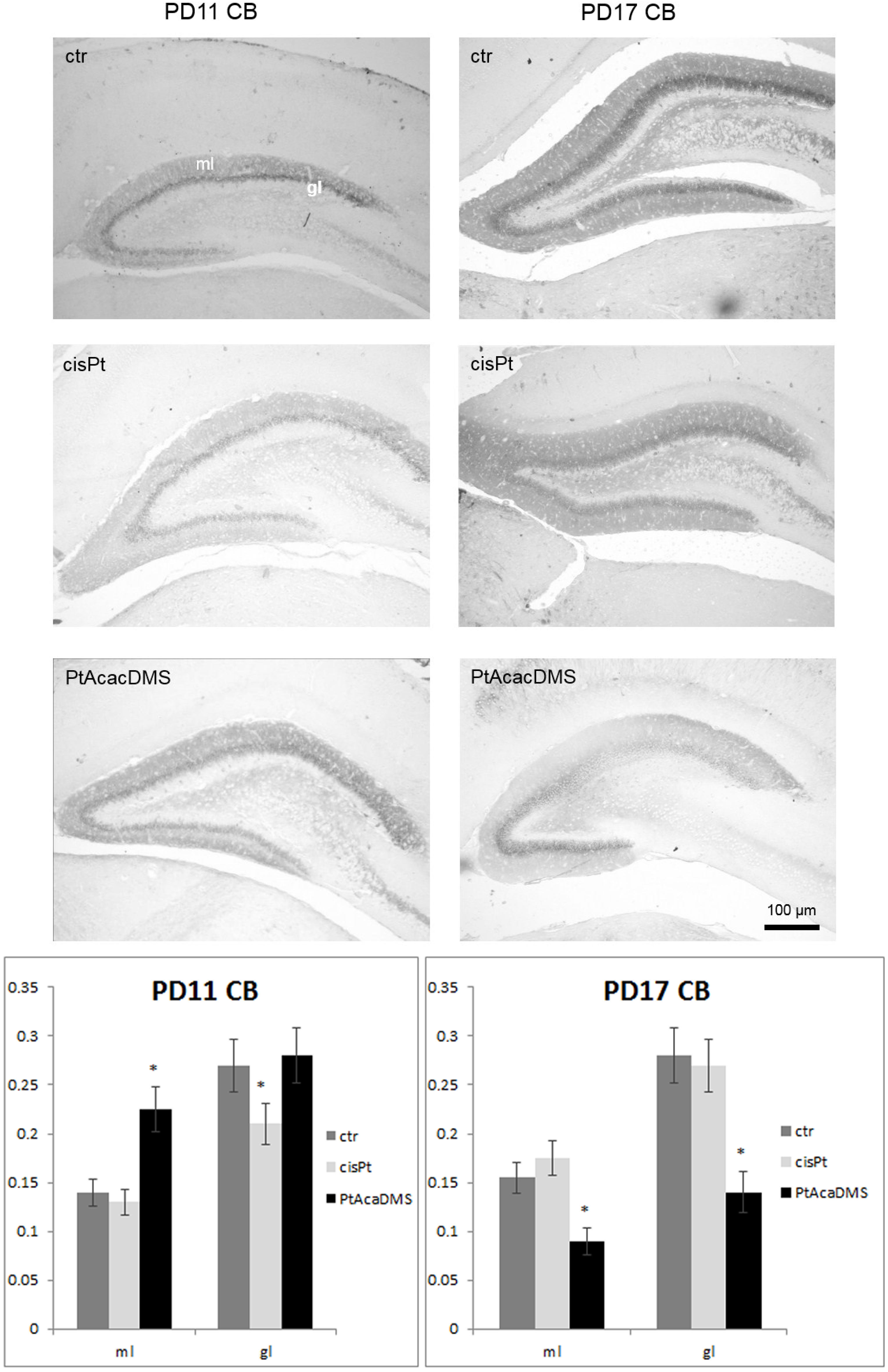{kind=link}
{kind=link}
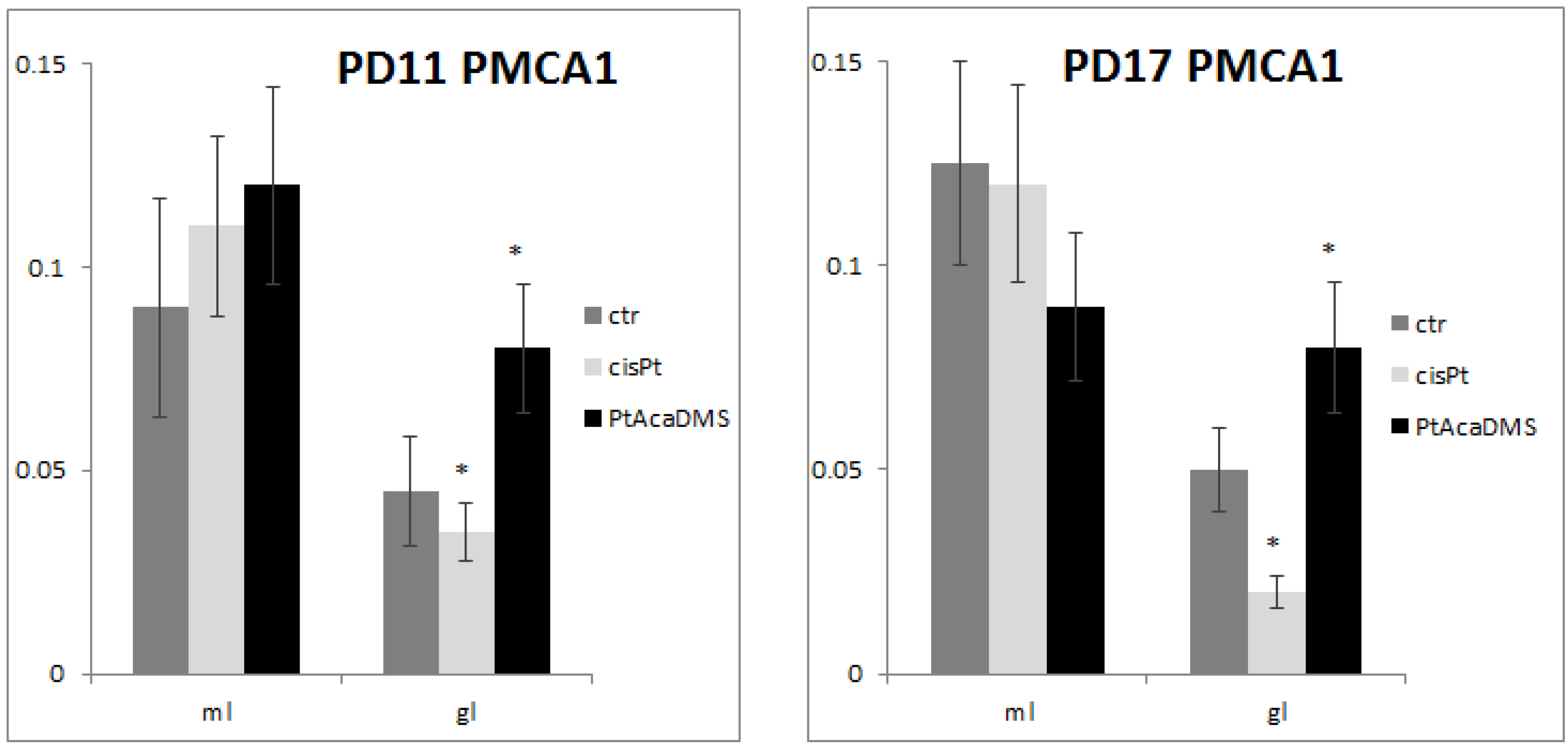{kind=link}
| Days of treatment | cisPt | PtAcacDMS |
|---|---|---|
| PD11 | Decreased Ca2+ buffering: CB ↓ | Steady Ca2+ buffering: CB = |
| Decreased Ca2+ efflux: PMCA1 ↓ | Decreased Ca2+ efflux: PMCA1 ↓ | |
| PD17 | Decreased Ca2+ buffering: CB ↓ | Steady Ca2+ buffering: CB = |
| Decreased Ca2+ efflux: PMCA1 ↓ | Decreased Ca2+ efflux: PMCA1 ↓ |
| Days of treatment | cisPt | PtAcacDMS |
|---|---|---|
| PD11 | Decreased Ca2+ buffering: CB ↓ | Steady Ca2+ buffering: CB = |
| Decreased Ca2+ efflux: PMCA1 ↓ | Increased Ca2+ efflux: PMCA1 ↑ | |
| PD17 | Steady Ca2+ buffering: CB = | Decreased Ca2+ buffering: CB ↓ |
| Decreased Ca2+ efflux: PMCA1 ↓ | Increased Ca2+ efflux: PMCA1 ↑ |
3.4. Platinum Compounds and Morphological and Molecular Damages in the Immature Cerebellum and Hippocampus
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Screnci, D.; McKeage, M.J. Platinum neurotoxicity: Clinical profiles, experimental models and neuroprotective approaches. J. Inorg. Biochem. 1999, 77, 105–110. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Martinez, S.M.; Prieto-Garcia, L.; Prieto, M.; Lopez-Novoa, J.M.; Lopez Hernandez, F.J. Subcellular targets of cisplatin cytotoxicity: An integrated view. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, A.S.; Singh, N. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology 2012, 291, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.J.; Gorlick, R. Chemotherapy resistance in osteosarcoma: Current challenges and future directions. Exp. Rev. Anticancer Ther. 2006, 6, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Ann. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Ricci, G.; de Maria, F.; Antonini, G.; Turella, P.; Bullo, A.; Stella, L.; Filomeni, G.; Federici, G.; Caccuri, A.M. 7-nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases: Mechanism of action of potential anticancer drugs. J. Biol. Chem. 2005, 280, 26397–26405. [Google Scholar] [CrossRef] [PubMed]
- Pasetto, L.M.; D’Andrea, M.R.; Brandes, A.A.; Rossi, E.; Monfardini, S. The development of platinum compounds and their possible combination. Crit. Rev. Oncol. Hematol. 2006, 60, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Mormiroli, P. Chemotherapy-induced peripheral neurotoxicity. Nat. Rev. 2010, 6, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Kyritsis, A.P.; Makatsoris, T.; Kalofonus, H.P. Chemotherapy-induced peripheral neuropathy in adults: A comprehensive update of the literature. Cancer Manag. Res. 2014, 6, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.S. Chemotherapy-related cognitive impairment: The breast cancer experience. Oncol. Nurs. Forum 2012, 39, E31–E40. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, K.D.; Hutchinson, A.D.; Wilson, A.D.; Nettelbeck, T. A meta-analysis of the effects of chemotherapy on cognition in patients with cancer. Cancer Treat. Rev. 2013, 39, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Seigers, R.; Fardell, J.E. Neurobiological basis of chemotherapy-induced cognitive impairment: A review of rodent research. Neurosci. Biobehav. Rev. 2011, 35, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.K.; de Angelis, L.M. Neurologic complications of cancer chemotherapy. Semin. Oncol. 2006, 33, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Sterzing, F.; Grehn, F.; Dinkel, G.; Krempien, R.; Hartung, G.; Debus, J.; Harms, W. Severe reversible toxic encephalopathy induced by cisplatin in a patient with cervical carcinoma receiving combined radiochemotherapy. Strahlenther. Onkol. 2007, 183, 487–489. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, K.; Asakura, M.; Minami, T.; Okazaki, Y.; Kadota, E.; Hashimoto, S. Toxicity of cisplatin to the central nervous system of male rabbits. Biol. Trace Elem. Res. 2000, 74, 223–225. [Google Scholar] [CrossRef]
- Abou-Elghait, A.; El-Gamal, D.A.; Abdel-Sameea, A.R.; Mohamed, A.A. Effect of cisplatin on the cerebellar cortex and spinal cord of adult male albino rat and the possible role of vitamin E: Light and electron microscopic study. Egypt. J. Histol. 2010, 33, 202–212. [Google Scholar]
- Gong, X.; Schwartz, P.H.; Linskey, M.E.; Bota, D.A. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology 2011, 76, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Andres, A.L.; Gong, X.; Di, K.; Bota, D.A. Low-doses of cisplatin injure hippocampal synapses: A mechanism for “chemo” brain. Exp. Neurol. 2014, 255, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Prestayko, A.W.; D’Aoust, J.C.; Issell, B.F.; Crooke, S.T. Cisplatin (cis diamine-dichloroplatinum II). Cancer Treat. Rev. 1979, 6, 17–35. [Google Scholar] [CrossRef]
- Gilchrist, L.S.; Marais, L.; Tanner, L. Comparison of two chemotherapy-induced peripheral neuropathy measurement approaches in children. Support Care Cancer 2014, 22, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Sioka, C.; Kyritsis, A. Central and peripheral nervous system toxicity of common chemotherapeutic agents. Cancer Chemother. Pharmacol. 2009, 63, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Rodier, P.M. Chronology of neuron development: Animal studies and their clinical implications. Dev. Med. Child Neurol. 1980, 22, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Rodier, P.M. Developing brain as a target of toxicity. Environ. Health Perspect. 1995, 103, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Rodier, P.M. Environmental causes of Central Nervous System maldevelopment. Pediatrics 2004, 113, 1076–1083. [Google Scholar] [PubMed]
- Ferguson, S.A. Neuroanatomical and functional alterations resulting from early postnatal cerebellar insults in rodents. Pharmacol. Biochem. Behav. 1996, 55, 663–671. [Google Scholar] [CrossRef]
