
A Statistical Exploration of QSAR Models in Cancer Risk Assessment: A Case Study on Pesticide-Active Substances and Metabolites




Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Information and Data Preprocessing Phase
- Positive: if one strain result was positive;
- Negative: if all the strain results were negative;
- Equivocal: if some of the strain results in the same experimental study were equivocal, they could not be classified into one of the other categories.
2.2. The Danish QSAR Software
- CASE Ultra (MultiCASE Inc., Beachwood, OH, USA): A system of statistical models based on fragments. The methodology decomposes the training set structures into all possible fragments between 2 and 10 heavy (non-hydrogen) atoms. The system generates simple linear chains, branched fragments, and complex substructures from combinations of multiple fragments. A structural fragment is identified as a positive alert if it resembles fragments from chemicals exhibiting toxicity for a given endpoint. Conversely, fragments are deemed non-hazardous (deactivating) if they are similar to those in chemicals deemed safe for the endpoint. Once the lists of positive and deactivating alerts are defined, CASE Ultra develops local (Q)SAR models for each alert to explain the activity of the entire training set associated with that alert. Using a stepwise regression method, local (Q)SARs are constructed based on specific molecular descriptors. The combination of positive and deactivating alerts (though not all alerts will necessarily have an associated local (Q)SAR) forms a global (Q)SAR model for a given endpoint. The results available in the Danish QSAR database refer to MultiCASE CASE Ultra v. 1.4.6.6.
- Leadscope (Leadscope Inc.; Columbus, OH, USA): Leadscope Predictive Data Miner is a software equipped with an extensive library of approximately 27,000 predefined structural features, with some derived from the training set. The software systematically analyzes chemical substructures and calculates molecular descriptors to correlate substances with their potential toxicological properties. Using statistical methods such as the χ2 test and Student’s t-test, Leadscope selects descriptors deemed most relevant for toxicological predictions. Subsequently, regression techniques, such as partial least squares regression (PLS) or partial logistic regression (PLR), are employed to construct predictive models for application to untested molecules. The results available in the Danish QSAR database refer to Leadscope Predictive Data Miner, a component of Leadscope Enterprise version 3.5.
- SciQSAR: SciQSAR software provides over 400 molecular descriptors and a variety of statistical tools to construct predictive models, including discriminant analysis models. This tool was used as implemented in the Danish (Q)SAR software [11].
- Battery: Some predictions are made using two or three independent models: CASE Ultra (CU), Leadscope Predictive Data Miner (LS), and SciQSAR (SQ). Based on the predictions of each model, a “battery prediction” is conducted using a Battery algorithm defined by the criteria reported in the DB user manual (https://qsardb.food.dtu.dk/db/index.html, accessed on 27 March 2025).
- Predictions from CASE Ultra (MultiCASE CASE Ultra version 1.4.) were available for the following categories:Male Rat, Female Rat, Rat (cumulative both sex prediction), Male Mouse, Female Mouse, Mouse (cumulative both sex prediction), Rodent (cumulative both sex and species prediction) and Liver-Specific Cancer in a Rat or Mouse.
- Predictions from Leadscope (Leadscope Enterprise version 3.5, unless otherwise specified) were available for the following categories:Male Rat, Female Rat, Rat (cumulative both sex prediction, Leadscope Enterprise version 3.1.1-10), Male Mouse, Female Mouse, Mouse (cumulative both sex prediction, Leadscope Enterprise version 3.1.1-10), Rodent (cumulative both sex and species prediction, Leadscope Enterprise version 3.1.1-10) and Liver-Specific Cancer in Rat or Mouse (Leadscope Enterprise version 3.1.1-10).
- Predictions from SciQSAR (SciQSAR version 3.1.00) and Battery calculation were available exclusively for Liver-Specific Cancer in a Rat or Mouse.
- Predictions performed in the Models Module of the Danish (Q)SAR Database were also limited to Liver-Specific Cancer in a Rat or Mouse.
