
Polyhexamethylene Guanidine Phosphate Enhanced Procoagulant Activity through Oxidative-Stress-Mediated Phosphatidylserine Exposure in Platelets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Platelets
2.3. Flow Cytometric Analysis
2.4. Measurement of Intracellular ATP Level
2.5. Detection of Phospholipid Translocation
2.6. Thrombin Generation Assay
2.7. Statistical Analysis
3. Results
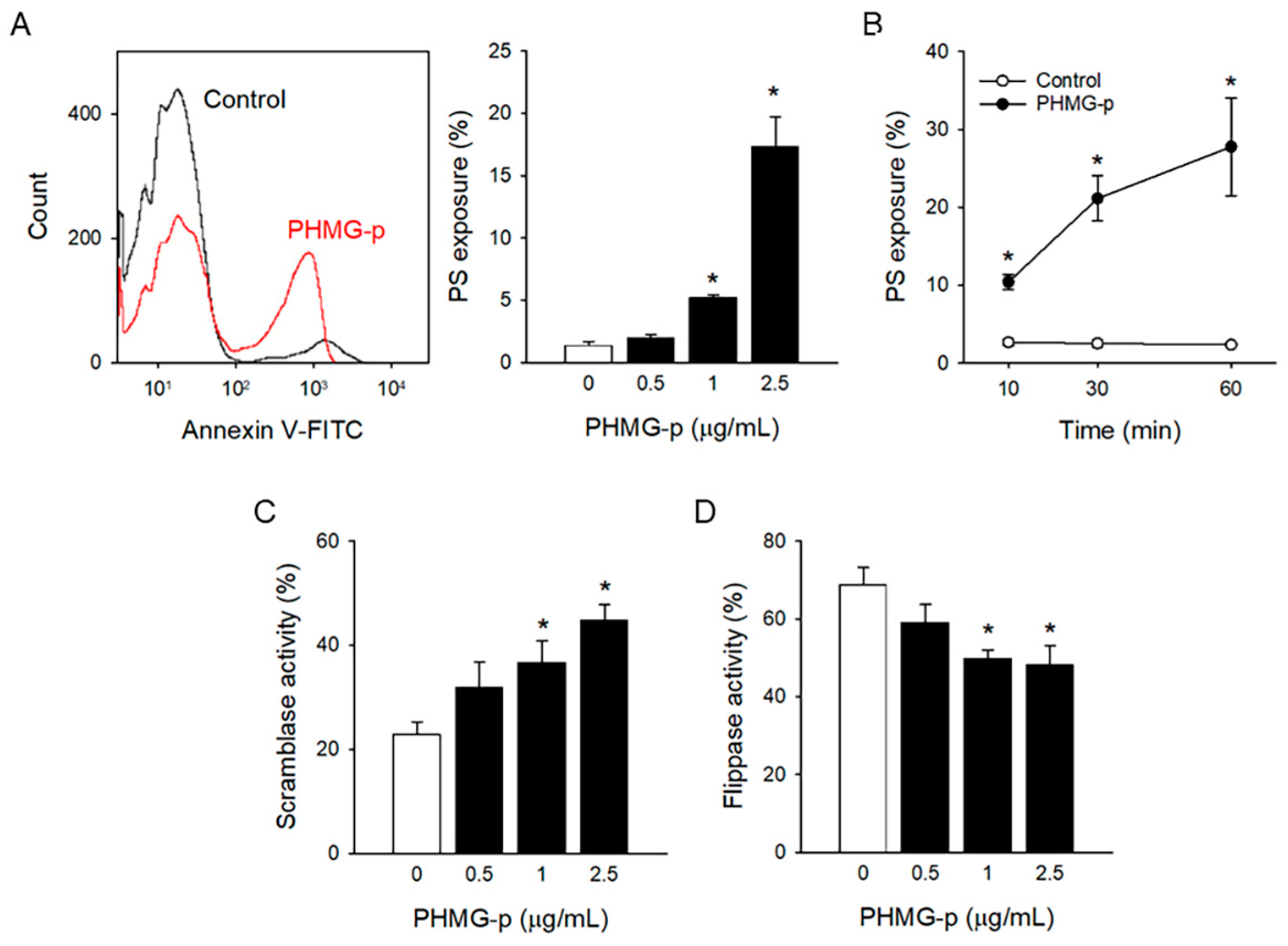
3.1. PHMG-p-Induced PS Exposure in Platelets
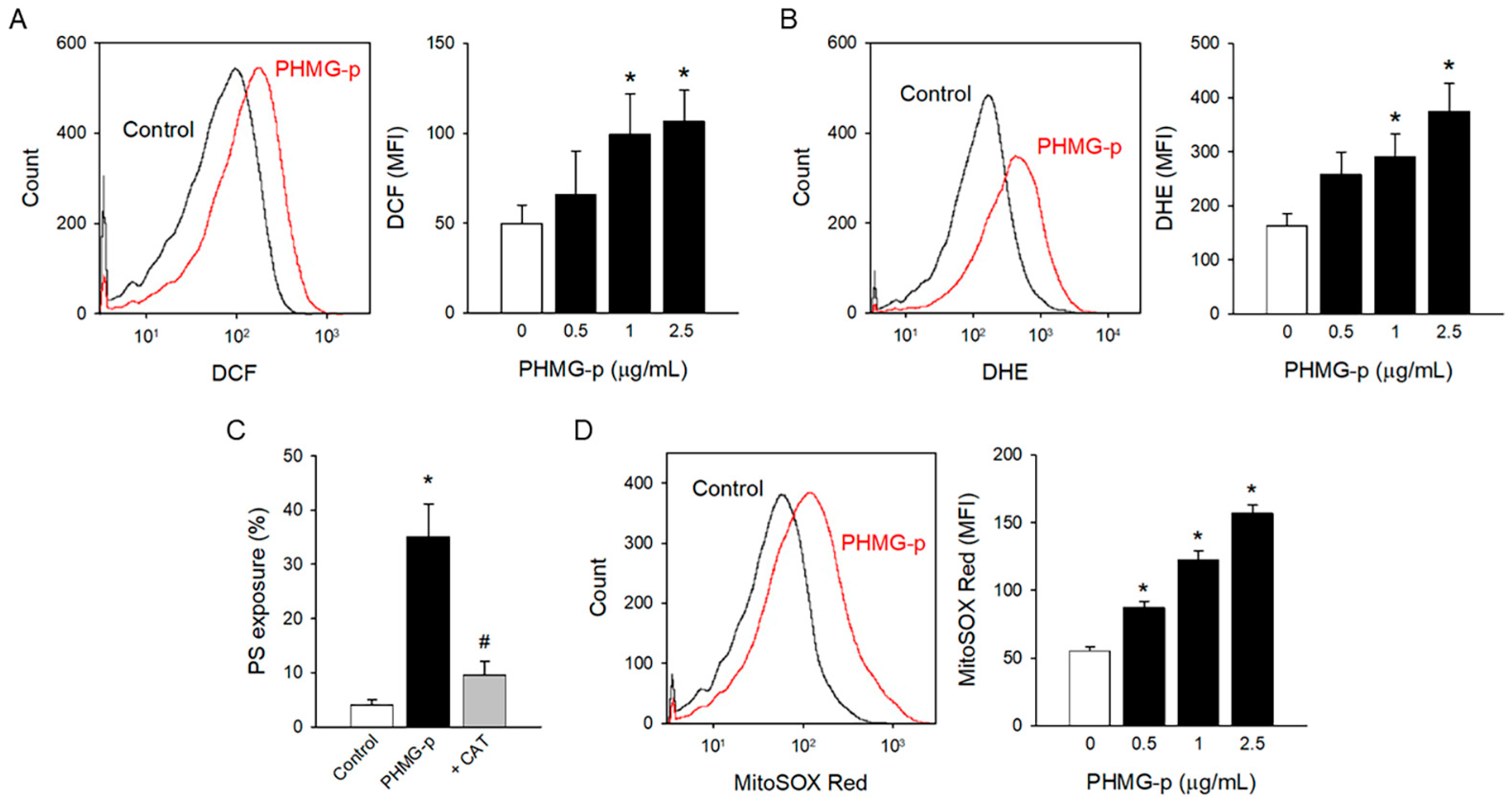
3.2. Oxidative Stress Induced by PHMG-p
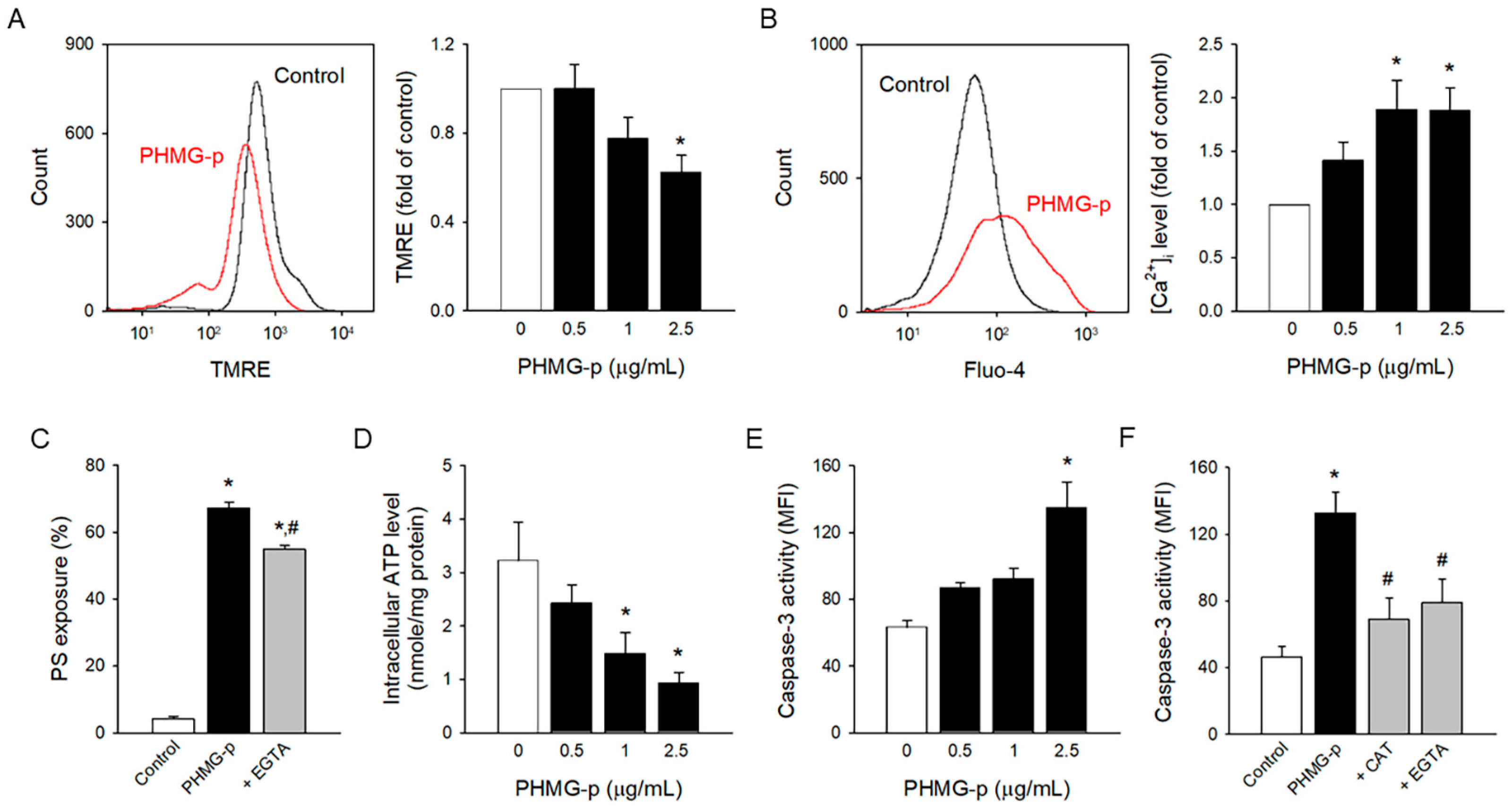
3.3. Intracellular Mechanism Underlying PHMG-p-Induced PS Exposure
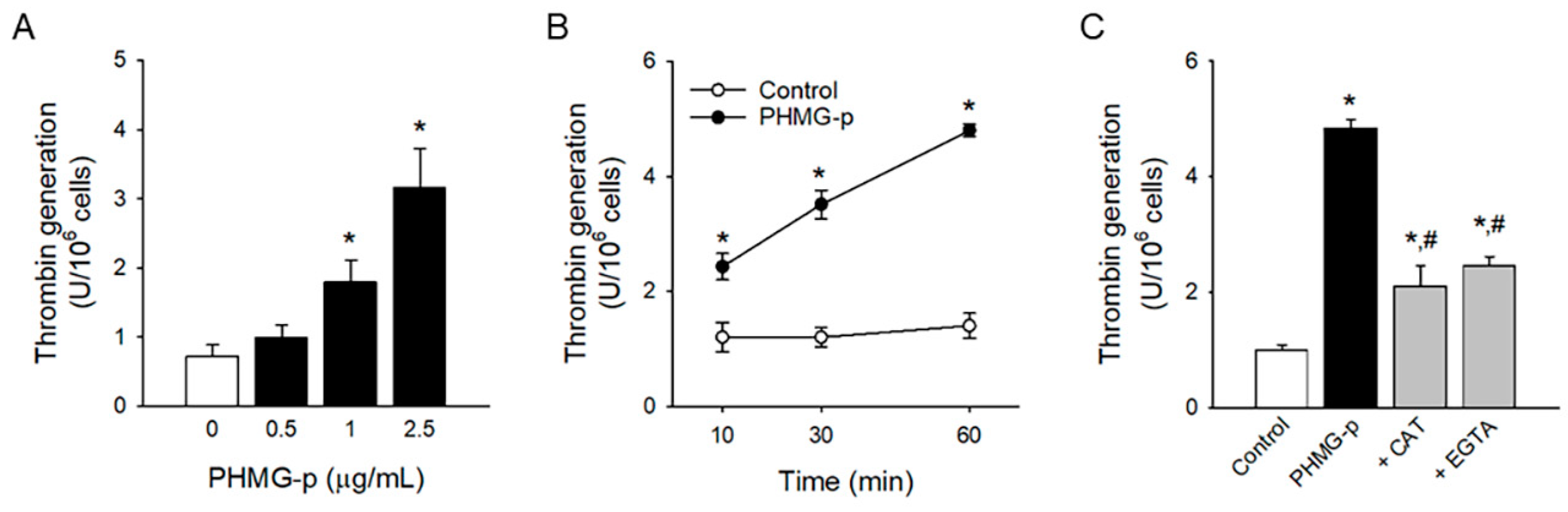
3.4. Procoagulant Effects of PHMG-p
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohta, S.; Misawa, Y.; Miyamoto, H.; Makino, M.; Nagai, K.; Shiraishi, T.; Nakagawa, Y.; Yamato, S.; Tachikawa, E.; Zenda, H. A comparative study of characteristics of current-type and conventional-type cationic bactericides. Biol. Pharm. Bull. 2001, 24, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, A.; Kratzer, C.; Graninger, W.; Georgopoulos, A. Antimicrobial and toxicological profile of the new biocide Akacid plus. J. Antimicrob. Chemother. 2006, 58, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Oule, M.K.; Quinn, K.; Dickman, M.; Bernier, A.M.; Rondeau, S.; De Moissac, D.; Boisvert, A.; Diop, L. Akwaton, polyhexamethylene-guanidine hydrochloride-based sporicidal disinfectant: A novel tool to fight bacterial spores and nosocomial infections. J. Med. Microbiol. 2012, 61, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Vitt, A.; Sofrata, A.; Slizen, V.; Sugars, R.V.; Gustafsson, A.; Gudkova, E.I.; Kazeko, L.A.; Ramberg, P.; Buhlin, K. Antimicrobial activity of polyhexamethylene guanidine phosphate in comparison to chlorhexidine using the quantitative suspension method. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Oule, M.K.; Azinwi, R.; Bernier, A.M.; Kablan, T.; Maupertuis, A.M.; Mauler, S.; Nevry, R.K.; Dembele, K.; Forbes, L.; Diop, L. Polyhexamethylene guanidine hydrochloride-based disinfectant: A novel tool to fight meticillin-resistant Staphylococcus aureus and nosocomial infections. J. Med. Microbiol. 2008, 57, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Cationic antimicrobial polymers and their assemblies. Int. J. Mol. Sci. 2013, 14, 9906–9946. [Google Scholar] [CrossRef]
