
New QSAR Models to Predict Human Transthyretin Disruption by Per- and Polyfluoroalkyl Substances (PFAS): Development and Application



Abstract

1. Introduction
2. Materials and Methods
2.1. Modelled Datasets and Data Curation
2.2. Calculation of Molecular Descriptors and Dataset Splitting for External Validation
2.3. QSAR Models Development
2.3.1. Classification-Based QSARs
2.3.2. Regression-Based QSARs
2.3.3. External Validation
2.4. Applicability Domains
2.4.1. LDA-QSARs Applicability Domain
2.4.2. MLR-QSARs Applicability Domain
2.5. OECD List of PFAS
3. Results and Discussion
3.1. LDA-QSAR
3.2. MLR-QSAR
3.3. Case Study: Screening the Potential hTTR Disruption of the PFAS Included in the OECD List
3.4. Comparison with Previous Similar Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | Accuracy |
| AD | Applicability domain |
| AMW | Average molecular weight |
| ANSA | 8-Anilino-1-naphtalenesulfonic acid |
| AOP | Adverse outcome pathway |
| AUC | Area under the curve |
| CASRN | Chemical Abstracts Service Registration Number |
| DTC | Decision tree classifier |
| EC50 | Effect concentration 50% |
| ED | Endocrine disruption |
| EURL ECVAM | European Union Reference Laboratory for Alternatives to Animal Testing |
| FN | False negative |
| FP | False positive |
| HPT | Hypothalamic–pituitary–thyroid |
| hTTR | Human transthyretin |
| IC50 | Inhibitory concentration 50% |
| kNN | k-Nearest neighbor |
| LC50 | Lethal concentration 50% |
| LOEC | Lowest observed effect concentration |
| LDA | Linear discriminant analysis |
| MAE | Mean absolute error |
| MIE | Molecular initiating event |
| MLR | Multiple linear regression |
| MR | Misclassification rate |
| NAMs | New approach methodologies |
| OECD | Organisation for Economic Co-operation and Development |
| OLS | Ordinary least squares |
| P | Precision |
| PCA | Principal component analysis |
| PFAS | Per- and polyfluoroalkyl substances |
| QSAR | Quantitative structure–activity relationship |
| RLBA | Radiolabeled [125I]-T4 in vitro binding assay |
| ROC | Receiver operating characteristic |
| RP | Relative competitive potency |
| RPF | Relative potency factor |
| SMILES | Simplified molecular input line entry system |
| SN | Sensitivity |
| SP | Specificity |
| T4 | Thyroxine |
| TH | Thyroid hormone |
| THSDCs | Thyroid hormone system-disrupting chemicals |
| US EPA | United States Environmental Protection Agency |
References
- Evich, M.G.; Davis, M.J.B.; McCord, J.P.; Acrey, B.; Awkerman, J.A.; Knappe, D.R.U.; Lindstrom, A.B.; Speth, T.F.; Tebes-Stevens, C.; Strynar, M.J.; et al. Per- and Polyfluoroalkyl Substances in the Environment. Science 2022, 375, 512. [Google Scholar] [CrossRef] [PubMed]
- Brunn, H.; Arnold, G.; Körner, W.; Rippen, G.; Steinhäuser, K.G.; Valentin, I. PFAS: Forever Chemicals—Persistent, Bioaccumulative and Mobile. Reviewing the Status and the Need for Their Phase out and Remediation of Contaminated Sites. Environ. Sci. Eur. 2023, 35, 20. [Google Scholar] [CrossRef]
- De Silva, A.O.; Armitage, J.M.; Bruton, T.A.; Dassuncao, C.; Heiger-Bernays, W.; Hu, X.C.; Kärrman, A.; Kelly, B.; Ng, C.; Robuck, A.; et al. PFAS Exposure Pathways for Humans and Wildlife: A Synthesis of Current Knowledge and Key Gaps in Understanding. Environ. Toxicol. Chem. 2021, 40, 631–657. [Google Scholar] [CrossRef]
- Andrews, D.Q.; Stoiber, T.; Temkin, A.M.; Naidenko, O.V. Discussion. Has the Human Population Become a Sentinel for the Adverse Effects of PFAS Contamination on Wildlife Health and Endangered Species? Sci. Total Environ. 2023, 901, 165939. [Google Scholar] [CrossRef]
- Jane, L.; Espartero, L.; Yamada, M.; Ford, J.; Owens, G.; Prow, T.; Juhasz, A. Health-Related Toxicity of Emerging per- and Polyfluoroalkyl Substances: Comparison to Legacy PFOS and PFOA. Environ. Res. 2022, 212, 113431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Naveed, H.; Chen, K.; Chen, L. Toxicity of Per- and Polyfluoroalkyl Substances and Their Substitutes to Terrestrial and Aquatic Invertebrates—A Review. Toxics 2025, 13, 47. [Google Scholar] [CrossRef]