2.3. Data Analysis Strategy
2.3.1. Statistical Methods
2.3.2. Principal Component Analysis
2.3.3. Variable Clustering Analysis
2.3.4. Correlation Analysis
3. Results and Discussion
3.1. Danish (Q)SAR Predictions
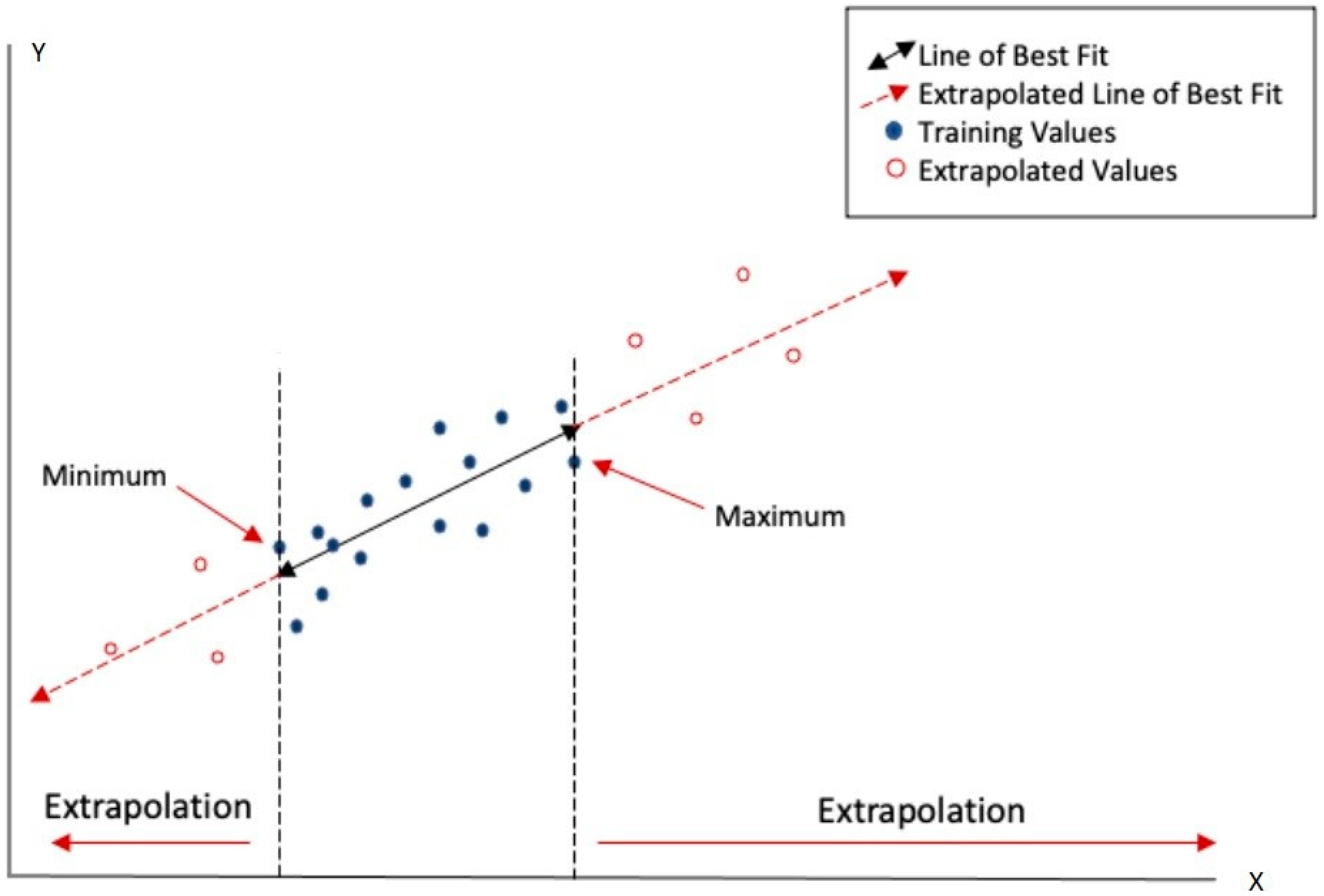
3.2. Applicability Domain and Uncertainty Sources
- Parameter uncertainty: model parameters are often calculated rather than experimentally measured, introducing unwanted (and difficult to detect) errors. Parametric variability: input variables, such as chemical descriptors, might differ depending on the software used for their calculation.
- Structural uncertainty: each model has biases associated with the core dataset used for its development.
- Algorithmic uncertainty: errors or numerical approximations in algorithms can affect the accuracy of results.
- Experimental uncertainty: errors in the experimental data used to build or validate the models directly impact the reliability of (Q)SAR predictions.
