
Speciation in Metal Toxicity and Metal-Based Therapeutics

Abstract
:1. Introduction
2. Importance of Speciation: The Example of Ni
3. Definitions and Terminology: Speciation and Some Imprecise Terms
3.1. Definition Related to Speciation
3.2. Structural Considerations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Atomic number | Name | Oxidation states |
|---|---|---|---|
| V | 23 | Vanadium | IV/V |
| Cr | 24 | Chromium | III/VI |
| Mn | 25 | Manganese | II/III/IV |
| Fe | 26 | Iron | 0/II/III |
| Co | 27 | Cobalt | II/III |
| Ni | 28 | Nickel | II/IV |
| Cu | 29 | Copper | 0/I/II |
| Zn | 30 | Zinc | 0/II |
| As | 33 | Arsenic | III/V |
| Se | 34 | Selenium | II/IV/VI |
| Mo | 42 | Molybdenum | II/III/IV/VI |
| Pd | 46 | Palladium | II/IV |
| Ag | 47 | Silver | 0/I/II |
| Sn | 50 | Tin | II/IV |
| Sb | 51 | Antimony | III/V |
| Te | 52 | Tellurium | 0/II/IV/VI |
| Pt | 78 | Platinum | II/IV |
| Au | 79 | Gold | 0/I/III |
| Hg | 80 | Mercury | 0/I/II |
| Tl | 81 | Thallium | I/III |
| Pb | 82 | Lead | II/IV |
| U | 92 | Uranium | III/VI |
| Pu | 94 | Plutonium | III/IV/V/VI |
3.3. Imprecise Terms
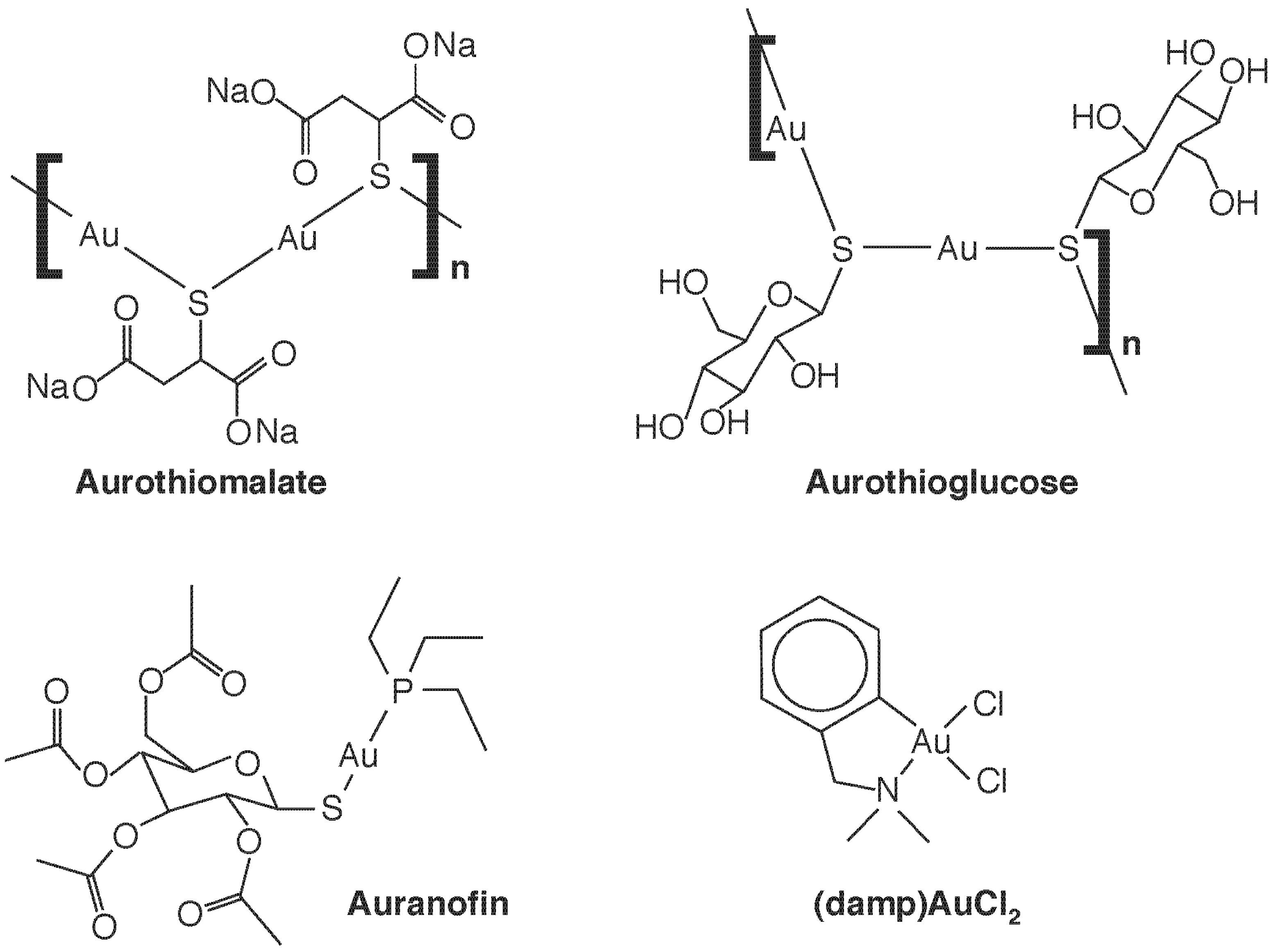
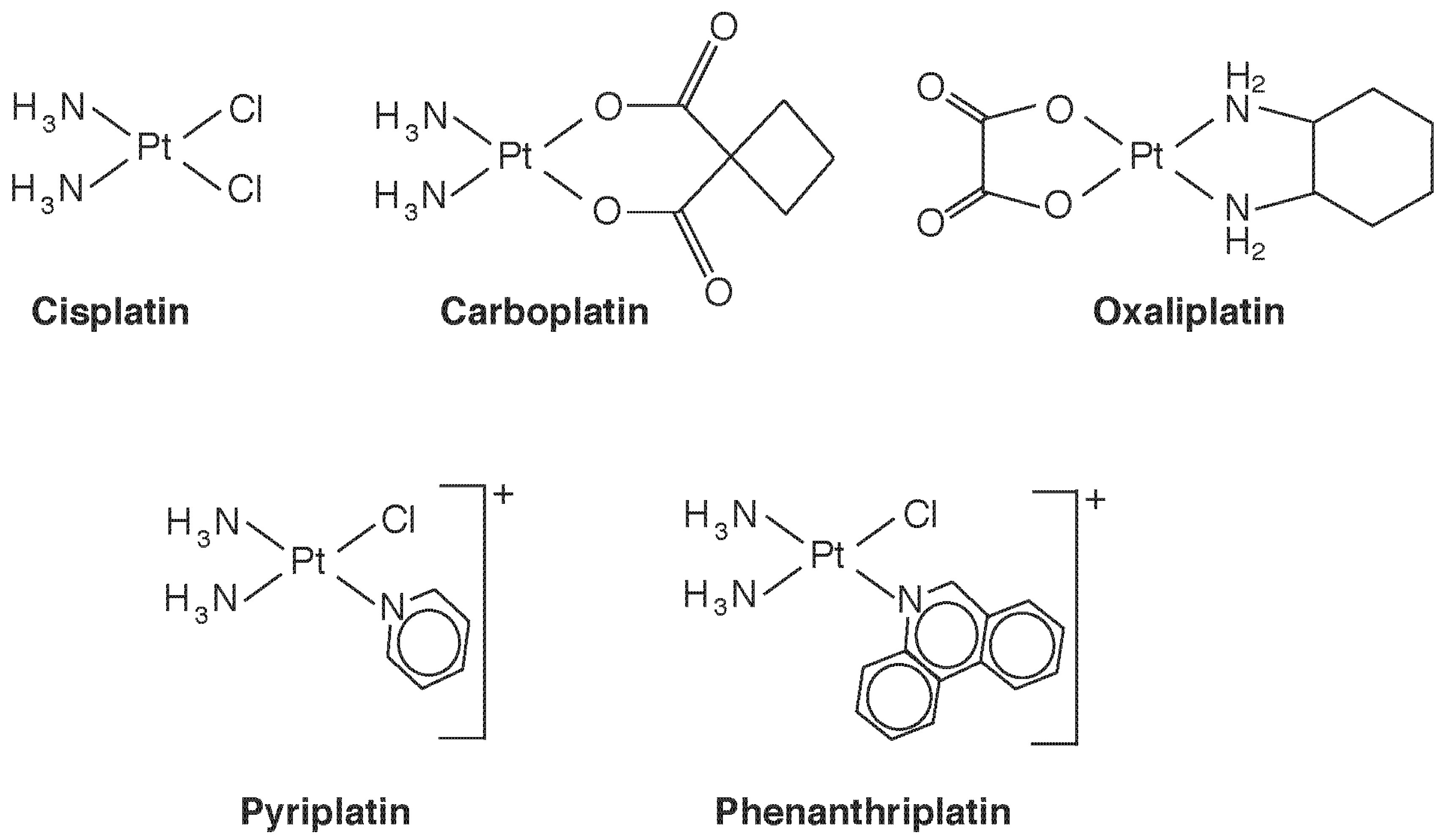
4. Speciation-Based Metal Therapy: Au and Pt