- Fujii, T. Transgenerational effects of maternal exposure to chemicals on the functional development of the brain in the offspring. Cancer Causes Control 1997, 8, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Fonnum, F.; Lock, E.A. Cerebellum as a target for toxic substances. Toxicol. Lett. 2000, 112–113, 9–16. [Google Scholar] [CrossRef]
- Roberts, J.J.; Pera, J.J., Jr. Platinum, Gold, and other Metal Chemotherapeutic Agents: Chemistry and Biochemistry; Lippard, S.J., Ed.; American Chemical Society: Washington, DC, USA, 1983; pp. 2–25. [Google Scholar]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Pérez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, M.G.; O’Neill, C.; Robertson, D.; Kelland, L.R.; Harrap, K.R. cisDiammine dichloroplatinum(II)-induced cell death through apoptosis in sensitive and resistant human ovarian carcinoma cell lines. Cancer Chemother. Pharmacol. 1996, 37, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Henkels, K.M.; Turchi, J.J. Induction of apoptosis in cisplatin-sensitive and -resistant human ovarian cancer cell lines. Cancer Res. 1997, 57, 4488–4492. [Google Scholar] [PubMed]
- De Pascali, S.A.; Papadia, P.; Ciccarese, A.; Pacifico, C.; Fanizzi, F.P. First examples of βdiketonate platinum II complexes with sulfoxide ligands. Eur. J. Inorg. Chem. 2005, 5, 788–796. [Google Scholar] [CrossRef]
- De Pascali, S.A.; Papadia, P.; Capoccia, S.; Marchiò, L.; Lanfranchi, M.; Ciccarese, A.; Fanizzi, F.P. Hard/soft selectivity in ligand substitution reactions of β-diketonate platinum(II) complexes. Dalton Trans. 2009, 37, 7786–7795. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Calabriso, N.; de Pascali, S.A.; Urso, L.; Ciccarese, A.; Fanizzi, F.P.; Migoni, D.; Marsigliante, S. New platinum(II) complexes containing both an O,O′-chelated acetylacetonate ligand and a sulfur ligand in the platinum coordination sphere induce apoptosis in HeLa cervical carcinoma cells. Biochem. Pharmacol. 2007, 74, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Calabriso, N.; Fanizzi, F.P.; de Pascali, S.A.; Urso, L.; Ciccarese, A.; Migoni, D. Marsigliante, [Pt(O,O′-acac)(γ-acac)(DMS)], a new Pt compound exerting fast cytotoxicity in MCF-7 breast cancer cells via the mitochondrial apoptotic pathway. Br. J. Pharmacol. 2008, 153, 34–49. [Google Scholar] [CrossRef] [PubMed]
- De Pascali, S.A.; Lugoli, F.; de Donno, A.; Fanizzi, F.P. Mutagenic tests confirm that new acetylacetonate Pt(II) complexes induce apoptosis in cancer cells interacting with nongenomic biological targets. Met. Based Drugs. 2011, 2011, 763436. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Calabriso, N.; Vetrugno, C.; Fanizzi, F.P.; de Pascali, S.A.; Storelli, C.; Marsigliante, S. The platinum (II) complex [Pt(O,O′-acac)(gamma-acac)(DMS)] alters the intracellular calcium homeostasis in MCF-7 breast cancer cells. Biochem. Pharmacol. 2011, 81, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Piccolini, V.M.; Santin, G.; Bottone, M.G.; de Pascali, S.A.; Migoni, D.; Iadarola, P.; Fanizzi, F.P.; Bernocchi, G. The developmental neurotoxicity study of platinum compounds: Effects of cisplatin versus a novel Pt(II) complex on rat cerebellum. Neurotoxicol. Teratol. 