3.3. Statistical Analysis
3.3.1. Principal Component Analysis
- -
- Separate columns (variables) were created for carcinogenic predictions from different models, assigning the following values: one for negative predictions, two for equivocal predictions, and three for positive predictions.
- -
- For each column defined above, an additional variable was created to include information about the AD. Specifically, a value of 0 was assigned to OUT OF DOMAIN predictions, and a value of 1 was assigned to IN DOMAIN predictions.
- The relative independence of the predictions from Leadscope and CASE Ultra systems (associated with different components).
- The uniqueness of Liver-Specific Cancer in a Rat or Mouse.
- PCdom1 exhibits strong positive correlations with R_CASE_DOM1, R_CASE_DOM, RF_CASE_DOM, RM_CASE_DOM, FM_CASE_DOM, LS_R_M_BATT_DOM, LS_R_M_CASE_DOM, and M_CASE_DOM. The strong correlation of R_CASE_DOM1 with PCdom1 (0.791) suggests that this AD is particularly influential in shaping PCdom1.
- PCdom1 also shows a strong negative correlation with MM_LEAD_DOM, indicating that the ADs of the two models are complementary. Substances falling within the domain of one model are generally outside the domain of the other. This observation raises a very important point for model integration, which we will discuss in the following.
- PCdom2 is primarily associated with R_LEAD_DOM, MM_CASE_DOM, RF_LEAD_DOM, and M_LEAD_DOM.
- PCdom3 has strong positive correlations with the applicability domains of RM_LEAD_DOM and R_LEAD_DOM1.
- R_CASE_DOM1 has a very strong correlation with PCdom1 (0.791), making this component highly representative of the AD of CASE Ultra for Rodent (Rodent) that, in turn, can be considered to occupy a central position among the different (Q)SAR models.
- RF_CASE_DOM and RM_CASE_DOM (the ADs of CASE Ultra for a Female Rat and Male Rat) also show strong positive correlations with PCdom1 (0.618 and 0.614, respectively). This highlights the substantial internal coherence of CASE Ultra for the Rodent, Female Rat, and Male Rat, distinguishing them significantly from the ADs defining subsequent components (PCdom2 and PCdom3).
- MM_LEAD_DOM has a significant negative correlation (−0.587) with PCdom1. This implies that when MM_LEAD_DOM increases, the value of PCdom1 decreases, confirming the complementarity of ADs of CASE Ultra and Leadscope that can be leveraged for the integration of the two methods.
- Most other variables exhibit moderate positive correlations with PCdom1, indicating some association but one that is not as strong as the variables mentioned above.
3.3.2. Variable Clustering Analysis
- CASE Ultra predictions, which are further clustered as follows:
- Mouse, Female Mouse, and Male Mouse.
- Rodent, Rat, Female Rat, and Male Rat.
- Predictions from various models (SciQSAR, Leadscope, CASE Ultra, Battery) targeting liver-specific endpoints (a Rat or Mouse).
- Leadscope predictions are further grouped as follows:
- Rodent, Mouse, and Rat.
- Female Mouse, Male Mouse, Female Rat, and Male Rat.
- Predictions from CASE Ultra tend to cluster by species, suggesting that this model may incorporate species-specific biological features. Within species-based clusters, additional finer distinctions appear based on sex.
- Predictions for liver-specific endpoints are clearly separated from other variables (notably, they are not differentiated by sex or gender).
- Predictions from the Leadscope cluster differ from CASE Ultra, suggesting differences in how these models prioritize or interpret variables such as species and sex.
- Distinct Algorithms or Methodologies: The models may utilize different computational approaches, leading to divergent outputs.
- Diverse Training Datasets: CASE Ultra and Leadscope might rely on different datasets or inclusion criteria that influence the predictions.
- Different Objectives: Despite both focusing on carcinogenicity, the models may prioritize specific chemical classes or mechanisms differently.
- Accuracy and Reliability: One model may demonstrate greater consistency or reliability, leading to clearer clustering distinctions.