- Gilbert, P.; Moore, L.E. Cationic antiseptics: Diversity of action under a common epithet. J. Appl. Microbiol. 2005, 99, 703–715. [Google Scholar] [CrossRef]
- Zhou, Z.; Zheng, A.; Zhong, J. Interactions of biocidal guanidine hydrochloride polymer analogs with model membranes: A comparative biophysical study. Acta Biochim. Biophys. Sin. 2011, 43, 729–737. [Google Scholar] [CrossRef]
- Aleshina, E.Y.; Yudanova, T.N.; Skokova, I.F. Production and properties of polyvinyl alcohol spinning solutions containing protease C and polyhexamethylene guanidine. Fibre Chem. 2001, 33, 421–423. [Google Scholar] [CrossRef]
- National Industrial Chemical Notification and Assessment Scheme (NICNAS). Full Public Report Polyhexamethyleneguanidine Phosphate; Department of Health and Aged Care, Austrailian Government: Sydney, Australia, 2003. [Google Scholar]
- Ju, Y.J.; Lee, S.; Sheen, S.; Choi, D.W.; Leem, J.H.; Lee, S.Y. A comprehensive study of deaths due to exposure to humidifier disinfectant in Korea: Focusing on medical records, assessment of exposure to humidifier disinfectants, and causes of death. Epidemiol. Health 2021, 43, e2021091. [Google Scholar] [CrossRef]
- The Korean Society of Preventive Medicine. A Study on Health Damage of Humidifier Disinfectants Using National Helath Insurance Big Data; NIER-SP2018-302; National Institute of Environmental Research: Incheon, Republic of Korea, 2019. [Google Scholar]
- Paek, D. Assessment of Information System of Humidifier Disinfectant Damage Relief Management—Investigation and Recognition of Damage; NIER-SP2018-304; National Institute of Environmental Research: Incheon, Republic of Korea, 2019. [Google Scholar]