4.1. Gold Compounds

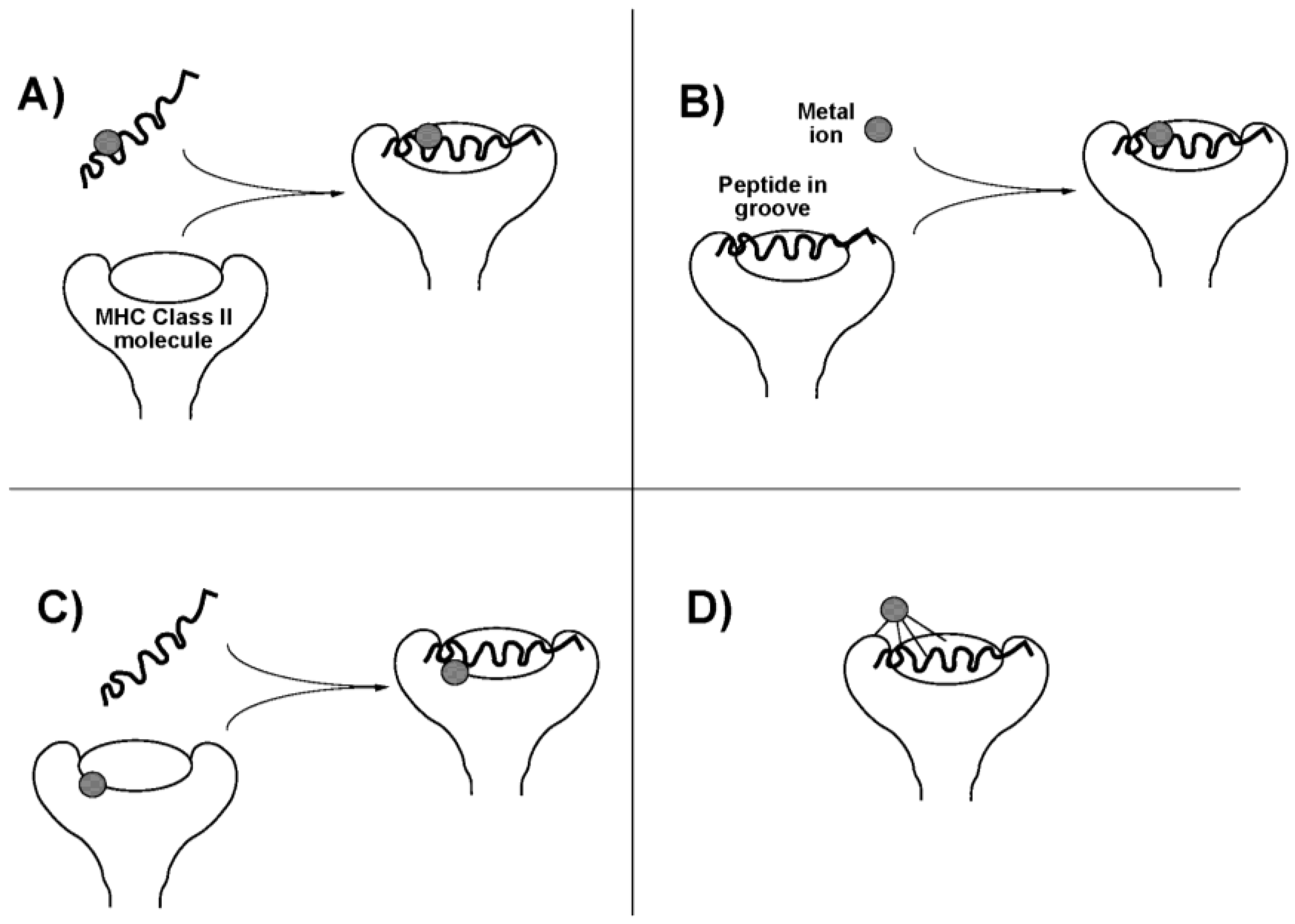
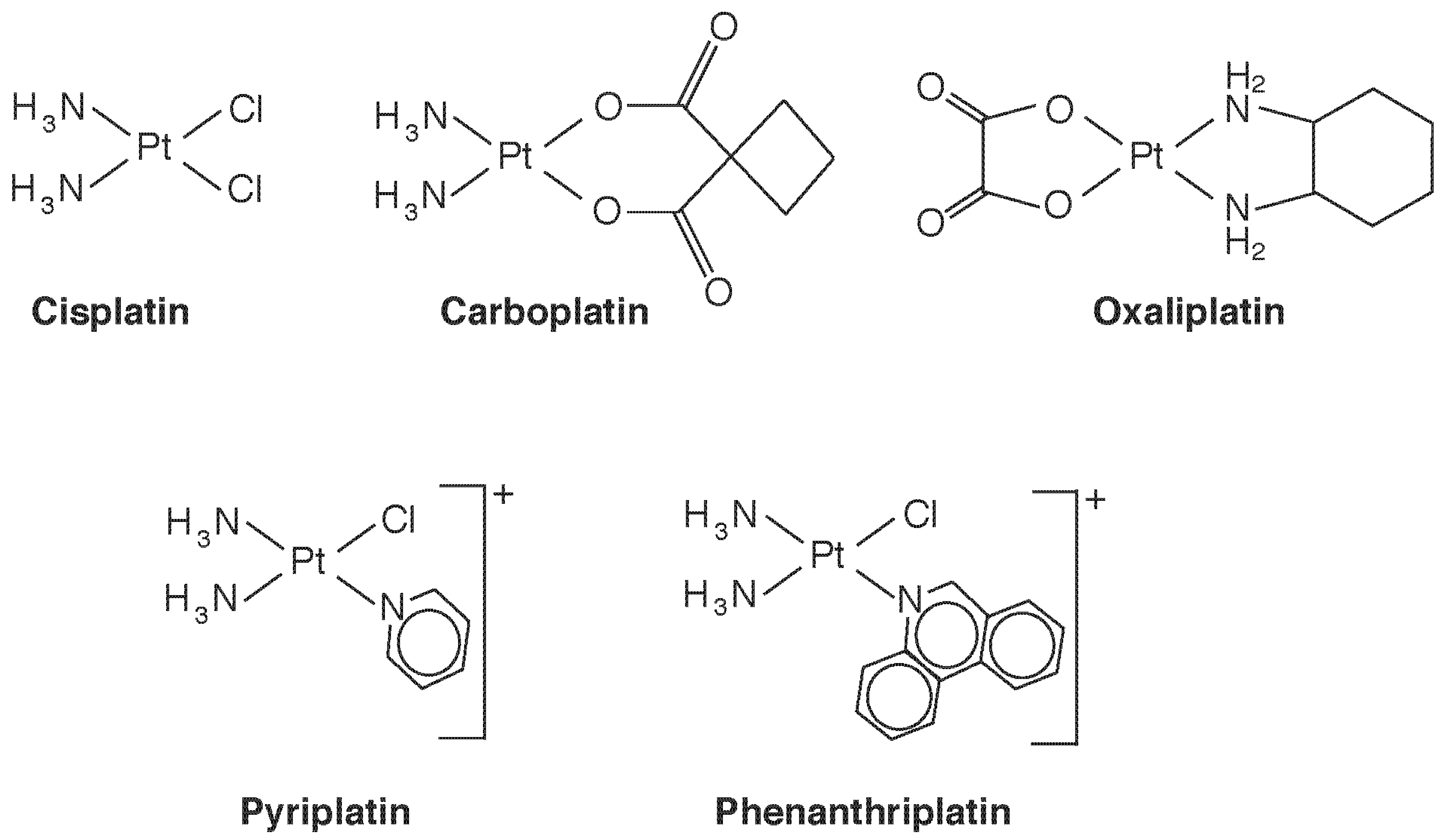
4.2. Platinum Compounds
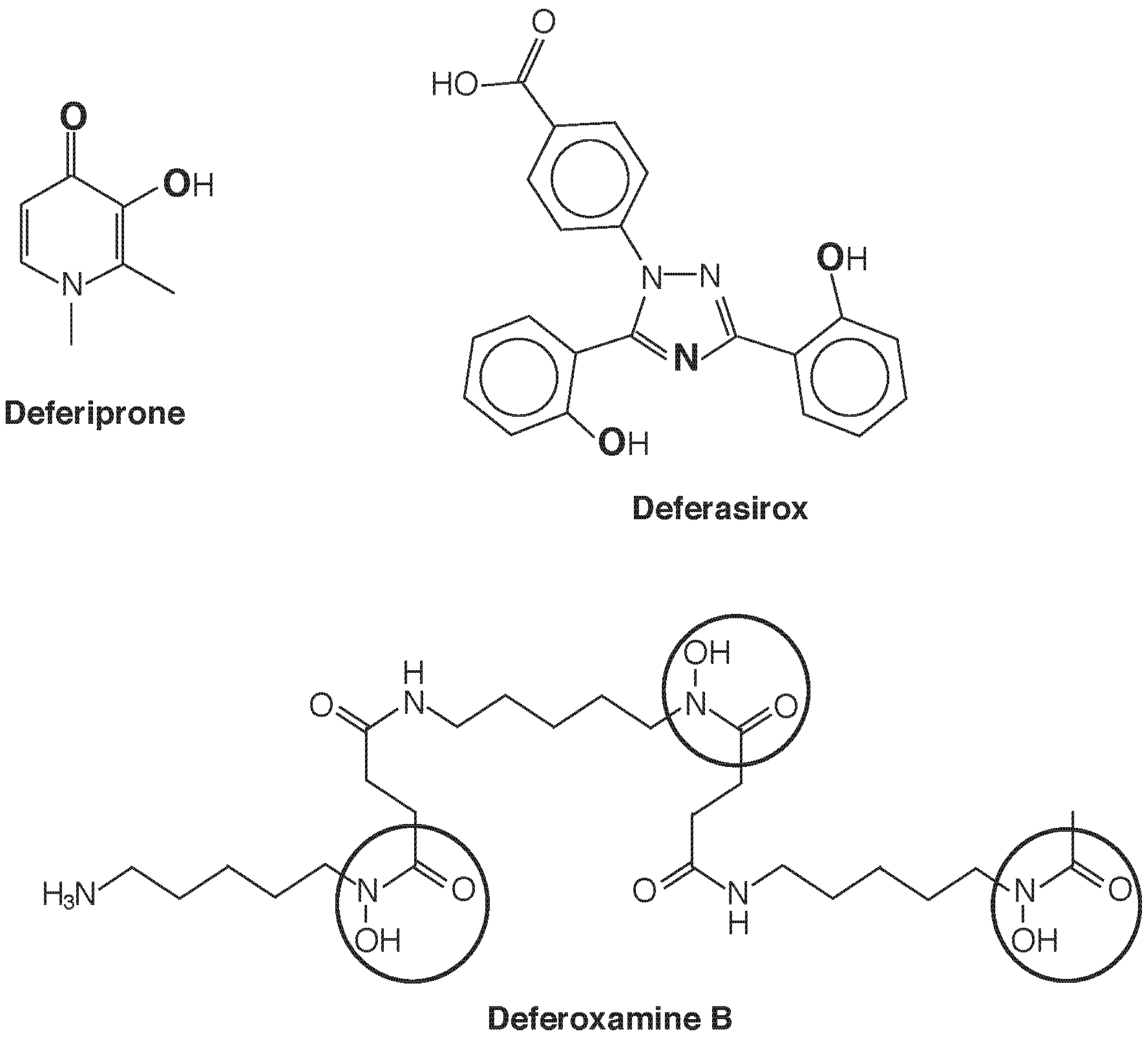
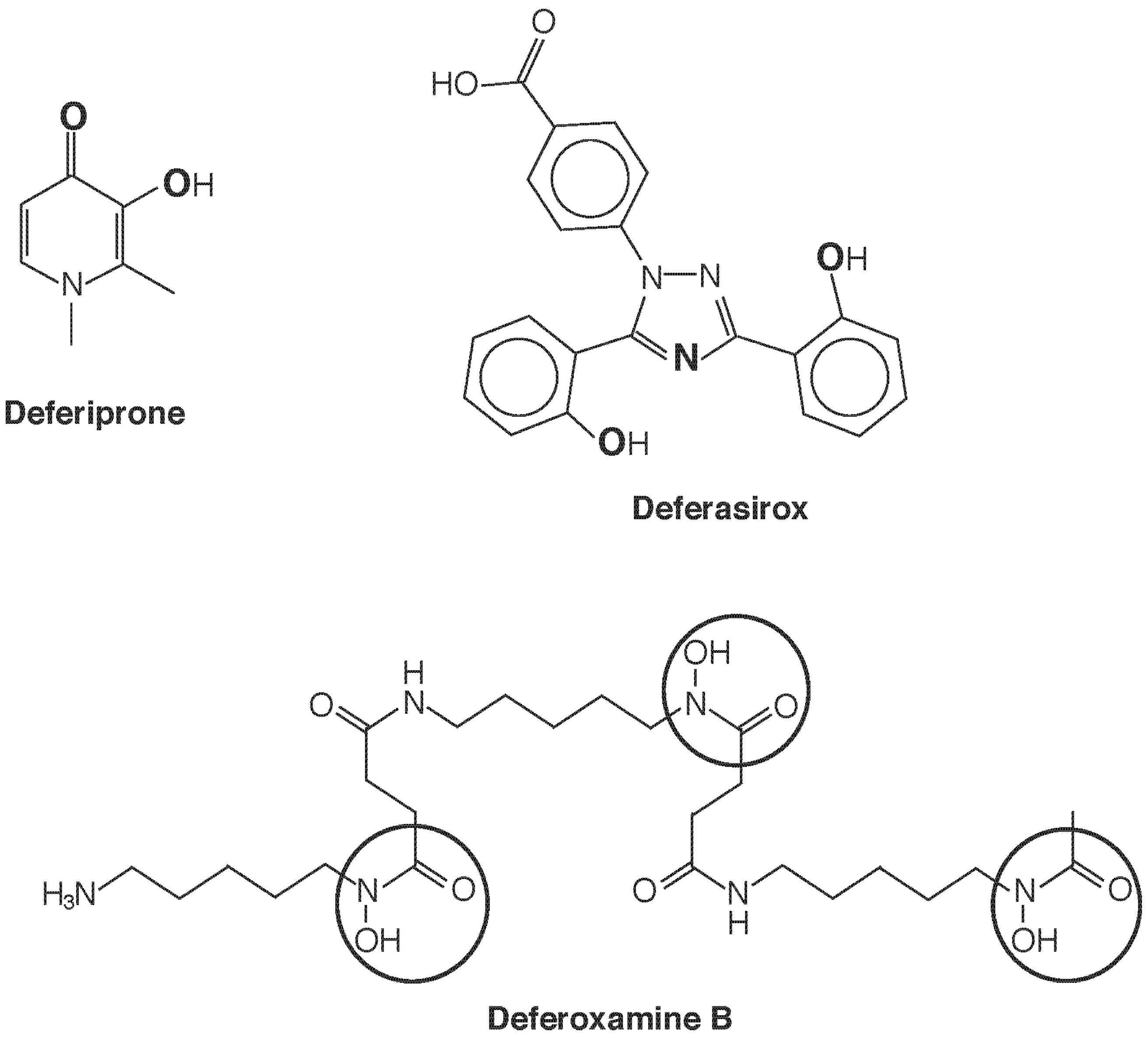
5. Chelation Therapy as an Exercise in Speciation: Fe and U
5.1. Chelation for Iron Overload
5.2. Chelation for Uranium Poisoning
6. Conclusions
Conflict of Interest
References
- International Agency for Research on Cancer. Chromium, nickel and welding. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1990; Volume 49, pp. 257–445. [Google Scholar]
- Apostoli, P.; Cornelis, R.; Duffus, J.H.; Hoet, P.; Lison, D.; Templeton, D.M. Elemental Speciation in Human Health Risk Assessment. IPCS, Environmental Health Criteria 234; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Sunderman, F.W., Jr.; Dingle, B.; Hopfer, S.M.; Swift, T. Acute nickel toxicity in electroplating workers who accidently ingested a solution of nickel sulfate and nickel chloride. Am. J. Ind. Med. 1988, 14, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Peana, M.; Medici, S.; Nurchi, V.M.; Crisponi, G.; Zoroddu, M.A. Nickel binding sites in histone proteins: Spectroscopic and structural characterization. Coord. Chem. Rev. 2013, 257, 2737–2751. [Google Scholar] [CrossRef]