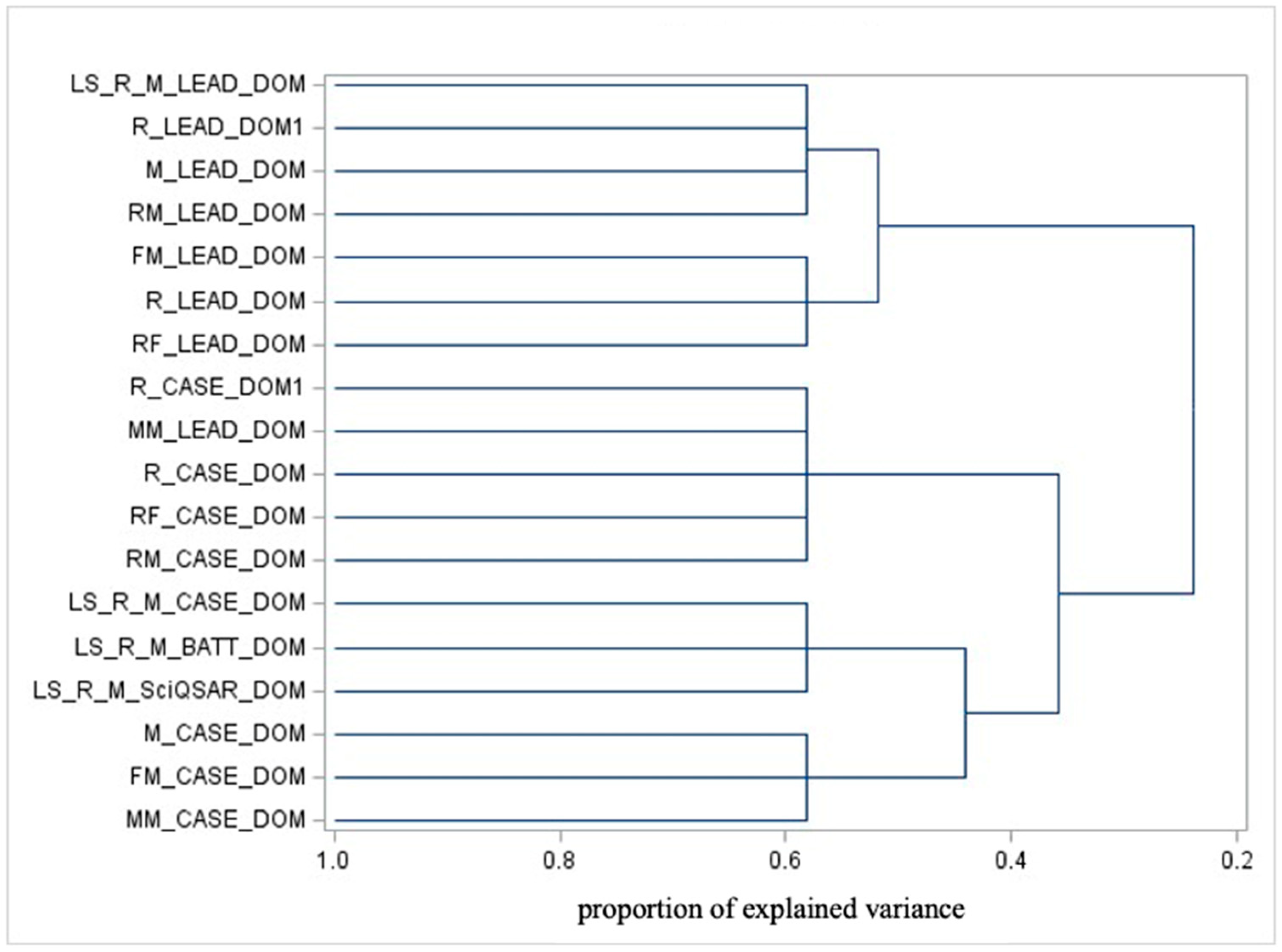
3.3.3. Domain Clustering
- CASE Ultra’s applicability domain vs. liver-specific domains, which can be further divided into the following:Specific instances of CASE Ultra and liver-specific domains (except for liver-specific Leadscope, which clusters with general Leadscope AD).
- Leadscope’s AD.
- -
- The distinction between CASE Ultra and Liver-Specific domains reflects differences in their focus or training sets. Liver-specific models likely emphasize specific mechanisms and subsets of substances.
- -
- The separation of Leadscope’s domain from the others underscores its unique training set, focus, or methodology.
- -
- Leadscope’s liver-specific domain clustering with the general Leadscope domain may indicate shared foundational characteristics coming from identical model specifications.
3.3.4. Correlation Analysis
- RM_CASE_DOM vs. RM_CASE: Strong negative correlation (−0.763, p < 0.0001), suggesting that as RM_CASE increases, RM_CASE_DOM decreases (or vice versa).