- Paek, D.; Koh, Y.; Park, D.U.; Cheong, H.K.; Do, K.H.; Lim, C.M.; Hong, S.J.; Kim, Y.H.; Leem, J.H.; Chung, K.H.; et al. Nationwide Study of Humidifier Disinfectant Lung Injury in South Korea, 1994–2011. Incidence and Dose-Response Relationships. Ann. Am. Thorac. Soc. 2015, 12, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Cho, H.J.; Lee, E.; Choi, Y.J.; Kim, Y.H.; Lee, J.L.; Lee, Y.J.; Hong, S.J. Rate of humidifier and humidifier disinfectant usage in Korean children: A nationwide epidemiologic study. Environ. Res. 2017, 155, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Park, D.U.; Ryu, S.H.; Lim, H.K.; Kim, S.K.; Choi, Y.Y.; Ahn, J.J.; Lee, E.; Hong, S.B.; Do, K.H.; Cho, J.L.; et al. Types of household humidifier disinfectant and associated risk of lung injury (HDLI) in South Korea. Sci. Total Environ. 2017, 596–597, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.E.; Hong, S.B.; Do, K.H.; Kim, H.J.; Chung, S.; Lee, E.; Choi, J.; Hong, S.J. Humidifier disinfectant lung injury, how do we approach the issues? Environ. Health Toxicol. 2016, 31, e2016019. [Google Scholar] [CrossRef]
- Hong, M.; Ju, M.J.; Yoon, J.; Lee, W.; Lee, S.; Jo, E.K.; Choi, S.Y.; Yang, W.; Choi, Y.H. Exposures to humidifier disinfectant and various health conditions in Korean based on personal exposure assessment data of claimants for compensation. BMC Public Health 2023, 23, 1800. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, H.H.; Cho, K.H. Acute cardiovascular toxicity of sterilizers, PHMG, and PGH: Severe inflammation in human cells and heart failure in zebrafish. Cardiovasc. Toxicol. 2013, 13, 148–160. [Google Scholar] [CrossRef]
- Do, V.Q.; Seo, Y.S.; Park, J.M.; Yu, J.; Duong, M.T.H.; Nakai, J.; Kim, S.K.; Ahn, H.C.; Lee, M.Y. A mixture of chloromethylisothiazolinone and methylisothiazolinone impairs rat vascular smooth muscle by depleting thiols and thereby elevating cytosolic Zn(2+) and generating reactive oxygen species. Arch. Toxicol. 2021, 95, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Shin, Y.; Kim, E.H.; Lee, Y.; Kim, S.; Kim, H.S.; Kim, H.C.; Leem, J.H.; Kim, H.R.; Bae, O.N. Functional and dynamic mitochondrial damage by chloromethylisothiazolinone/methylisothiazolinone (CMIT/MIT) mixture in brain endothelial cell lines and rat cerebrovascular endothelium. Toxicol. Lett. 2022, 366, 45–57. [Google Scholar] [CrossRef]