2011, 33, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Büsselberg, D. Anti-cancer drugs interfere with intracellular calcium signaling. Neurotoxicology 2009, 30, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Arain, M.; Haque, M.; Johal, L.; Mathur, P.; Nel, W.; Rais, A.; Sandhu, R.; Sharma, S. Maturation of the adolescent brain. Neuropsychiatr. Dis. Treat. 2013, 9, 449–461. [Google Scholar] [PubMed]
- Altman, J.; Bayer, S.A. Development of the Cerebellar System in Relation to its Evolution, Structure, and Functions; CRC Press: Boca Raton, FL, USA, 1997. [Google Scholar]
- Hashimoto, M.; Hibi, M. Development and evolution of cerebellar neural circuits. Dev. Growth Differ. 2012, 54, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Danglot, L.; Triller, A.; Marty, S. The development of hippocampal interneurons in rodents. Hippocampus 2006, 16, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G. Adult Neurogenesis; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Piccolini, V.M.; Bottone, M.G.; Bottiroli, G.; de Pascali, S.A.; Fanizzi, F.P.; Bernocchi, G. Platinum drugs and neurotoxicity: Effects on intracellular calcium homeostasis. Cell Biol. Toxicol. 2013, 29, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Biran, V.; Verney, C.; Ferriero, D.M. Perinatal cerebellar injury in human and animal models. Neurol. Res. Int. 2012, 2012, 858929. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R. The cerebellum and neuropsychiatric disorders. Psychiatry Res. 2012, 198, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.A. Development of the hippocampal region in the rat II. Morphogenesis during embryonic and early postnatal life. J. Comp. Neurol. 1980, 190, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Altman, J. Morphological development of the rat cerebellum and some of its mechanisms. In The Cerebellum-New Vistas; Palay, S.L., Chan-Palay, V., Eds.; Springer: Berlin, Germany, 1982; pp. 8–49. [Google Scholar]
- Altman, J.; Bayer, A.S. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J. Comp. Neurol. 1990, 301, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Cantile, C.; Tamagno, I.; Merighi, A. Apoptosis in the mammalian CNS: Lessons from animal models. Vet. J. 2005, 170, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L.; Castagna, C.; Merighi, A. Neuronal cell death: An overview of its different forms in central and peripheral neurons. Methods Mol. Biol. 2015, 1254, 1–18. [Google Scholar] [PubMed]
- Zhang, L.; Goldman, J.E. Generation of cerebellar interneurons from dividing progenitors in white matter. Neuron 1996, 16, 47–54. [Google Scholar] [CrossRef]
- Sudarov, A.; Joyner, A.L. Cerebellum morphogenesis: The foliation pattern is orchestrated by multi-cellular anchoring centers. Neural Dev. 2007, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Piccolini, V.M.; Bernocchi, G. Postnatal development of the central nervous system: Anomalies in the formation of cerebellum fissures. Anat. Rec. 2010, 293, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F.; Buszaki, G. Interneurons of the hippocampus. Hippocampus 1996, 6, 347–470. [Google Scholar] [CrossRef]