- RM_LEAD_DOM vs. RM_LEAD: Moderate negative correlation (−0.471, p = 0.0043).
- RF_CASE_DOM vs. RF_CASE: Strong negative correlation (−0.605, p = 0.0001).
- RF_LEAD_DOM vs. RF_LEAD: Moderate negative correlation (−0.480, p = 0.0035).
- R_CASE_DOM vs. R_CASE: Strong negative correlation (−0.746, p < 0.0001).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Applicability domain |
| ASs | Active substances |
| CPDB | Carcinogenic Potency Database |
| CU | CASE Ultra |
| Danish EPA | Danish Environmental Protection Agency |
| DTU | Technical University of Denmark |
| ECHA | European Chemicals Agency |
| ECVAM | Genotoxicity & Carcinogenicity Database by the European Reference Laboratory for Alternatives to Animal Testing |
| EFSA | European Food Safety Authority |
| EQ | Equivocal |
| IN | In the domain |
| ISS | Italian National Institute of Health |
| ISSBIOC | Biocides and Plant Protection Database by ISS |
| ISSCAN | Carcinogenicity & Mutagenicity Database by ISS |
| LS | Leadscope Predictive Data Miner |
| MET | Metabolites |
| ML | Machine learning |
| MM/MF | Mouse Male/Mouse Female |
| MN_ID | Unique identifier of substances under analysis |
| NAMs | New approach methodologies |
| ND | No data available |
| NEG | Negative result |
| OECD | Organisation for Economic Cooperation and Development |
| OUT | Out of the domain |
| PCA | Principal Component Analysis |
| PINNs | Physics-Informed Neural Networks |
| PLR | Partial logistic regression |
| PLS | Partial least squares regression |
| POS | Positive result |
| RM/RF | Rat Male/Rat Female |
| (Q)SAR | (Quantitative) structure–activity relationship |
| SQ | SciQSAR |
| WoE | Weight of evidence |
References
- Tannenbaum, J.; Bennett, B.T. Russell and Burch’s 3Rs then and now: The need for clarity in definition and purpose. J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 1–13. [Google Scholar]
- Jean-Quartier, C.; Jeanquartier, F.; Jurisica, I.; Holzinger, A. In silico cancer research towards 3R. BMC Cancer 2018, 18, 408. [Google Scholar] [CrossRef] [PubMed]
- ECHA. New Approach Methodologies in Regulatory Science Proceedings of a Scientific Workshop. Helsinki, April 2016. Available online: https://echa.europa.eu/documents/10162/21838212/scientific_ws_proceedings_en.pdf/a2087434-0407-4705-9057-95d9c2c2cc57 (accessed on 27 March 2025).
- ECHA, Non-Animal Approaches. Current Status of Regulatory Applicability Under The REACH, CLP and Biocidal Products Regulations. Available online: https://echa.europa.eu/documents/10162/22931011/non_animal_approcches_en.pdf/87ebb68f-2038-f597-fc33-f4003e9e7d7d (accessed on 3 November 2024).
- ECHA. The Use of Alternatives to Testing on Animals for the REACH Regulation. June 2023. Available online: https://echa.europa.eu/documents/10162/23919267/230530_117_3_alternatives_test_animals_2023_en.pdf/9cfc291e-9baf-ffa2-466c-2bc2c6f06b8e?t=1685428213290 (accessed on 3 November 2024).
- Macmillan, D.S.; Bergqvist, A.; Burgess-Allen, E.; Callan, I.; Dawick, J.; Carrick, B.; Ellis, G.; Ferro, R.; Goyak, K.; Smulders, C.; et al. The last resort requirement under REACH: From principle to practice. Regul. Toxicol. Pharmacol. 2024, 147, 105557. [Google Scholar] [CrossRef]
- Gissi, A.; Tcheremenskaia, O.; Bossa, C.; Battistelli, C.L.; Browne, P. The OECD (Q)SAR Assessment Framework: A tool for increasing regulatory uptake of computational approaches. Comput. Toxicol. 2024, 31, 100326. [Google Scholar] [CrossRef]
- OECD. (Q)SAR Assessment Framework: Guidance for the Regulatory Assessment of (Quantitative) Structure Activity Relationship Models and Predictions, Second Edition. Series on Testing and Assessment No. 405. 2024. Available online: https://www.oecd.org/en/publications/q-sar-assessment-framework-guidance-for-the-regulatory-assessment-of-quantitative-structure-activity-relationship-models-and-predictions-second-edition_bbdac345-en.html (accessed on 27 March 2025).