- Coperchini, F.; Croce, L.; Ricci, G.; Magri, F.; Rotondi, M.; Imbriani, M.; Chiovato, L. Thyroid Disrupting Effects of Old and New Generation PFAS. Front. Endocrinol. 2021, 11, 612320. [Google Scholar] [CrossRef]
- Zoeller, R.T.; Tan, S.W.; Tyl, R.W. General Background on the Hypothalamic-Pituitary-Thyroid (HPT) Axis. Crit. Rev. Toxicol. 2007, 37, 11–53. [Google Scholar] [CrossRef]
- Bassett, J.H.D.; Williams, G.R. Role of Thyroid Hormones in Skeletal Development and Bone Maintenance. Endocr. Rev. 2016, 37, 135–187. [Google Scholar] [CrossRef]
- Klein, I.; Ojamaa, K. Thyroid Hormone and the Cardiovascular System. N. Eng. J. Med. 2001, 344, 501–509. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.-Y.; Brent, G.A. Thyroid Hormone Regulation of Metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef]
- Schroeder, A.C.; Privalsky, M.L. Thyroid Hormones, T3 and T4, in the Brain. Front. Endocrinol. 2014, 5, 40. [Google Scholar] [CrossRef]
- Haigis, A.-C.; Vergauwen, L.; LaLone, C.A.; Villeneuve, D.L.; O’Brien, J.M.; Knapen, D. Cross-Species Applicability of an Adverse Outcome Pathway Network for Thyroid Hormone System Disruption. Toxicol. Sci. 2023, 195, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Noyes, P.D.; Friedman, K.P.; Browne, P.; Haselman, J.T.; Gilbert, M.E.; Hornung, M.W.; Barone, S.; Crofton, K.M.; Laws, S.C.; Stoker, T.E.; et al. Evaluating Chemicals for Thyroid Disruption: Opportunities and Challenges with in Vitro Testing and Adverse Outcome Pathway Approaches. Environ. Health Perspect. 2019, 127, 095001. [Google Scholar] [CrossRef] [PubMed]
- Gião, T.; Saavedra, J.; Cotrina, E.; Quintana, J.; Llop, J.; Arsequell, G.; Cardoso, I. Undiscovered Roles for Transthyretin: From a Transporter Protein to a New Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2075. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wu, Y.; Tao, G.; Xu, J.; Du, Z.; Wu, M.; Gu, T.; Xiong, J.; Xiao, S.; Wei, X.; et al. Association between PFAS Exposure and Thyroid Health: A Systematic Review and Meta-Analysis for Adolescents, Pregnant Women, Adults and Toxicological Evidence. Sci. Total Environ. 2024, 953, 175958. [Google Scholar] [CrossRef]
- De Escobar, G.M.; Obregón, M.J.; del Rey, F.E. Maternal Thyroid Hormones Early in Pregnancy and Fetal Brain Development. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 225–248. [Google Scholar] [CrossRef]
- Rodrigues, V.G.; Henrique, G.; Sousa-Vidal, É.K.; de Souza, R.M.M.; Tavares, E.F.C.; Mezzalira, N.; Marques, T.d.O.; Alves, B.M.; Pinto, J.A.A.; Irikura, L.N.N.; et al. Thyroid under Attack: The Adverse Impact of Plasticizers, Pesticides, and PFASs on Thyroid Function. Endocrines 2024, 5, 430–453. [Google Scholar] [CrossRef]
- Moog, N.K.; Entringer, S.; Heim, C.; Wadhwa, P.D.; Kathmann, N.; Buss, C. Influence of Maternal Thyroid Hormones during Gestation on Fetal Brain Development. Neuroscience 2017, 342, 68–100. [Google Scholar] [CrossRef]
- Coperchini, F.; Teliti, M.; Greco, A.; Croce, L.; Rotondi, M. Per-Polyfluoroalkyl Substances (PFAS) as Thyroid Disruptors: Is There Evidence for Multi-Transgenerational Effects? Expert Rev. Endocrinol. Metab. 2024, 19, 307–315. [Google Scholar] [CrossRef]
- Bernasconi, C.; Langezaal, I.; Bartnicka, J.; Asturiol, D.; Bowe, G.; Coecke, S.; Kienzler, A.; Liska, R.; Milcamps, A.; Munoz-Pineiro, M.A.; et al. Validation of a Battery of Mechanistic Methods Relevant for the Detection of Chemicals That Can Disrupt the Thyroid Hormone System; Publications Office of the European Union: Luxembourg, 2023; EUR 31456 EN; Available online: https://publications.jrc.ec.europa.eu/repository/handle/JRC132532 (accessed on 18 June 2024).