- Ito, M. Cerebellar circuitry as a neuronal machine. Prog. Neurobiol. 2006, 78, 272–303. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.; Han, R.; Yang, Y.; Mayer-Pröschel, M.; Noble, M. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J. Biol. 2006, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, C. Calcium signaling and the development of specific neuronal connections. Prog. Brain Res. 2009, 75, 443–452. [Google Scholar]
- Komuro, H.; Kumada, T. Ca2+ transients control CNS neuronal migration. Cell Calcium 2005, 37, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.M.; Fuertes, M.A.; Alonso, C.; Perez, J.M. Is cisplatin-induced cell death always produced by apoptosis? Mol. Pharmacol. 2001, 59, 657–663. [Google Scholar] [PubMed]
- Kawai, Y.; Nakao, T.; Kunimura, N.; Kohda, Y.; Gemba, M. Relationship of intracellular calcium and oxygen radicals to cisplatin-related renal cell injury. J. Pharmacol. Sci. 2006, 100, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Huang, Y. Intracellular free calcium concentration and cisplatin resistance in human lung adenocarcinoma A549 cells. Biosci. Rep. 2000, 20, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Splettstoesser, F.; Florea, A.M.; Büsselberg, D. IP(3) receptor antagonist, 2-APB, attenuates cisplatin induced Ca2+-influx in HeLa-S3 cells and prevents activation of calpain and induction of apoptosis. Br. J. Pharmacol. 2007, 151, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Büsselberg, D. Occurrence, use and potential toxic effects of metals and metal compounds. Biometals 2006, 19, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Persechini, A.; Moncrief, N.D.; Kretsinger, R.H. The EF-hand family of calcium-modulated proteins. Trends Neurosci. 1989, 12, 462–467. [Google Scholar] [CrossRef]
- Schwaller, B.; Meyer, M.; Schiffmann, S. “New” functions for “old” proteins: The role of the calcium binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum 2002, 1, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Bastianelli, E. Distribution of calcium-binding proteins in the cerebellum. Cerebellum 2003, 2, 242–262. [Google Scholar] [CrossRef] [PubMed]
- Jinno, S.; Kosaka, T. Cellular architecture of the mouse hippocampus: A quantitative aspect of chemically defined GABAergic neurons with stereology. Neurosci. Res. 2006, 56, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Baimbridge, K.G.; Celio, M.R.; Rogers, J.H. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992, 15, 303–308. [Google Scholar] [CrossRef]
- Celio, M.R. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience 1990, 35, 375–475. [Google Scholar] [CrossRef]
- Celio, M.; Pauls, T.; Schwaller, B. (Eds.) Guidebook to the Calcium Binding Proteins; Oxford University Press: Oxford, UK, 1996.