- Landolph, J.R.; Dews, P.M.; Ozburn, L.; Evans, D.P. Metal-induced gene expression and neoplastic transformation. In Toxicology of Metals; Chang, L.W., Ed.; CRC Lewis Purlishers: Boca Raton, FL, USA, 1996; pp. 321–329. [Google Scholar]
- Templeton, D.M.; Xu, S.X.; Stuhne-Sekalec, L. Isotope-specific analysis of Ni by ICP-MS: Applications of stable isotope tracers to biokinetic studies. Sci. Total Environ. 1994, 148, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, G.D.; Søderberg, U.; Jørgensen, P.J.; Templeton, D.M.; Rasmussen, S.; Andersen, K.E.; Grandjean, P. Absorption and retention of nickel from drinking water in relation to food intake and nickel sensitivity. Toxicol. Appl. Pharmacol. 1999, 154, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Templeton, D.M.; Ariese, F.; Cornelis, R.; Danielsson, L.-G.; Muntau, H.; van Leeuwen, H.; Lobinski, R. Guidelines for terms related to chemical speciation and fractionation of elements: Definitions, structural aspects, and methodological approaches. Pure Appl. Chem. 2000, 72, 1453–1470. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Medici, S.; Ledda, A.; Nurchi, V.M.; Lachowicz, J.I.; Peana, M. Toxicity of nanoparticles. Curr. Med. Chem. 2015, 22, 3837–3853. [Google Scholar]