- Ao, J.; Tang, W.; Liu, X.; Ao, Y.; Zhang, Q.; Zhang, J. Polyfluoroalkyl Phosphate Esters (PAPs) as PFAS Substitutes and Precursors: An Overview. J. Hazard Mater. 2024, 464, 133018. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Ng, C.A. High-Throughput Screening of Protein Interactions with per- and Polyfluoroalkyl Substances (PFAS) Used in Photolithography. J. Hazard Mater. 2025, 487, 137235. [Google Scholar] [CrossRef]
- Fenton, S.E.; Ducatman, A.; Boobis, A.; DeWitt, J.C.; Lau, C.; Ng, C.; Smith, J.S.; Roberts, S.M. Per- and Polyfluoroalkyl Substance Toxicity and Human Health Review: Current State of Knowledge and Strategies for Informing Future Research. Environ. Toxicol. Chem. 2021, 40, 606–630. [Google Scholar] [CrossRef]
- European Chemicals Agency. Key Areas of Regulatory Challenge; European Chemicals Agency: Helsinki, Finland, 2024; Available online: https://echa.europa.eu/documents/10162/17228/key_areas_regulatory_challenges_2024.pdf/e9b997e1-794e-cab7-8503-ba4385070730?t=1718103205169 (accessed on 20 January 2025).
- Organisation for Economic Co-operation and Development. Revised Guidance Document 150 on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption-Introduction; OECD Series on Testing and Assessment, No. 150; OECD Publishing: Paris, France, 2018; pp. 19–39. [Google Scholar] [CrossRef]
- Barton-Maclaren, T.S.; Wade, M.; Basu, N.; Bayen, S.; Grundy, J.; Marlatt, V.; Moore, R.; Parent, L.; Parrott, J.; Grigorova, P.; et al. Innovation in Regulatory Approaches for Endocrine Disrupting Chemicals: The Journey to Risk Assessment Modernization in Canada. Environ. Res. 2022, 204, 112225. [Google Scholar] [CrossRef]
- Judson, R.S.; Paul Friedman, K.; Houck, K.; Mansouri, K.; Browne, P.; Kleinstreuer, N.C. New Approach Methods for Testing Chemicals for Endocrine Disruption Potential. Curr. Opin. Toxicol. 2018, 9, 40–47. [Google Scholar] [CrossRef]
- Ramhøj, L.; Axelstad, M.; Baert, Y.; Cañas-Portilla, A.I.; Chalmel, F.; Dahmen, L.; De La Vieja, A.; Evrard, B.; Haigis, A.-C.; Hamers, T.; et al. New Approach Methods to Improve Human Health Risk Assessment of Thyroid Hormone System Disruption–a PARC Project. Front. Toxicol. 2023, 5, 1189303. [Google Scholar] [CrossRef] [PubMed]
- Dharpure, R.; Pramanik, S.; Pradhan, A. In Silico Analysis Decodes Transthyretin (TTR) Binding and Thyroid Disrupting Effects of per- and Polyfluoroalkyl Substances (PFAS). Arch. Toxicol. 2023, 97, 755–768. [Google Scholar] [CrossRef]
- Tiburtini, G.A.; Bertarini, L.; Bersani, M.; Dragani, T.A.; Rolando, B.; Binello, A.; Barge, A.; Spyrakis, F. In Silico Prediction of the Interaction of Legacy and Novel Per- and Poly-Fluoroalkyl Substances (PFAS) with Selected Human Transporters and of Their Possible Accumulation in the Human Body. Arch. Toxicol. 2024, 98, 3035–3047. [Google Scholar] [CrossRef]
- Cousins, I.T.; DeWitt, J.C.; Glüge, J.; Goldenman, G.; Herzke, D.; Lohmann, R.; Miller, M.; Ng, C.A.; Scheringer, M.; Vierke, L.; et al. Strategies for Grouping Per- and Polyfluoroalkyl Substances (PFAS) to Protect Human and Environmental Health. Environ. Sci. Process. Impacts 2020, 22, 1444–1460. [Google Scholar] [CrossRef]
- Guelfo, J.L.; Korzeniowski, S.; Mills, M.A.; Anderson, J.; Anderson, R.H.; Arblaster, J.A.; Conder, J.M.; Cousins, I.T.; Dasu, K.; Henry, B.J.; et al. Environmental Sources, Chemistry, Fate, and Transport of Per- and Polyfluoroalkyl Substances: State of the Science, Key Knowledge Gaps, and Recommendations Presented at the August 2019 SETAC Focus Topic Meeting. Environ. Toxicol. Chem. 2021, 40, 3234–3260. [Google Scholar] [CrossRef]
- Kar, S.; Sepúlveda, M.S.; Roy, K.; Leszczynski, J. Endocrine-Disrupting Activity of per- and Polyfluoroalkyl Substances: Exploring Combined Approaches of Ligand and Structure Based Modeling. Chemosphere 2017, 184, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Kovarich, S.; Papa, E.; Li, J.; Gramatica, P. QSAR Classification Models for the Screening of the Endocrine-Disrupting Activity of Perfluorinated Compounds. SAR QSAR Environ. Res. 2012, 23, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Sosnowska, A.; Mudlaff, M.; Mombelli, E.; Behnisch, P.; Zdybel, S.; Besselink, H.; Kuckelkorn, J.; Bulawska, N.; Kepka, K.; Kowalska, D.; et al. Identification of New PFAS for Severe Interference with Thyroid Hormone Transport: A Combined in Vitro/Silico Approach. J. Hazard Mater. 2025, 491, 137949. [Google Scholar] [CrossRef] [PubMed]
- HyperChem, Hypercube, Inc.: 1115 NW 4th Street, Gainesville, Florida 32601, USA.