- Raskob, G.E.; Angchaisuksiri, P.; Blanco, A.N.; Buller, H.; Gallus, A.; Hunt, B.J.; Hylek, E.M.; Kakkar, A.; Konstantinides, S.V.; McCumber, M.; et al. Thrombosis: A major contributor to global disease burden. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2363–2371. [Google Scholar] [CrossRef]
- Alkarithi, G.; Duval, C.; Shi, Y.; Macrae, F.L.; Ariens, R.A.S. Thrombus Structural Composition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2370–2383. [Google Scholar] [CrossRef]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791. [Google Scholar] [CrossRef]
- Nieswandt, B.; Pleines, I.; Bender, M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 92–104. [Google Scholar] [CrossRef]
- Agbani, E.O.; Poole, A.W. Procoagulant platelets: Generation, function, and therapeutic targeting in thrombosis. Blood 2017, 130, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Campbell, R.A. Procoagulant platelets: Novel players in thromboinflammation. Am. J. Physiol. Cell Physiol. 2022, 323, C951–C958. [Google Scholar] [CrossRef] [PubMed]
- Podoplelova, N.A.; Sveshnikova, A.N.; Kotova, Y.N.; Eckly, A.; Receveur, N.; Nechipurenko, D.Y.; Obydennyi, S.I.; Kireev, I.I.; Gachet, C.; Ataullakhanov, F.I.; et al. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood 2016, 128, 1745–1755. [Google Scholar] [CrossRef]
- Wang, J.; Yu, C.; Zhuang, J.; Qi, W.; Jiang, J.; Liu, X.; Zhao, W.; Cao, Y.; Wu, H.; Qi, J.; et al. The role of phosphatidylserine on the membrane in immunity and blood coagulation. Biomark. Res. 2022, 10, 4. [Google Scholar] [CrossRef]
- Nagata, S.; Sakuragi, T.; Segawa, K. Flippase and scramblase for phosphatidylserine exposure. Curr. Opin. Immunol. 2020, 62, 31–38. [Google Scholar] [CrossRef]
- Kim, H.R.; Lee, K.; Park, C.W.; Song, J.A.; Shin, D.Y.; Park, Y.J.; Chung, K.H. Polyhexamethylene guanidine phosphate aerosol particles induce pulmonary inflammatory and fibrotic responses. Arch. Toxicol. 2016, 90, 617–632. [Google Scholar] [CrossRef]
- Kim, H.R.; Shin, D.Y.; Chung, K.H. The role of NF-kappaB signaling pathway in polyhexamethylene guanidine phosphate induced inflammatory response in mouse macrophage RAW264.7 cells. Toxicol. Lett. 2015, 233, 148–155. [Google Scholar] [CrossRef]