- Templeton, D.M. Biomedical aspects of trace element speciation. Fresenius J. Anal. Chem. 1999, 363, 505–511. [Google Scholar] [CrossRef]
- Templeton, D.M. Elemental speciation in clinical sciences. In Analytical Techniques for Clinical Chemistry: Methods and Applications; Caroli, S., Záray, G., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 159–177. [Google Scholar]
- Remelli, M.; Peana, M.; Medici, S.; Delogu, L.G.; Zoroddu, M.A. Interaction of divalent cations with peptide fragments from Parkinson’s disease genes. Dalton Trans. 2013, 42, 5964–5974. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.P. The symbiosis of metal and protein functions. Eur. J. Biochem. 1985, 150, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Duffus, J.H. “Heavy metals”—A meaningless term? Pure Appl. Chem. 2002, 74, 793–807. [Google Scholar] [CrossRef]
- Duffus, J.H.; Nordberg, M.; Templeton, D.M. Glossary of terms used in toxicology, 2nd ed. Pure Appl. Chem. 2007, 79, 1153–1341. [Google Scholar]
- Benedek, T.G. The history of gold therapy for tuberculosis. J. Hist. Med. Allied Sci. 2004, 59, 50–89. [Google Scholar] [CrossRef] [PubMed]
- Fricker, S.P. Medical uses of gold compounds: Past, present and future. Gold Bull. 1996, 29, 53–60. [Google Scholar] [CrossRef]