- Dragon, Version 5.5 (2007) and Version 6.0 (2011).
- Mauri, A. alvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Roy, K., Ed.; Springer: New York, NY, USA, 2020; pp. 801–820. ISBN 978-1-07-160150-1. [Google Scholar]
- Weiss, J.M.; Andersson, P.L.; Lamoree, M.H.; Leonards, P.E.G.; van Leeuwen, S.P.J.; Hamers, T. Competitive Binding of Poly- and Perfluorinated Compounds to the Thyroid Hormone Transport Protein Transthyretin. Toxicol. Sci. 2009, 109, 206–216. [Google Scholar] [CrossRef]
- Evangelista, M.; Chirico, N.; Papa, E. In Silico Models for the Screening of Human Transthyretin Disruptors. J. Hazard Mater. 2024, 480, 136188. [Google Scholar] [CrossRef]
- Degitz, S.J.; Olker, J.H.; Denny, J.S.; Degoey, P.P.; Hartig, P.C.; Cardon, M.C.; Eytcheson, S.A.; Haselman, J.T.; Mayasich, S.A.; Hornung, M.W. In Vitro Screening of per- and Polyfluorinated Substances (PFAS) for Interference with Seven Thyroid Hormone System Targets across Nine Assays. Toxicol. Vitr. 2024, 95, 105762. [Google Scholar] [CrossRef]
- Organisation for Economic Co-operation and Development. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD Series on Testing and Assessment, No. 69; OECD Publishing: Paris, France, 2014. [Google Scholar] [CrossRef]
- Yang, X.; Ou, W.; Xi, Y.; Chen, J.; Liu, H. Emerging Polar Phenolic Disinfection Byproducts Are High-Affinity Human Transthyretin Disruptors: An in Vitro and in Silico Study. Environ. Sci. Technol. 2019, 53, 7019–7028. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Organisation for Economic Co-operation and Development. Summary Report on the New Comprehensive Global Database of Per- and Polyfluoroalkyl Substances (PFASs); OECD Series on Risk Management of Chemicals; OECD Publishing: Paris, France, 2018. [Google Scholar] [CrossRef]
- Kwon, H.; Ali, Z.A.; Wong, B.M. Harnessing Semi-Supervised Machine Learning to Automatically Predict Bioactivities of Per- and Polyfluoroalkyl Substances (PFASs). Environ. Sci. Technol. Lett. 2023, 10, 1017–1022. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Papa, E.; Kovarich, S.; Gramatica, P. QSAR Prediction of the Competitive Interaction of Emerging Halogenated Pollutants with Human Transthyretin. SAR QSAR Environ. Res. 2013, 24, 333–349. [Google Scholar] [CrossRef]
- Xi, Y.; Yang, X.; Zhang, H.; Liu, H.; Watson, P.; Yang, F. Binding Interactions of Halo-Benzoic Acids, Halo-Benzenesulfonic Acids and Halo-Phenylboronic Acids with Human Transthyretin. Chemosphere 2020, 242, 125135. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ou, W.; Zhao, S.; Wang, L.; Chen, J.; Kusko, R.; Hong, H.; Liu, H. Human Transthyretin Binding Affinity of Halogenated Thiophenols and Halogenated Phenols: An in Vitro and in Silico Study. Chemosphere 2021, 280, 130627. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ou, W.; Zhao, S.; Xi, Y.; Wang, L.; Liu, H. Rapid Screening of Human Transthyretin Disruptors through a Tiered in Silico Approach. ACS Sustain. Chem. Eng. 2021, 9, 5661–5672. [Google Scholar] [CrossRef]