- Garcia-Segura, L.M.; Baetens, D.; Roth, J.; Norman, A.W.; Orci, L. Immunohistochemical mapping of calcium-binding protein immunoreactivity in the rat central nervous system. Brain Res. 1984, 296, 75–86. [Google Scholar] [CrossRef]
- Ulfig, N.; Chan, W.Y. Differential expression of calcium-binding proteins in the red nucleus of the developing and adult human brain. Anat. Embryol. 2001, 203, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Nagerl, U.V.; Mody, I.; Jeub, M.; Lie, A.A.; Elger, C.E.; Beck, H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca2+ influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J. Neurosci. 2000, 20, 1831–1836. [Google Scholar] [PubMed]
- Maskey, D.; Pradhan, J.; Aryal, B.; Lee, C.M.; Choi, I.Y.; Park, K.S.; Kim, S.B.; Kim, H.G.; Kim, M.J. Chronic 835-MHz radiofrequency exposure to mice hippocampus alters the distribution of calbindin and GFAP immunoreactivity. Brain Res. 2010, 1346, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Enderlin, S.; Norman, A.W.; Celio, M.R. Ontogeny of the calcium binding protein calbindin D-28k in the rat nervous system. Anat. Embryol. 1987, 177, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Hogan, D.; Berman, N.E. Transient expression of calbindin-D28k immunoreactivity in layer V pyramidal neurons during postnatal development of kitten cortical areas. Brain Res. Dev. Brain Res. 1993, 74, 177–192. [Google Scholar] [CrossRef]
- Friauf, E. Distribution of calcium-binding protein calbindin-D28k in the auditory system of adult and developing rats. J. Comp. Neurol. 1994, 349, 193–211. [Google Scholar] [CrossRef] [PubMed]
- Pisu, M.B.; Roda, E.; Avella, D.; Bernocchi, G. Developmental plasticity of rat cerebellar cortex after cisplatin injury: Inhibitory synapses and differentiating Purkinje neurons. Neuroscience 2004, 129, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Avella, D.; Pisu, M.B.; Roda, E.; Gravati, M.; Bernocchi, G. Reorganization of the rat cerebellar cortex during postnatal development following cisplatin treatment. Exp. Neurol. 2006, 201, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Keuker, J.I.; Rochford, C.D.; Witter, M.P.; Fuchs, E. A cytoarchitectonic study of the hippocampal formation of the tree shrew (Tupaia belangeri). J. Chem. Neuroanat. 2003, 26, 1–15. [Google Scholar] [CrossRef]
- Magloczky, Z.; Freund, T.F. Selective neuronal death in the contralateral hippocampus following unilateral kainate injections into the CA3 subfield. Neuroscience 1993, 56, 317–335. [Google Scholar] [CrossRef]
- Ciaroni, S.; Cecchini, T.; Ferri, P.; Cuppini, R.; Ambrogini, P.; Santi, S.; Benedetti, S.; del Grande, P.; Papa, S. Neural precursor proliferation and newborn cell survival in the adult rat dentate gyrus are affected by vitamin E deficiency. Neurosci. Res. 2002, 44, 369–377. [Google Scholar] [CrossRef]
- Brandt, M.D.; Jessberger, S.; Steiner, B.; Kronenberg, G.; Reuter, K.; Bick-Sander, A.; von der Behrens, W.; Kempermann, G. Transient calretinin expression defines early postmitotic step of neuronal differentiation in adult hippocampal neurogenesis of mice. Mol. Cell. Neurosci. 2003, 24, 603–613. [Google Scholar] [CrossRef]
- Soriano, E.; del Rio, J.A.; Martinez, A.; Super, H. Organization of the embryonic and early postnatal murine hippocampus. I. Immunocytochemical characterization of neuronal populations in the subplate and marginal zone. J. Comp. Neurol. 1994, 342, 571–595. [Google Scholar] [CrossRef] [PubMed]
- Abraham, H.; Veszpremi, B.; Kravjak, A.; Kovacs, K.; Gomori, E.; Seress, L. Ontogeny of calbindin immunoreactivity in the human hippocampal formation with a special emphasis on granule cells of the dentate gyrus. Int. J. Dev. Neurosci. 2009, 27, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Braun, K. Calcium-binding proteins in avian and mammalian central nervous system: Localization, development and possible functions. Prog. Histochem. Cytochem. 1990, 21, 1–64. [Google Scholar] [CrossRef]