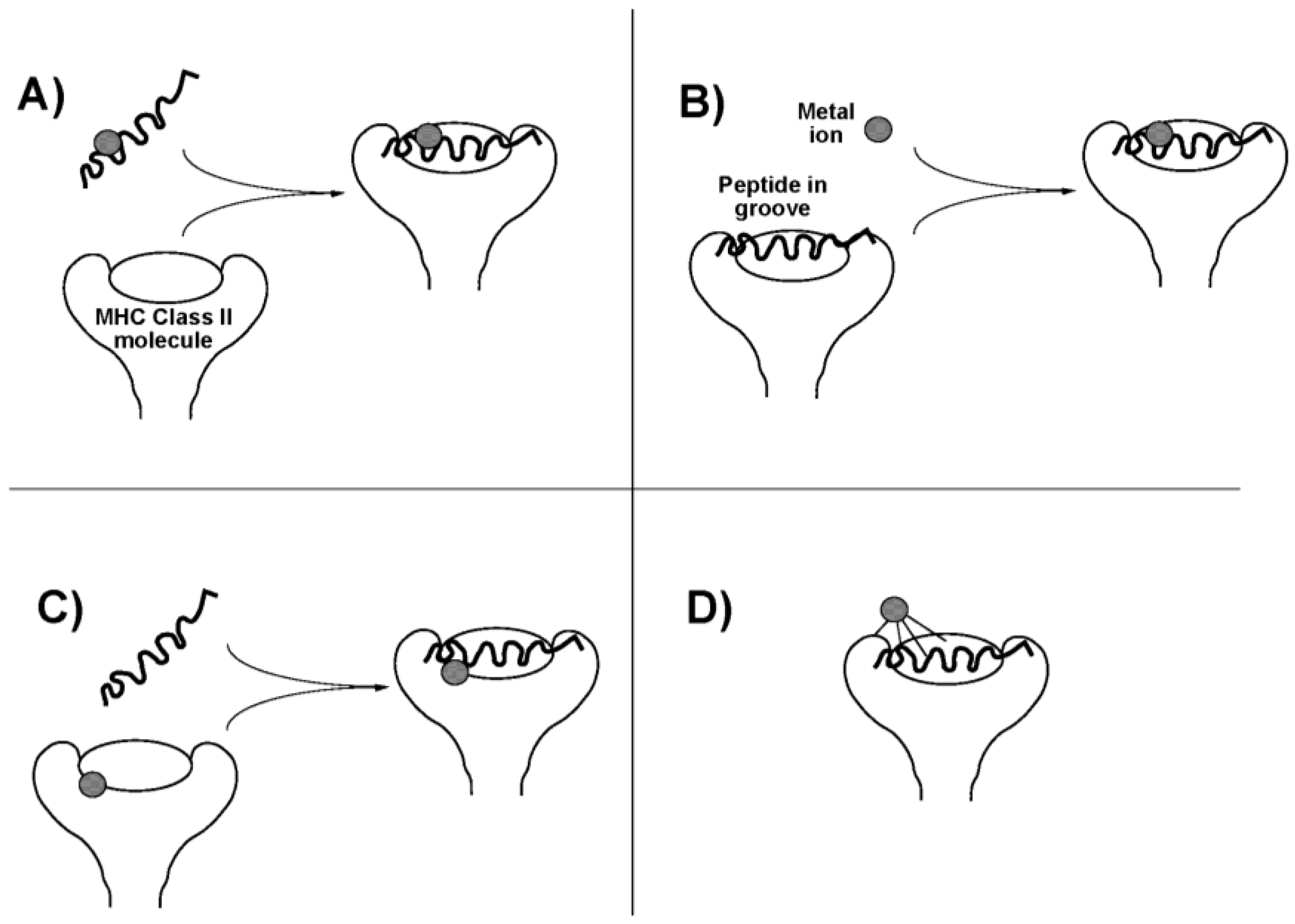
- Templeton, D.M. Mechanisms of immunosensitization to metals. Pure Appl. Chem. 2004, 76, 1255–1268. [Google Scholar] [CrossRef]
- Romagnoli, P.; Spinas, G.A.; Sinigaglia, F. Gold-specific T cells in rheumatoid arthritis patients treated with gold. J. Clin. Invest. 1992, 89, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Sinigaglia, F. The molecular basis of metal recognition by T cells. J. Invest. Dermatol. 1994, 102, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Griem, P.; Takahashi, K.; Kalbacher, H.; Gleichmann, E. The antirheumatic drug disodium aurothiomalate inhibits CD4+ T-cell recognition of peptides containing two or more cysteine residues. J. Immunol. 1995, 155, 1575–1587. [Google Scholar] [PubMed]
- Eiter, L.C.; Hall, N.W.; Day, C.S.; Saluta, G.; Kucera, G.L.; Bierbach, L. Gold(I) Analogues of a platinum–acridine antitumor agent are only moderately cytotoxic but show potent cctivity against Mycobacterium tuberculosis. J. Med. Chem. 2009, 52, 6519–6522. [Google Scholar] [CrossRef] [PubMed]