- Williams, A.J.; Grulke, C.M.; Edwards, J.; McEachran, A.D.; Mansouri, K.; Baker, N.C.; Patlewicz, G.; Shah, I.; Wambaugh, J.F.; Judson, R.S.; et al. The CompTox Chemistry Dashboard: A Community Data Resource for Environmental Chemistry. J. Cheminform. 2017, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Chirico, N.; McLachlan, M.S.; Li, Z.; Papa, E. In Silico Approaches for the Prediction of the Breakthrough of Organic Contaminants in Wastewater Treatment Plants. Environ. Sci. Process. Impacts 2024, 26, 400–410. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A New Software for the Development, Analysis, and Validation of QSAR MLR Models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Wold, S.; Esbensen, K.; Geladi, P. Principal Component Analysis. Chemometr. Intell. Lab. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Rücker, C.; Rücker, G.; Meringer, M. Y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Cawley, G.C.; Talbot, N.L.C. On Over-Fitting in Model Selection and Subsequent Selection Bias in Performance Evaluation. J. Mach. Learn. Res. 2010, 11, 2079–2107. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning. Data Mining, Inference, and Prediction, 2nd ed.; Springer: New York, NY, USA, 2009. [Google Scholar]
- Todeschini, R.; Ballabio, D.; Grisoni, F. Beware of Unreliable Q2! A Comparative Study of Regression Metrics for Predictivity Assessment of QSAR Models. J. Chem. Inf. Model. 2016, 56, 1905–1913. [Google Scholar] [CrossRef]
- Klingspohn, W.; Mathea, M.; ter Laak, A.; Heinrich, N.; Baumann, K. Efficiency of Different Measures for Defining the Applicability Domain of Classification Models. J. Cheminform. 2017, 9, 44. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Cherkasov, A.; Li, J.; Gramatica, P.; Hansen, K.; Schroeter, T.; Müller, K.-R.; et al. Applicability Domains for Classification Problems: Benchmarking of Distance to Models for Ames Mutagenicity Set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef] [PubMed]
- Mathea, M.; Klingspohn, W.; Baumann, K. Chemoinformatic Classification Methods and Their Applicability Domain. Mol. Inform. 2016, 35, 160–180. [Google Scholar] [CrossRef] [PubMed]