- Park, J.S.; Park, Y.J.; Kim, H.R.; Chung, K.H. Polyhexamethylene guanidine phosphate-induced ROS-mediated DNA damage caused cell cycle arrest and apoptosis in lung epithelial cells. J. Toxicol. Sci. 2019, 44, 415–424. [Google Scholar] [CrossRef]
- Song, J.; Jung, K.J.; Yang, M.J.; Han, S.C.; Lee, K. Assessment of acute and repeated pulmonary toxicities of oligo(2-(2-ethoxy)ethoxyethyl guanidium chloride in mice. Toxicol. Res. 2021, 37, 99–113. [Google Scholar] [CrossRef]
- Fuentes, E.; Araya-Maturana, R.; Urra, F.A. Regulation of mitochondrial function as a promising target in platelet activation-related diseases. Free Radic. Biol. Med. 2019, 136, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Choo, H.J.; Saafir, T.B.; Mkumba, L.; Wagner, M.B.; Jobe, S.M. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Arauna, D.; Fuentes, F.; Araya-Maturana, R.; Palomo, I.; Alarcon, M.; Sebastian, D.; Zorzano, A.; Fuentes, E. Mitoquinone (MitoQ) Inhibits Platelet Activation Steps by Reducing ROS Levels. Int. J. Mol. Sci. 2020, 21, 6192. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, F.; Chen, X.; Luo, M.; Hu, L.; Lu, Y. The role of mitochondria-derived reactive oxygen species in hyperthermia-induced platelet apoptosis. PLoS ONE 2013, 8, e75044. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Xie, R.; Liu, R.; Lu, Y. Mitochondria-derived reactive oxygen species play an important role in Doxorubicin-induced platelet apoptosis. Int. J. Mol. Sci. 2015, 16, 11087–11100. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and reactive oxygen species: Physiology and pathophysiology. Int. J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef]
- Arachiche, A.; Kerbiriou-Nabias, D.; Garcin, I.; Letellier, T.; Dachary-Prigent, J. Rapid procoagulant phosphatidylserine exposure relies on high cytosolic calcium rather than on mitochondrial depolarization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1883–1889. [Google Scholar] [CrossRef]
- Obydennyy, S.I.; Sveshnikova, A.N.; Ataullakhanov, F.I.; Panteleev, M.A. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 2016, 14, 1867–1881. [Google Scholar] [CrossRef] [PubMed]