- Lippard, S.J.; Todd, R.C. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Pruefer, F.G.; Lizarraga, F.; Maldonado, V.; Melendez-Zajgla, J. Participation of Omi Htra2 serine–protease activity in the apoptosis induced by cisplatin on SW480 colon cancer cells. J. Chemother. 2008, 20, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Pinato, O.; Musetti, C.; Sissi, C. Pt-based drugs: the spotlight will be on proteins. Metallomics 2014, 6, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Barry, N.P.E.; Sadler, P.J. 100 Years of metal coordiation chemistry: from Alfred Werner to anticancer metallodrugs. Pur Appl. Chem. 2014, 86, 1897–1910. [Google Scholar] [CrossRef]
- Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Crisponi, G.; Zoroddu, M.A. Noble metals in medicine: Latest advances. Coord. Chem. Rev. 2015, 284, 329–350. [Google Scholar] [CrossRef]
- Templeton, D.M. Genetic defects in iron and copper trafficking. In Endogenous Toxins: Target for Disease Treatment and Prevention; O’Brien, P.J., Bruce, W.R., Eds.; Wiley-VHC: Weinheim, Germany, 2010; Volume 1, pp. 395–418. [Google Scholar]
- Stuhne-Sekalec, L.; Xu, S.X.; Parkes, J.G.; Olivieri, N.F.; Templeton, D.M. Speciation of tissue and cellular iron with on-line detection by inductively coupled plasma-mass spectrometry. Anal. Biochem. 1992, 205, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Baes, C.F., Jr.; Mesmer, R.E. The Hydrolysis of Cations; Robert E. Krieger: Malabar, FL, USA, 1986. [Google Scholar]