- Organisation for Economic Co-operation and Development. Lists of PFOS, PFAS, PFOA, PFCA, Related Compounds and Chemicals That May Degrade to PFCA; OECD Series on Risk Management, No. 21; OECD Publishing: Paris, France, 2007; Available online: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono(2006)15 (accessed on 20 January 2025).
- Emonts, J.; Buyel, J.F. An Overview of Descriptors to Capture Protein Properties—Tools and Perspectives in the Context of QSAR Modeling. Comput. Struct. Biotechnol. J. 2023, 21, 3234–3247. [Google Scholar] [CrossRef]
- Li, F.; Wang, P.; Fan, T.; Zhang, N.; Zhao, L.; Zhong, R.; Sun, G. Prioritization of the Ecotoxicological Hazard of PAHs towards Aquatic Species Spanning Three Trophic Levels Using 2D-QSTR, Read-across and Machine Learning-Driven Modelling Approaches. J. Hazard Mater. 2024, 465, 133410. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Rational Drug Design for Anti-Cancer Chemotherapy: Multi-Target QSAR Models for the in Silico Discovery of Anti-Colorectal Cancer Agents. Bioorg. Med. Chem. 2012, 20, 4848–4855. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Moreau, G.; Broto, P. The Autocorrelation of a Topological Structure: A New Molecular Descriptor. Nouv. J. Chim. 1980, 4, 359–360. [Google Scholar]
- King, J.W. A Z-Weighted Information Content Index. Int. J. Quantum Chem. 1989, 36, 165–170. [Google Scholar] [CrossRef]
- Weinebeck, A.; Kaminski, S.; Murrenhoff, H.; Leonhard, K. A New QSPR-Based Prediction Model for Biofuel Lubricity. Tribol. Int. 2017, 115, 274–284. [Google Scholar] [CrossRef]
- Jia, T.; Liu, W.; Keller, A.A.; Gao, L.; Xu, X.; Wu, W.; Wang, X.; Yu, Y.; Zhao, G.; Li, B.; et al. Potential Impact of Organophosphate Esters on Thyroid Eye Disease Based on Machine Learning and Molecular Docking. Sci. Total Environ. 2024, 957, 177835. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.-M.; Qin, W.-P.; Cao, L.-Y.; Zhang, J.; Yang, Y.; Wan, B.; Guo, L.-H. Binding Interactions of Perfluoroalkyl Substances with Thyroid Hormone Transport Proteins and Potential Toxicological Implications. Toxicol. 2016, 366–367, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, J.; Garcia, R.; Salabert, M.T.; Soler, R. Charge Indexes. New Topological Descriptors. J. Chem. Inf. Comput. Sci. 1994, 34, 520–525. [Google Scholar] [CrossRef]
- Carlier, M.P.; Cenijn, P.H.; Baygildiev, T.; Irwan, J.; Escher, S.E.; van Duursen, M.B.M.; Hamers, T. Profiling the Endocrine-Disrupting Properties of Triazines, Triazoles, and Short-Chain PFAS. Toxicol. Sci. 2024, 202, 250–264. [Google Scholar] [CrossRef]
- Langberg, H.A.; Choyke, S.; Hale, S.E.; Koekkoek, J.; Cenijn, P.H.; Lamoree, M.H.; Rundberget, T.; Jartun, M.; Breedveld, G.D.; Jenssen, B.M.; et al. Effect-Directed Analysis Based on Transthyretin Binding Activity of Per- and Polyfluoroalkyl Substances in a Contaminated Sediment Extract. Environ. Toxicol. Chem. 2024, 43, 245–258. [Google Scholar] [CrossRef]