- Benfenati, E.; Chaudhry, Q.; Gini, G.; Dorne, J.L. Integrating in silico models and read-across methods for predicting toxicity of chemicals: A step-wise strategy. Environ. Int. 2019, 131, 105060. [Google Scholar] [CrossRef]
- EFSA PPR Panel. Guidance on the establishment of the residue definition for dietary risk assessment EFSA Panel on Plant Protection Products and their Residues (PPR). EFSA J. 2016, 14, 12. [Google Scholar] [CrossRef]
- Technical University of Denmark. Danish (Q)SAR database/(Q)SAR Models. Division of Diet, Disease Prevention and Toxicology, National Food Institute, Technical University of Denmark. Available online: https://qsar.food.dtu.dk/ (accessed on 27 March 2025).
- Bossa, C.; Benigni, R.; Tcheremenskaia, O.; Battistelli, C.L. (Q)SAR methods for predicting genotoxicity and carcinogenicity: Scientific rationale and regulatory frameworks. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1800, pp. 447–473. [Google Scholar] [CrossRef]
- Battistelli, C.L.; Bossa, C. Development, Use, and Validation of (Q)SARs for Predicting Genotoxicity and Carcinogenicity: Experiences from Italian National Institute of Health Activities. In Computational Toxicology: Methods and Protocols; Springer: New York, NY, USA, 2025; pp. 231–247. [Google Scholar] [CrossRef]
- Madden, J.C.; Enoch, S.J.; Paini, A.; Cronin, M.T.D. A Review of In Silico Tools as Alternatives to Animal Testing: Principles, Resources and Applications. Altern. Lab. Anim. 2020, 48, 146–172. [Google Scholar] [CrossRef]
- OECD. OECD Principles for the Validation for Regulatory Purposes of (Quantitative) Structure-Activity Relationship Models. 2004. Available online: https://www.oecd.org/content/dam/oecd/en/topics/policy-sub-issues/assessment-of-chemicals/oecd-principles-for-the-validation-for-regulatory-purposes-of-quantitative-structure-activity-relationship-models.pdf (accessed on 27 March 2025).
- Hardy, A.; Benford, D.; Halldorsson, T.; Jeger, M.J.; Knutsen, H.K.; More, S.; Naegeli, H.; Noteborn, H.; Ockleford, C.; Ricci, A. Guidance on the use of the weight of evidence approach in scientific assessments. EFSA J. 2017, 15, e04971. [Google Scholar] [CrossRef]
- ICH M7(R2). ICH M7(R2) Guideline on Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk Step 5 July 2023. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-m7r2-guideline-assessment-and-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit-potential-carcinogenic-risk-step-5_en.pdf (accessed on 20 December 2024).
- Benigni, R.; Battistelli, C.L.; Bossa, C.; Giuliani, A.; Fioravanzo, E.; Bassan, A.; Fuart Gatnik, M.; Rathman, J.; Yang, C.; Tcheremenskaia, O. Evaluation of the applicability of existing (Q)SAR models for predicting the genotoxicity of pesticides and similarity analysis related with genotoxicity of pesticides for facilitating of grouping and read across. EFSA Support. Publ. 2019, 16, 1598E. [Google Scholar] [CrossRef]
- Giuliani, A. The application of principal component analysis to drug discovery and biomedical data. Drug Discov. Today 2017, 22, 1069–1076. [Google Scholar] [CrossRef]
- Cox, D.R. The Analysis of Multivariate Binary Data. Appl. Stat. 1972, 21, 113. [Google Scholar] [CrossRef]
- Richman, M.B. Rotation of principal components. J. Climatol. 1986, 6, 293–335. [Google Scholar] [CrossRef]
- Corton, J.C.; Peters, J.M.; Klaunig, J.E. The PPARα-dependent rodent liver tumor response is not relevant to humans: Addressing misconceptions. Arch. Toxicol. 2019, 92, 83–119. [Google Scholar] [CrossRef] [PubMed]
- de Winter, J.C.F.; Gosling, S.D.; Potter, J. Comparing the Pearson and Spearman correlation coefficients across distributions and sample sizes: A tutorial using simulations and empirical data. Psychol. Methods 2016, 21, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C. Quantitative approach to biochemical structure-activity relationships. Acc. Chem. Res. 1969, 2, 232–239. [Google Scholar] [CrossRef]
- The Royal Society. Science in the Age of AI: How Artificial Intelligence is Changing the Nature and Method of Scientific Research. 2024. Available online: https://royalsociety.org/-/media/policy/projects/science-in-the-age-of-ai/science-in-the-age-of-ai-report.pdf (accessed on 27 March 2025).