- Leytin, V.; Allen, D.J.; Mutlu, A.; Gyulkhandanyan, A.V.; Mykhaylov, S.; Freedman, J. Mitochondrial control of platelet apoptosis: Effect of cyclosporin A, an inhibitor of the mitochondrial permeability transition pore. Lab. Investig. 2009, 89, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Kroemer, G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 2013, 23, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016, 23, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Paliienko, K.O.; Veklich, T.O.; Shatursky, O.Y.; Shkrabak, O.A.; Pastukhov, A.O.; Galkin, M.O.; Krisanova, N.V.; Chunikhin, A.J.; Rebriev, A.V.; Lysytsya, A.V.; et al. Membrane action of polyhexamethylene guanidine hydrochloride revealed on smooth muscle cells, nerve tissue and rat blood platelets: A biocide driven pore-formation in phospholipid bilayers. Toxicol. Vitr. 2019, 60, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Tomasiak, M.; Stelmach, H.; Rusak, T.; Ciborowski, M.; Radziwon, P. The involvement of Na+/K(+)-ATPase in the development of platelet procoagulant response. Acta Biochim. Pol. 2007, 54, 625–639. [Google Scholar] [CrossRef]
- Song, J.; Jung, K.J.; Yoon, S.J.; Lee, K.; Kim, B. Polyhexamethyleneguanidine phosphate induces cytotoxicity through disruption of membrane integrity. Toxicology 2019, 414, 35–44. [Google Scholar] [CrossRef]
- Jung, H.N.; Zerin, T.; Podder, B.; Song, H.Y.; Kim, Y.S. Cytotoxicity and gene expression profiling of polyhexamethylene guanidine hydrochloride in human alveolar A549 cells. Toxicol. Vitr. 2014, 28, 684–692. [Google Scholar] [CrossRef]
- Park, E.J.; Park, S.J.; Kim, S.; Lee, K.; Chang, J. Lung fibroblasts may play an important role in clearing apoptotic bodies of bronchial epithelial cells generated by exposure to PHMG-P-containing solution. Toxicol. Lett. 2018, 286, 108–119. [Google Scholar] [CrossRef]
- Kim, S.H.; Kwon, D.; Lee, S.; Ki, S.H.; Jeong, H.G.; Hong, J.T.; Lee, Y.H.; Jung, Y.S. Polyhexamethyleneguanidine Phosphate-Induced Cytotoxicity in Liver Cells Is Alleviated by Tauroursodeoxycholic Acid (TUDCA) via a Reduction in Endoplasmic Reticulum Stress. Cells 2019, 8, 1023. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M. Procoagulant platelets: Are they necrotic? Blood 2010, 116, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, E.C.; Ramstrom, S.; Thaler, J.; Lordkipanidze, M.; COAGAPO study group. Consensus report on markers to distinguish procoagulant platelets from apoptotic platelets: Communication from the Scientific and Standardization Committee of the ISTH. J. Thromb. Haemost. 2023, 21, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Abbasian, N.; Millington-Burgess, S.L.; Chabra, S.; Malcor, J.D.; Harper, M.T. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv. 2020, 4, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Schoenwaelder, S.M.; Yuan, Y.; Josefsson, E.C.; White, M.J.; Yao, Y.; Mason, K.D.; O’Reilly, L.A.; Henley, K.J.; Ono, A.; Hsiao, S.; et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 2009, 114, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lim, K.M.; Noh, J.Y.; Kim, K.; Kang, S.; Chang, Y.K.; Shin, S.; Chung, J.H. Doxorubicin-induced platelet procoagulant activities: An important clue for chemotherapy-associated thrombosis. Toxicol. Sci. 2011, 124, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Song, J.S.; Lim, K.M.; Kang, S.; Noh, J.Y.; Kim, K.; Bae, O.N.; Chung, J.H. Procoagulant and prothrombotic effects of the herbal medicine, Dipsacus asper and its active ingredient, dipsacus saponin C, on human platelets. J. Thromb. Haemost. 2012, 10, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, S.; Park, J.E.; Shim, H.E.; Lee, C.H.; Shin, H.S.; Lee, S.Y.; Jeon, J. Study on biological distribution of polyhexamethylene guanidine (PHMG), a toxic household chemical, using radiolabeling and molecular imaging tools. Environ. Eng. Res. 2022, 27, 210393. [Google Scholar] [CrossRef]
- Yu, Y.; Leng, T.; Yun, D.; Liu, N.; Yao, J.; Dai, Y.; Yang, P.; Chen, X. Global analysis of the rat and human platelet proteome—The molecular blueprint for illustrating multi-functional platelets and cross-species function evolution. Proteomics 2010, 10, 2444–2457. [Google Scholar] [CrossRef]
- Oshinowo, O.; Copeland, R.; Sakurai, Y.; Fay, M.E.; Petrich, B.G.; Leong, T.; Brainard, B.; Lam, W.A. Significant differences in single-platelet biophysics exist across species but attenuate during clot formation. Blood Adv. 2021, 5, 432–437. [Google Scholar] [CrossRef]