- Zhang, J.; Kamstra, J.H.; Ghorbanzadeh, M.; Weiss, J.M.; Hamers, T.; Andersson, P.L. In Silico Approach To Identify Potential Thyroid Hormone Disruptors among Currently Known Dust Contaminants and Their Metabolites. Environ. Sci. Technol. 2015, 49, 10099–10107. [Google Scholar] [CrossRef]
- Xin, Y.; Ren, X.-M.; Ruan, T.; Li, C.-H.; Guo, L.-H.; Jiang, G. Chlorinated Polyfluoroalkylether Sulfonates Exhibit Similar Binding Potency and Activity to Thyroid Hormone Transport Proteins and Nuclear Receptors as Perfluorooctanesulfonate. Environ. Sci. Technol. 2018, 52, 9412–9418. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| n | ACC | MR | SN | SP | P | AUC | MRBOOTSTRAP | Random Range | Random Descriptors Nature | Selected Molecular Descriptors | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Training | 82 | 0.89 | 0.11 | 0.92 | 0.84 | 0.90 | 0.85 | 0.32 ± 2.7 × 10−3 | 3.8 × 10−3 | 4.6 × 10−3 | GATS3e, ATSC6p, GATS8m, MIC2 |
| Test | 39 | 0.85 | 0.15 | 0.88 | 0.80 | 0.88 | 0.85 | - | - | - | - |
| n | R2 | MAE | Q2loo | Q2F3 | R2YS | MAEBOOTSTRAP | Random Range | Random Descriptors Nature | Selected Molecular Descriptors | |
|---|---|---|---|---|---|---|---|---|---|---|
| Training | 43 | 0.81 | 0.30 | 0.77 | - | 0.072 | 0.58 ± 5.7 × 10−3 | 8.3 × 10−10 | 3.3 × 10−10 | piPC5, GGI9, AATSC0e |
| Test | 20 | 0.77 | 0.26 | - | 0.82 | - | - | - | - | - |
| LDA-QSAR | MLR-QSAR | |||||
|---|---|---|---|---|---|---|
| Structure Category | Total (%) | Inside AD | Total (%) | Inside AD | ||
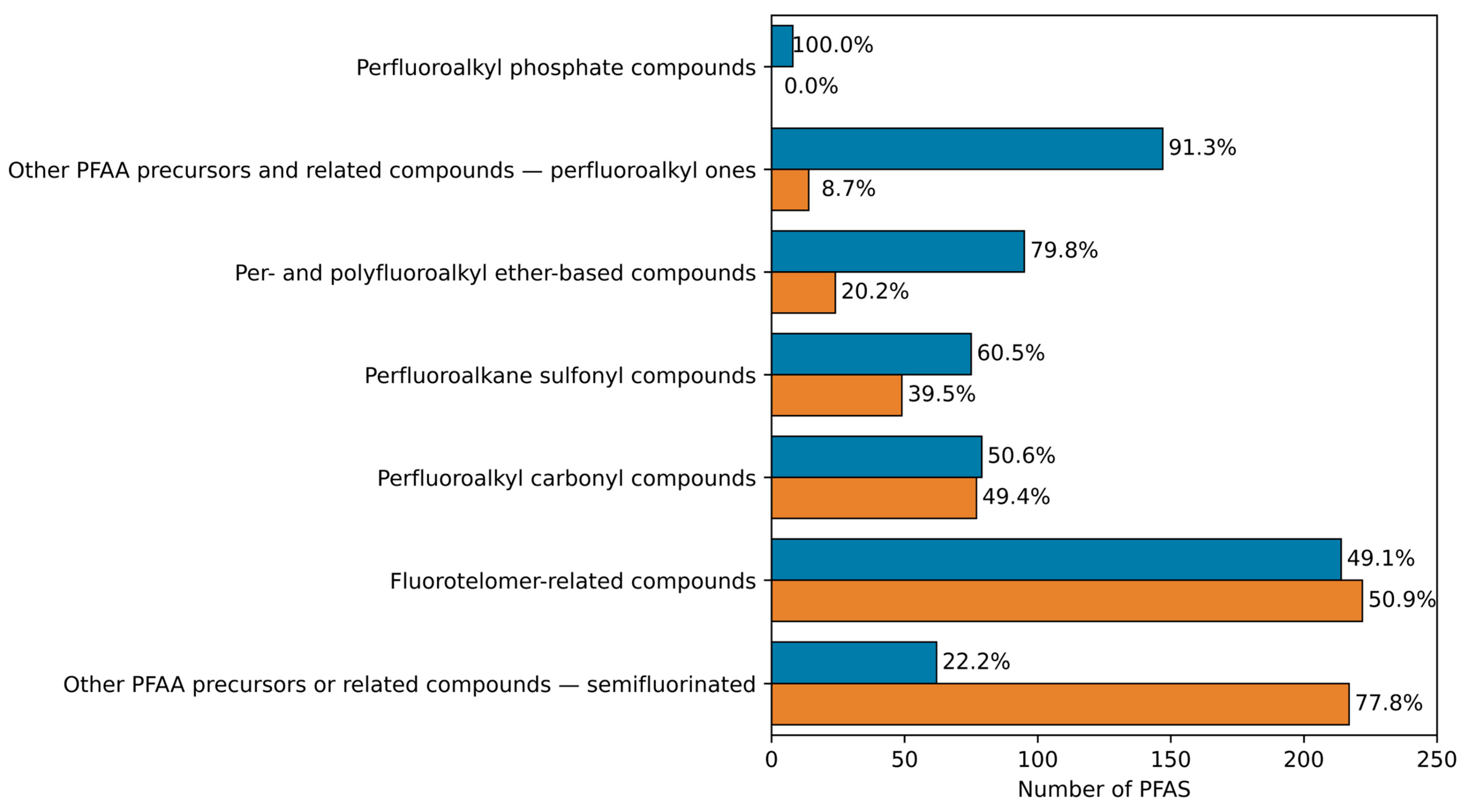
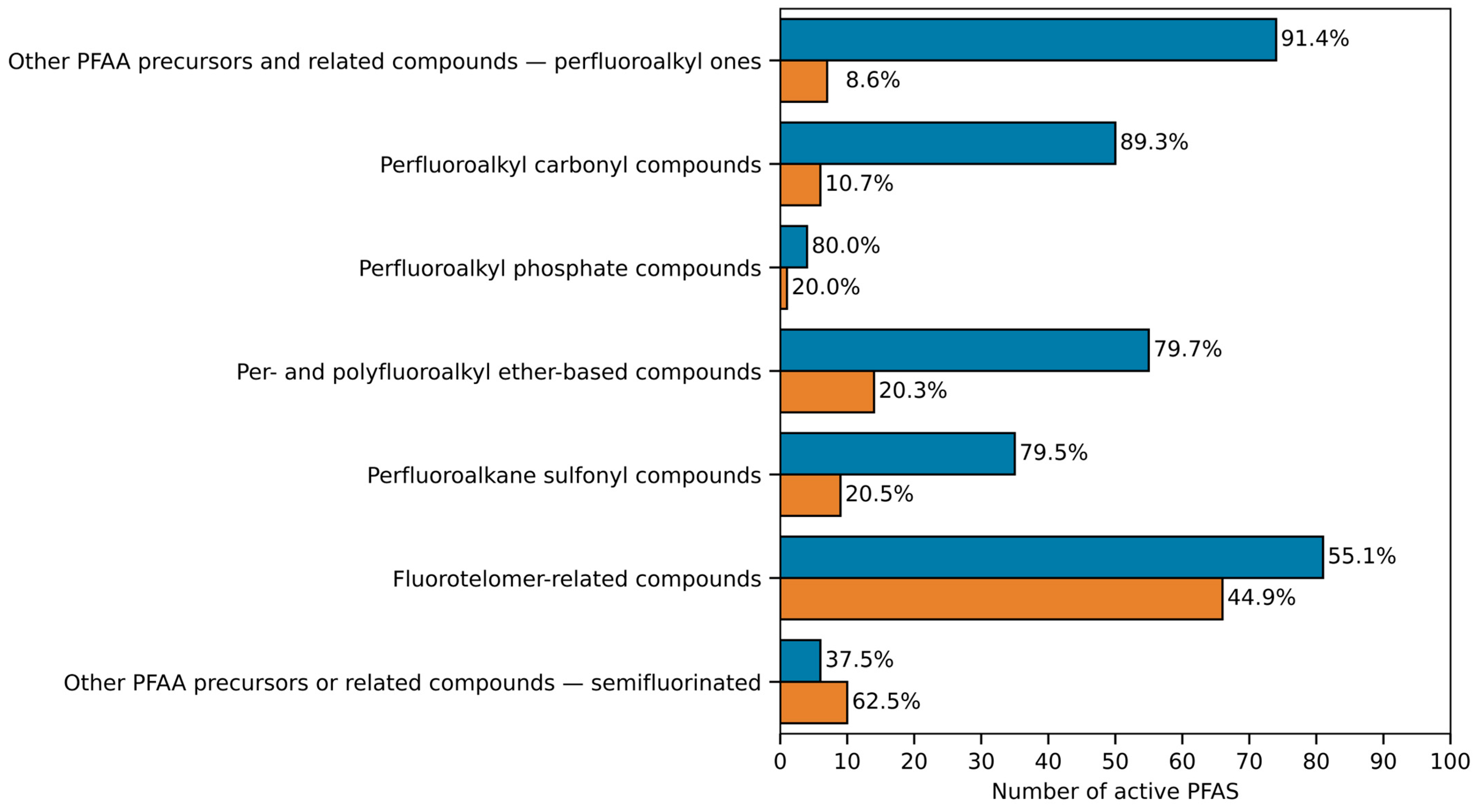
| - | - | Number of PFAS (%) | Number of Structural Subcategories | - | Number of PFAS (%) | Number of Structural Subcategories |
| Fluorotelomer—related compounds | 1086 (37.0) | 436 (40.1) | 24 | 214 (31.5) | 147 (68.7) | 19 |
| Other PFAA precursors or related compounds—semifluorinated | 686 (23.4) | 279 (40.7) | 8 | 62 (9.1) | 16 (25.8) | 6 |
| Perfluoroalkyl carbonyl compounds | 359 (12.2) | 156 (43.5) | 9 | 79 (11.6) | 56 (70.9) | 6 |
| Per- and polyfluoroalkyl ether-based compounds | 280 (9.6) | 119 (42.5) | 18 | 95 (14.0) | 69 (72.6) | 15 |
| Perfluoroalkane sulfonyl compounds | 271 (9.2) | 124 (45.8) | 9 | 75 (11.0) | 44 (58.7) | 7 |
| Other PFAA precursors and related compounds—perfluoroalkyl ones | 240 (8.2) | 161 (67.1) | 10 | 147 (21.6) | 81 (55.1) | 10 |
| Perfluoroalkyl phosphate compounds | 12 (0.4) | 8 (66.7) | 2 | 8 (1.2) | 5 (62.5) | 2 |
| Total | 2934 (100) | 1283 (43.7) | - | 680 (100) | 418 (61.5) | - |