- Giuliani, A. System Science Can Relax the Tension Between Data and Theory. Systems 2024, 12, 474. [Google Scholar] [CrossRef]
- Cai, S.; Mao, Z.; Wang, Z.; Yin, M.; Karniadakis, G.E. Physics-informed neural networks (PINNs) for fluid mechanics: A review. Acta Mech. Sin. 2021, 37, 1727–1738. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| PCtest (Predictions) | Eigenvalue | Cumulative Variance Explained |
|---|---|---|
| PCtest1 | 3.833 | 0.213 |
| PCtest2 | 3.037 | 0.382 |
| PCtest3 | 2.568 | 0.524 |
| PCtest4 | 1.740 | 0.621 |
| PCtest5 | 1.165 | 0.686 |
| PCtest6 | 1.070 | 0.745 |
| PCdom (Domain) | Eigenvalue | Cumulative Variance Explained |
|---|---|---|
| PCdom1 | 4.294 | 0.239 |
| PCdom2 | 2.782 | 0.393 |
| PCdom3 | 1.946 | 0.501 |
| PCdom4 | 1.751 | 0.598 |
| PCdom5 | 1.281 | 0.670 |
| PCdom6 | 1.088 | 0.730 |
| PCtest1 | PCtest2 | PCtest3 | PCtest4 | PCtest5 | PCtest6 | |
|---|---|---|---|---|---|---|
| RM_CASE | 0.440 | 0.353 | −0.355 | −0.133 | 0.401 | −0.153 |
| RM_LEAD | 0.747 | −0.185 | −0.0433 | 0.220 | −0.104 | 0.111 |
| RF_CASE | 0.449 | 0.357 | −0.328 | −0.542 | −0.128 | 0.239 |
| RF_LEAD | 0.493 | −0.0318 | −0.227 | 0.483 | 0.320 | 0.313 |
| R_CASE | 0.436 | 0.351 | −0.395 | −0.602 | 0.110 | 0.133 |
| R_LEAD | 0.625 | −0.130 | −0.235 | 0.316 | −0.140 | −0.0583 |
| MM_CASE | 0.268 | 0.388 | 0.564 | 0.393 | −0.0574 | 0.171 |
| MM_LEAD | 0.712 | −0.200 | −0.0172 | 0.114 | 0.111 | −0.029 |
| FM_CASE | 0.168 | 0.345 | 0.865 | −0.0778 | 0.0672 | −0.0759 |
| FM_LEAD | 0.450 | −0.364 | 0.172 | 0.208 | 0.295 | 0.343 |
| M_CASE | 0.160 | 0.316 | 0.837 | −0.236 | 0.243 | −0.0777 |
| M_LEAD | 0.608 | 0.0707 | 0.146 | −0.276 | −0.421 | 0.081 |
| R_CASE1 | 0.480 | 0.690 | 0.0973 | −0.0705 | 0.107 | −0.0975 |
| R_LEAD1 | 0.551 | 0.119 | −0.0276 | 0.241 | −0.591 | −0.362 |
| LS_R_M_BATT | −0.385 | 0.634 | −0.383 | 0.224 | −0.0104 | 0.139 |
| LS_R_M_CASE | 0.0942 | 0.467 | −0.270 | 0.295 | 0.286 | −0.493 |
| LS_R_M_LEAD | −0.310 | 0.610 | 0.0271 | 0.192 | −0.216 | 0.535 |
| LS_R_M_SciQSAR | −0.250 | 0.761 | −0.144 | 0.315 | −0.104 | −0.0414 |
| PCdom1 | PCdom2 | PCdom3 | PCdom4 | PCdom5 | PCdom6 | |
|---|---|---|---|---|---|---|
| LS_R_M_SciQSAR_DOM | 0.348 | 0.412 | 0.049 | 0.476 | 0.139 | −0.171 |
| RM_CASE_DOM | 0.614 | −0.0263 | 0.277 | −0.141 | 0.501 | 0.322 |
| RM_LEAD_DOM | −0.187 | 0.459 | 0.694 | 0.129 | −0.127 | 0.189 |
| RF_CASE_DOM | 0.618 | 0.0727 | 0.0965 | 0.165 | −0.499 | 0.227 |
| RF_LEAD_DOM | −0.354 | 0.541 | −0.419 | 0.328 | −0.282 | 0.180 |