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411–422. [Google Scholar] [PubMed]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Specific iron chelators determine the route of ferritin degradation. Blood Cells Mol. Dis. 2009, 114, 4546–4551. [Google Scholar]
- Sheth, S. Iron chelation: an update. Curr. Opin. Hematol. 2014, 21, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Maggio, A.; Filosa, A.; Vitrano, A.; Aloj, G.; Kattamis, A.; Ceci, A.; Fucharoen, S.; Cianciulli, P.; Grady, R.W.; Prossomariti, L.; et al. Iron chelation therapy in thalassemia major: A systematic review with meta-analyses of 1520 patients included on randomized clinical trials. Blood Cells Mol. Dis. 2011, 47, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Arpa, J.; Delatycki, M.B.; Sang, K.H.L.Q.; Mariotti, C.; Munnich, A.; Sanz-Gallego, I.; Tai, G.; Tarnopolsky, M.A.; Taroni, F.; et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann. Neurol. 2014, 76, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Temraz, S.; Santini, V.; Musallam, K.; Taher, A. Iron overload and chelation therapy in myelodysplastic syndromes. Crit. Rev. Oncol. Hematol. 2014, 91, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Briner, W. The toxicity of depleted uranium. J. Environ. Res. Public Health 2010, 7, 303–313. [Google Scholar] [CrossRef]