| Present Model | Kar et al. [34] | Kovarich et al. * [35] | Sosnowska et al. ** [36] | |
|---|---|---|---|---|
| Endpoint | hTTR binding affinity | hTTR binding affinity | hTTR binding affinity | RPF |
| In vitro assay | ANSA-based [42] | RLBA [40] | RLBA [40] | TTR-TRβ CALUX [36] |
| Method | LDA | LDA | kNN | DTC |
| Dataset size | 121 | 24 | 19 | 44 |
| Training set size | 82 | 16 | 10 | 33 |
| Test set size | 39 | 8 | 9 | 11 |
| Number of descriptors | 4 | 3 | 2 | 2 |
| SN training | 0.92 | 1 | 0.83–1 | 0.96 |
| SN test | 0.88 | 1 | 1 | 1 |
| SP training | 0.84 | 0.83 | 0.75–1 | 1 |
| SP test | 0.80 | 1 | 0.75–1 | 0.50 |
| ACC training | 0.89 | 0.94 | 0.90–1 | 0.97 |
| ACC test | 0.85 | 1 | 0.90–1 | 0.91 |
| P training | 0.90 | 0.91 | N/A | 1 |
| P test | 0.88 | 1 | N/A | 0.90 |
| AUC training | 0.85 | 0.95 | N/A | N/A |
| AUC test | 0.85 | 1 | N/A | N/A |
| This Model | Kar et al. [34] | Sosnowska et al. [36] Approach 1 | Sosnowska et al. [36] Approach 2 * | |
|---|---|---|---|---|
| Endpoint | RP | IC50 | RPF | RPF |
| Method | MLR | MLR | MLR | MLR |
| In vitro assay | ANSA-based [42] | RLBA [40] | RLBA [40] | TTR-TRβ CALUX [36] |
| Dataset size | 63 | 15 | 35 | 35 |
| Training set size | 43 | 10 | 24 | 25 |
| Test set size | 20 | 5 | 11 | 10 |
| Number of descriptors | 3 | 2 | 3 | 4–5 |
| Ratio training set size/ number of descriptors | 14.3 | 5 | 8 | 5–6.3 |
| R2 | 0.81 | 0.86 | 0.77 | N/A |
| R2EXT | 0.77 | 0.64 | N/A | N/A |
| MAETR | 0.30 | N/A | 0.43 | N/A |
| MAETEST | 0.26 | 0.11 | 0.40 | 0.34–0.54 |
| Q2loo | 0.77 | 0.73 | 0.77 | 0.76–0.82 |
| Q2F3 | 0.82 | N/A | 0.81 | 0.76–0.82 |
| R2YS | 0.07 | N/A | 0.13 | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evangelista, M.; Chirico, N.; Papa, E. New QSAR Models to Predict Human Transthyretin Disruption by Per- and Polyfluoroalkyl Substances (PFAS): Development and Application. Toxics 2025, 13, 590. https://doi.org/10.3390/toxics13070590
Evangelista M, Chirico N, Papa E. New QSAR Models to Predict Human Transthyretin Disruption by Per- and Polyfluoroalkyl Substances (PFAS): Development and Application. Toxics. 2025; 13(7):590. https://doi.org/10.3390/toxics13070590
Chicago/Turabian StyleEvangelista, Marco, Nicola Chirico, and Ester Papa. 2025. "New QSAR Models to Predict Human Transthyretin Disruption by Per- and Polyfluoroalkyl Substances (PFAS): Development and Application" Toxics 13, no. 7: 590. https://doi.org/10.3390/toxics13070590
APA StyleEvangelista, M., Chirico, N., & Papa, E. (2025). New QSAR Models to Predict Human Transthyretin Disruption by Per- and Polyfluoroalkyl Substances (PFAS): Development and Application. Toxics, 13(7), 590. https://doi.org/10.3390/toxics13070590