| R_CASE_DOM | 0.703 | −0.174 | 0.254 | 0.0277 | −0.00828 | 0.349 |
| R_LEAD_DOM | −0.304 | 0.751 | 0.0338 | 0.0134 | −0.00811 | 0.0689 |
| MM_CASE_DOM | 0.444 | 0.588 | −0.114 | −0.320 | 0.0916 | −0.0158 |
| MM_LEAD_DOM | −0.587 | 0.257 | −0.208 | −0.052 | 0.291 | −0.290 |
| FM_CASE_DOM | 0.565 | 0.390 | −0.511 | −0.223 | 0.0700 | 0.0804 |
| FM_LEAD_DOM | −0.379 | 0.430 | −0.198 | 0.125 | 0.312 | 0.573 |
| M_CASE_DOM | 0.535 | 0.152 | −0.444 | −0.483 | 0.131 | −0.142 |
| M_LEAD_DOM | −0.164 | 0.508 | 0.0331 | −0.531 | −0.399 | −0.00694 |
| R_CASE_DOM1 | 0.791 | −0.114 | −0.115 | −0.0862 | 0.00396 | 0.046 |
| R_LEAD_DOM1 | −0.122 | 0.433 | 0.533 | −0.190 | 0.398 | −0.0998 |
| LS_R_M_BATT_DOM | 0.556 | 0.301 | 0.179 | 0.446 | 0.163 | −0.442 |
| LS_R_M_CASE_DOM | 0.505 | 0.261 | −0.293 | 0.594 | −0.0268 | −0.0583 |
| LS_R_M_LEAD_DOM | 0.383 | 0.347 | 0.406 | −0.267 | −0.330 | −0.245 |
| RM_CASE | RM_LEAD | RF_CASE | RF_LEAD | R_CASE | |
|---|---|---|---|---|---|
| RM_CASE_DOM | −0.763 <0.0001 | 0.022 0.8986 | 0.041 0.8144 | −0.0489 0.7843 | −0.313 0.0669 |
| RM_LEAD_DOM | 0.103 0.5555 | −0.471 0.0043 | 0.042 0.8088 | 0.099 0.5717 | 0.081 0.6447 |
| RF_CASE_DOM | −0.043 0.8078 | 0.100 0.5654 | −0.604 0.0001 | 0.00000 1.0000 | −0.4011 0.0169 |
| RF_LEAD_DOM | 0.272 0.1140 | −0.415 0.0131 | −0.041 0.8144 | −0.480 0.0035 | 0.207 0.2333 |
| R_CASE_DOM | −0.480 0.0035 | 0.270 0.1167 | −0.339 0.0461 | 0.148 0.3948 | −0.746 <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, S.; Bossa, C.; Battistelli, C.L.; Giuliani, A. A Statistical Exploration of QSAR Models in Cancer Risk Assessment: A Case Study on Pesticide-Active Substances and Metabolites. Toxics 2025, 13, 299. https://doi.org/10.3390/toxics13040299
Greco S, Bossa C, Battistelli CL, Giuliani A. A Statistical Exploration of QSAR Models in Cancer Risk Assessment: A Case Study on Pesticide-Active Substances and Metabolites. Toxics. 2025; 13(4):299. https://doi.org/10.3390/toxics13040299
Chicago/Turabian StyleGreco, Serena, Cecilia Bossa, Chiara Laura Battistelli, and Alessandro Giuliani. 2025. "A Statistical Exploration of QSAR Models in Cancer Risk Assessment: A Case Study on Pesticide-Active Substances and Metabolites" Toxics 13, no. 4: 299. https://doi.org/10.3390/toxics13040299
APA StyleGreco, S., Bossa, C., Battistelli, C. L., & Giuliani, A. (2025). A Statistical Exploration of QSAR Models in Cancer Risk Assessment: A Case Study on Pesticide-Active Substances and Metabolites. Toxics, 13(4), 299. https://doi.org/10.3390/toxics13040299