- Katz, S.E. The chemistry and toxicology of depleted uranium. Toxics 2014, 2, 50–78. [Google Scholar] [CrossRef]
- Lawrence, G.D.; Patel, K.S.; Nusbaum, A. Uranium toxicity and chelation therapy. Pure Appl. Chem. 2014, 86, 1105–1110. [Google Scholar] [CrossRef]
- Ortega, A.; Domingo, J.L.; Gómez, M.; Corbella, J. Treatment of experimental acute uranium poisoning by chelating agents. Pharmacol. Toxicol. 1989, 64, 247–251. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Templeton, D.M. Speciation in Metal Toxicity and Metal-Based Therapeutics. Toxics 2015, 3, 170-186. https://doi.org/10.3390/toxics3020170
Templeton DM. Speciation in Metal Toxicity and Metal-Based Therapeutics. Toxics. 2015; 3(2):170-186. https://doi.org/10.3390/toxics3020170
Chicago/Turabian StyleTempleton, Douglas M. 2015. "Speciation in Metal Toxicity and Metal-Based Therapeutics" Toxics 3, no. 2: 170-186. https://doi.org/10.3390/toxics3020170
APA StyleTempleton, D. M. (2015). Speciation in Metal Toxicity and Metal-Based Therapeutics. Toxics, 3(2), 170-186. https://doi.org/10.3390/toxics3020170