- Toor, B.; McGregor, J.L.; McGregor, L.; Clemetson, K.J. Comparison of the major membrane glycoproteins and proteins of human, rabbit and rat blood platelets. Thromb. Res. 1982, 26, 317–328. [Google Scholar] [CrossRef]
- Kinlough-Rathbone, R.L.; Rand, M.L.; Packham, M.A. Rabbit and rat platelets do not respond to thrombin receptor peptides that activate human platelets. Blood 1993, 82, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, R.; Dubois, C.; Leroyer, A.S.; Sabatier, F.; Dignat-George, F. Revisited role of microparticles in arterial and venous thrombosis. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 24–35. [Google Scholar] [CrossRef] [PubMed]
- Bidot, L.; Jy, W.; Bidot, C., Jr.; Jimenez, J.J.; Fontana, V.; Horstman, L.L.; Ahn, Y.S. Microparticle-mediated thrombin generation assay: Increased activity in patients with recurrent thrombosis. J. Thromb. Haemost. 2008, 6, 913–919. [Google Scholar] [CrossRef]
- Lee, Y.H.; Seo, D.S.; Lee, M.J.; Cha, H.G. Immunohistochemical characterization of oxidative stress in the lungs of rats exposed to the humidifier disinfectant polyhexamethylene guanidine hydrochloride. J. Toxicol. Pathol. 2019, 32, 311–317. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.H.; Kim, K. Polyhexamethylene Guanidine Phosphate Enhanced Procoagulant Activity through Oxidative-Stress-Mediated Phosphatidylserine Exposure in Platelets. Toxics 2024, 12, 50. https://doi.org/10.3390/toxics12010050
Choi JH, Kim K. Polyhexamethylene Guanidine Phosphate Enhanced Procoagulant Activity through Oxidative-Stress-Mediated Phosphatidylserine Exposure in Platelets. Toxics. 2024; 12(1):50. https://doi.org/10.3390/toxics12010050
Chicago/Turabian StyleChoi, Ju Hee, and Keunyoung Kim. 2024. "Polyhexamethylene Guanidine Phosphate Enhanced Procoagulant Activity through Oxidative-Stress-Mediated Phosphatidylserine Exposure in Platelets" Toxics 12, no. 1: 50. https://doi.org/10.3390/toxics12010050
APA StyleChoi, J. H., & Kim, K. (2024). Polyhexamethylene Guanidine Phosphate Enhanced Procoagulant Activity through Oxidative-Stress-Mediated Phosphatidylserine Exposure in Platelets. Toxics, 12(1), 50. https://doi.org/10.3390/toxics12010050