- Blatow, M.; Caputi, A.; Burnashev, N.; Monyer, H.; Rozov, A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron 2003, 38, 79–88. [Google Scholar] [CrossRef]
- Arabadzisz, D.; Ylinen, A.; Emri, Z. Increased inter-spike intervals and fast after-hyperpolarization of action potentials in rat hippocampal pyramidal cells accompanied with altered calbindin immunoreactivity 10–12 months after global forebrain ischemia. Neurosci. Lett. 2002, 331, 103–106. [Google Scholar] [CrossRef]
- Strehler, E.E.; Zacharias, D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [PubMed]
- Carafoli, E.; Brini, M. Calcium pumps: Structural basis for and mechanism of calcium transmembrane transport. Curr. Opin. Chem. Biol. 2000, 4, 152–161. [Google Scholar] [CrossRef]
- Burette, A.; Rockwood, J.M.; Strehler, E.E.; Weinberg, R.J. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J. Comp. Neurol. 2003, 467, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, K.A.; Bushong, E.A.; Mauer, A.S.; Strehler, E.E.; Weinberg, R.J.; Burette, A.C. Cellular and subcellular localization of the neuron-specific plasma membrane calcium ATPase PMCA1a in the rat brain. J. Comp. Neurol. 2010, 518, 3169–3183. [Google Scholar] [CrossRef] [PubMed]
- Mata, A.M.; Sepulveda, M.R. Plasma membrane Ca-ATPases in the nervous system during development and ageing. World J. Biol. Chem. 2010, 1, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Marcos, D.; Sepulveda, M.R.; Berrocal, M.; Mata, A.M. Ontogeny of ATP hydrolysis and isoform expression of the plasma membrane Ca2+-ATPase in mouse brain. BMC Neurosci. 2009, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, D.A.; DeMarco, S.J.; Strehler, E.E. mRNA expression of the four isoforms of the human plasma membrane Ca2+-ATPase in the human hippocampus. Brain Res. Mol. Brain Res. 1997, 45, 173–176. [Google Scholar] [CrossRef]
- Berliocchi, L.; Bano, D.; Nicotera, P. Ca2+ signals and death programmes in neurons. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2255–2258. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.L.; Strehler, E.E. Plasma membrane calcium ATPases as critical regulators of calcium homeostasis during neuronal cell function. Front. Biosci. 1999, 4, D869–D882. [Google Scholar] [CrossRef] [PubMed]
- Shull, G.E. Gene knockout studies of Ca2+-transporting ATPases. Eur. J. Biochem. 2000, 267, 5284–5290. [Google Scholar] [CrossRef] [PubMed]
- De Talamoni, N.; Smith, C.A.; Wasserman, R.H.; Beltramino, C.; Fullmer, C.S.; Penniston, J.T. Immunocytochemical localization of the plasma membrane calcium pump, calbindin-D28k, and parvalbumin in Purkinje cells of avian and mammalian cerebellum. Proc. Natl. Acad. Sci. USA 1993, 90, 11949–11953. [Google Scholar] [CrossRef]
- Stahl, W.L.; Eakin, T.J.; Owens, J.W., Jr.; Breininger, J.F.; Filuk, P.E.; Anderson, W.R. Plasma membrane Ca(2+)-ATPase isoforms: Distribution of mRNAs in rat brain by in situ hydridization. Brain Res. Mol. Brain Res. 1992, 16, 223–231. [Google Scholar] [CrossRef]
- Stauffer, T.P.; Guerini, D.; Celio, M.R.; Carafoli, E. Immunolocalization of the plasma membrane Ca2+ pump isoforms in the rat brain. Brain Res. 1997, 748, 21–29. [Google Scholar] [CrossRef]
- Burette, A.; Weinberg, R.J. Perisynaptic organization of plasma membrane calcium pumps in cerebellar cortex. J. Comp. Neurol. 2007, 500, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.R.; Hidalgo-Sanchez, M.; Marcos, D.; Mata, A.M. Developmental distribution of plasma membrane Ca2+-ATPase isoforms in chick cerebellum. Dev. Dyn. 2007, 236, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, D.A.; Kappen, C. Developmental expression of the four plasma membrane calcium ATPase (Pmca) genes in the mouse. Biochim. Biophys. Acta 1999, 1428, 397–405. [Google Scholar] [CrossRef]
- Filoteo, A.G.; Elwess, N.L.; Enyedi, A.; Caride, A.; Aung, H.H.; Penniston, J.T. Plasma membrane Ca2+ pump in rat brain. Patterns of alternative splices seen by isoform-specific antibodies. J. Biol. Chem. 1997, 272, 23741–23747. [Google Scholar] [CrossRef] [PubMed]
- Kip, S.N.; Gray, N.W.; Burette, A.; Canbay, A.; Weinberg, R.J.; Strehler, E.E. Changes in the expression of plasma membrane calcium extrusion systems during maturation of hippocampal neurons. Hippocampus 2006, 20, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Kurnellas, M.P.; Lee, A.K.; Li, H.; Deng, L.; Ehrlich, D.J.; Elkabes, S. Molecular alterations in the cerebellum of the plasma membrane calcium ATPase 2 (PMCA2)-null mouse indicate abnormalities in Purkinje neurons. Mol. Cell. Neurosci. 2007, 34, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Pisu, M.B.; Roda, E.; Guioli, S.; Avella, D.; Bottone, M.G.; Bernocchi, G. Proliferation and migration of granule cells in the developing rat cerebellum: Cisplatin effects. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2005, 287, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Scherini, E.; Bernocchi, G. CisDDP treatment and development of the rat cerebellum. Prog. Neurobiol. 1994, 42, 161–196. [Google Scholar] [CrossRef]
- Piccolini, V.M.; Cerri, S.; Romanelli, E.; Bernocchi, G. Interactions of neurotransmitter systems during postnatal development of the rat hippocampal formation: Effects of cisplatin. Exp. Neurol. 2012, 234, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Bernocchi, G.; Bottone, M.G.; Piccolini, V.M.; Dal Bo, V.; Santin, G.; de Pascali, S.A.; Migoni, D.; Fanizzi, F.P. Developing central nervous system and vulnerability to platinum compounds. Chemother. Res. Pract. 2011, 2011, 315418. [Google Scholar] [CrossRef] [PubMed]
- Piccolini, V.M.; Esposito, A.; Dal Bo, V.; Insolia, V.; Bottone, M.G.; de Pascali, S.A.; Fanizzi, F.P.; Bernocchi, G. Cerebellum neurotransmission during postnatal development: [Pt(O,O′-acac)(γ-acac)(DMS)] vs. cisplatin and neurotoxicity. Int. J. Dev. Neurosci. 2015, 40, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Roda, E.; Avella, D.; Pisu, M.B.; Bernocchi, G. Monoamine receptors and immature cerebellum cytoarchitecture after cisplatin injury. J. Chem. Neuroanat. 2007, 33, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.W. Cell death, Bcl-2, Bax, and the cerebellum. Cerebellum 2002, 1, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Bottone, M.G.; Dal Bo, V.; Piccolini, V.M.; Bottiroli, G.; de Pascali, S.A.; Fanizzi, F.P.; Bernocchi, G. Developmental expression of cellular prion protein and apoptotic molecules in the rat cerebellum: Effects of platinum compounds. J. Chem. Neuroanat. 2012, 46, 19–29. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernocchi, G.; Fanizzi, F.P.; De Pascali, S.A.; Piccolini, V.M.; Gasperini, C.; Insolia, V.; Bottone, M.G. Neurotoxic Effects of Platinum Compounds: Studies in vivo on Intracellular Calcium Homeostasis in the Immature Central Nervous System. Toxics 2015, 3, 224-248. https://doi.org/10.3390/toxics3020224
Bernocchi G, Fanizzi FP, De Pascali SA, Piccolini VM, Gasperini C, Insolia V, Bottone MG. Neurotoxic Effects of Platinum Compounds: Studies in vivo on Intracellular Calcium Homeostasis in the Immature Central Nervous System. Toxics. 2015; 3(2):224-248. https://doi.org/10.3390/toxics3020224
Chicago/Turabian StyleBernocchi, Graziella, Francesco P. Fanizzi, Sandra A. De Pascali, Valeria M. Piccolini, Caterina Gasperini, Violetta Insolia, and Maria Grazia Bottone. 2015. "Neurotoxic Effects of Platinum Compounds: Studies in vivo on Intracellular Calcium Homeostasis in the Immature Central Nervous System" Toxics 3, no. 2: 224-248. https://doi.org/10.3390/toxics3020224
APA StyleBernocchi, G., Fanizzi, F. P., De Pascali, S. A., Piccolini, V. M., Gasperini, C., Insolia, V., & Bottone, M. G. (2015). Neurotoxic Effects of Platinum Compounds: Studies in vivo on Intracellular Calcium Homeostasis in the Immature Central Nervous System. Toxics, 3(2), 224-248. https://doi.org/10.3390/toxics3020224