


Comparable Response Following Exposure to Biodiesel and Diesel Exhaust Particles in Advanced Multicellular Human Lung Models



 , , ,
, , ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. BDEP and DEP Generation, Sampling and Chemical Characterization
2.2. Cell Culture
2.2.1. Human Pulmonary Bronchial Epithelial Cell (PBEC)
2.2.2. THP-1 Derived Macrophage (MQ) and MQ-ALI Monocultures
2.2.3. PBEC-ALI and MQ Co-Culture
2.3. BDEP and DEP Concentration Determination
2.4. Exposure of PBEC-ALI, MQ-ALI and PBEC-ALI/MQ to BDEP or DEP
2.5. Reactive Oxygen Species (ROS) Measurement
2.6. Apoptosis and Cell Viability
2.7. Glutathione Measurement
2.8. ELISA
2.9. RT-qPCR
2.10. Macrophage Polarization Markers CD86 and CD206
2.11. Phospholipid Measurement
2.12. Histone Phosphorylation and DNA Damage Assay
2.13. Prostaglandin E2 Inhibition
2.14. Phagocytosis Assay
2.15. Statistical Analysis
3. Results
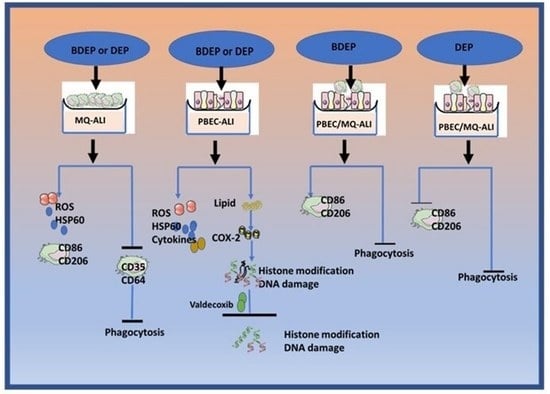
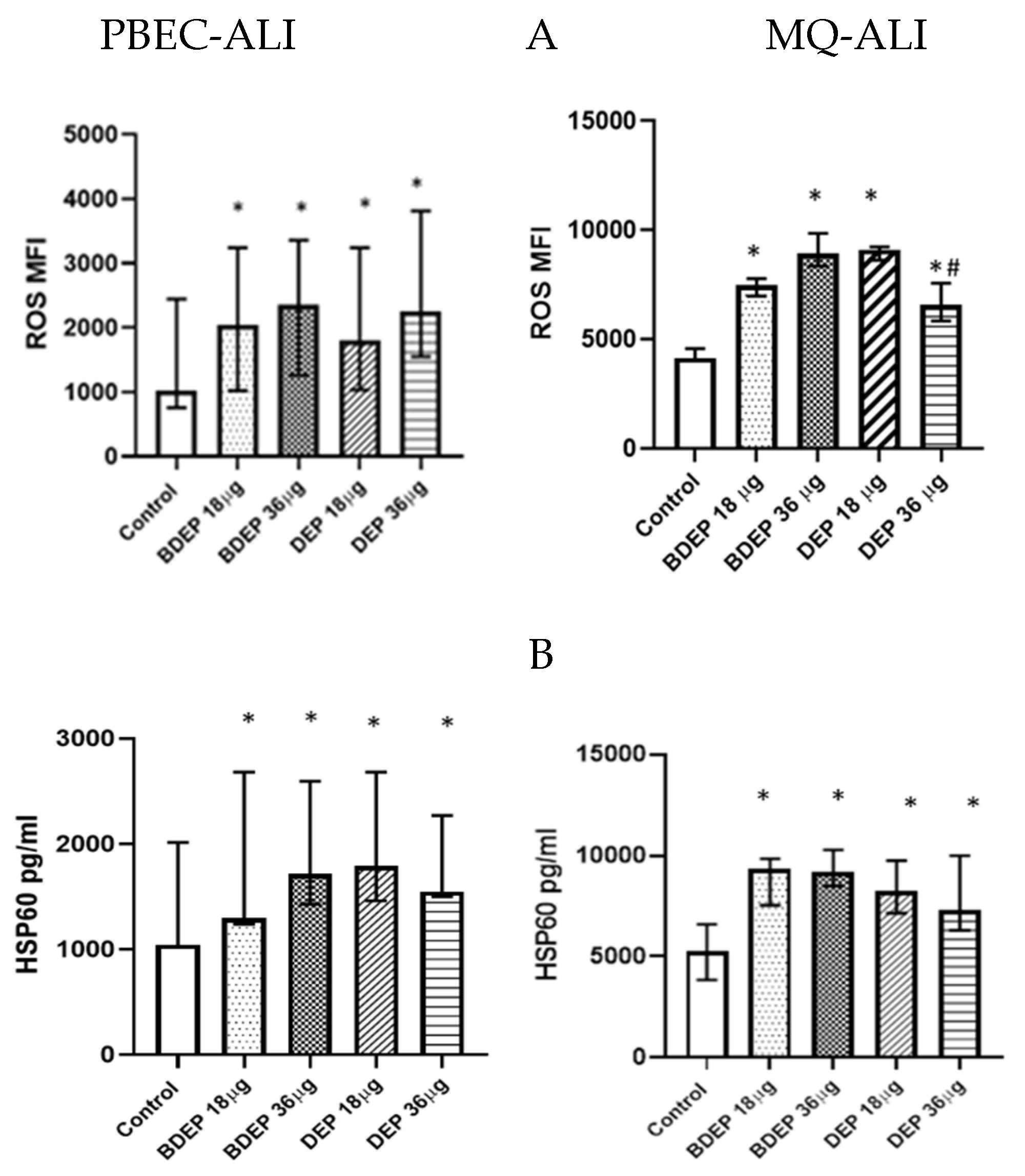
3.1. Oxidative Stress Induced by Exposure to BDEP and PDEP in PBEC-ALI and MQ-ALI
3.2. Cell Viability
3.3. Antioxidant Response against BDEP or DEP-Induced Oxidative Damage
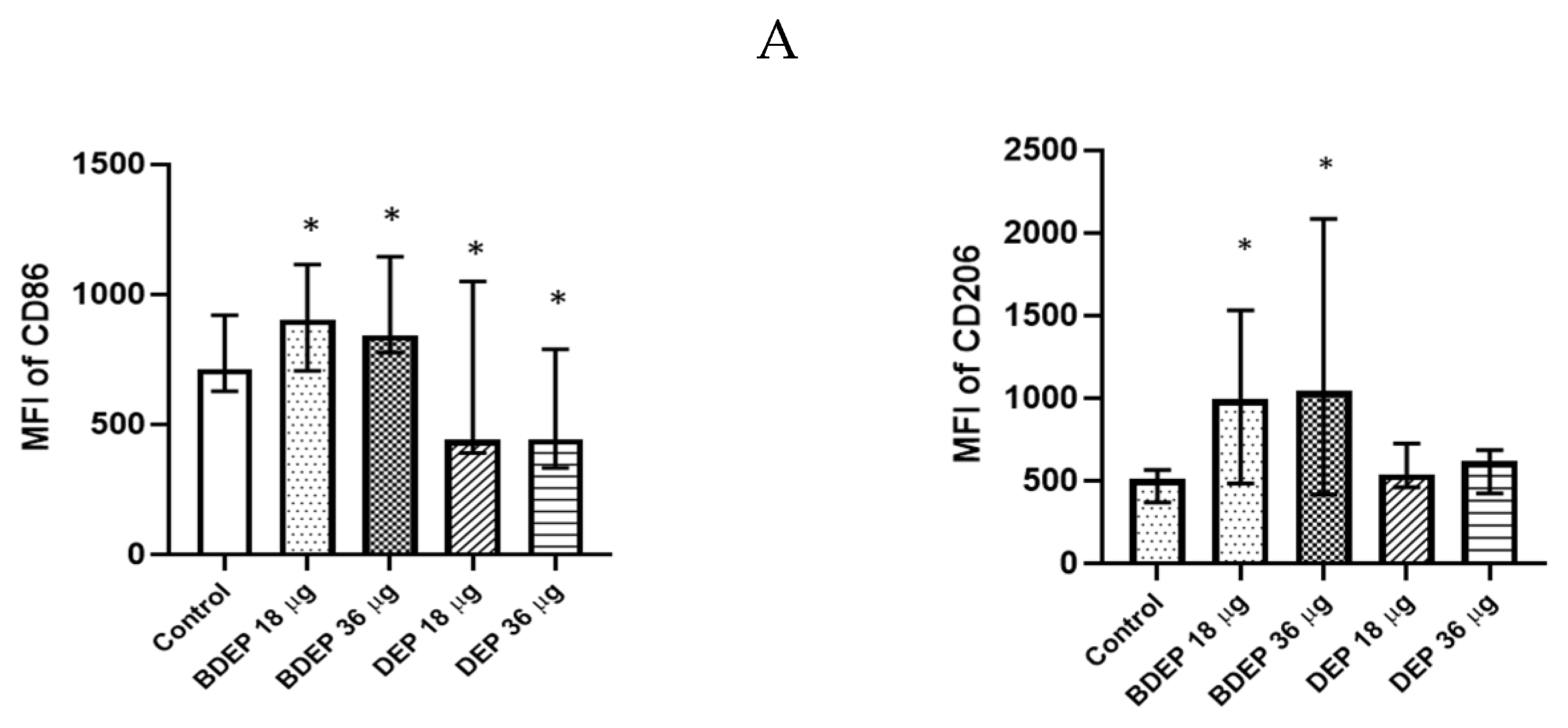
3.4. BDEP and DEP Induced MQ Polarization Surface Markers Expression
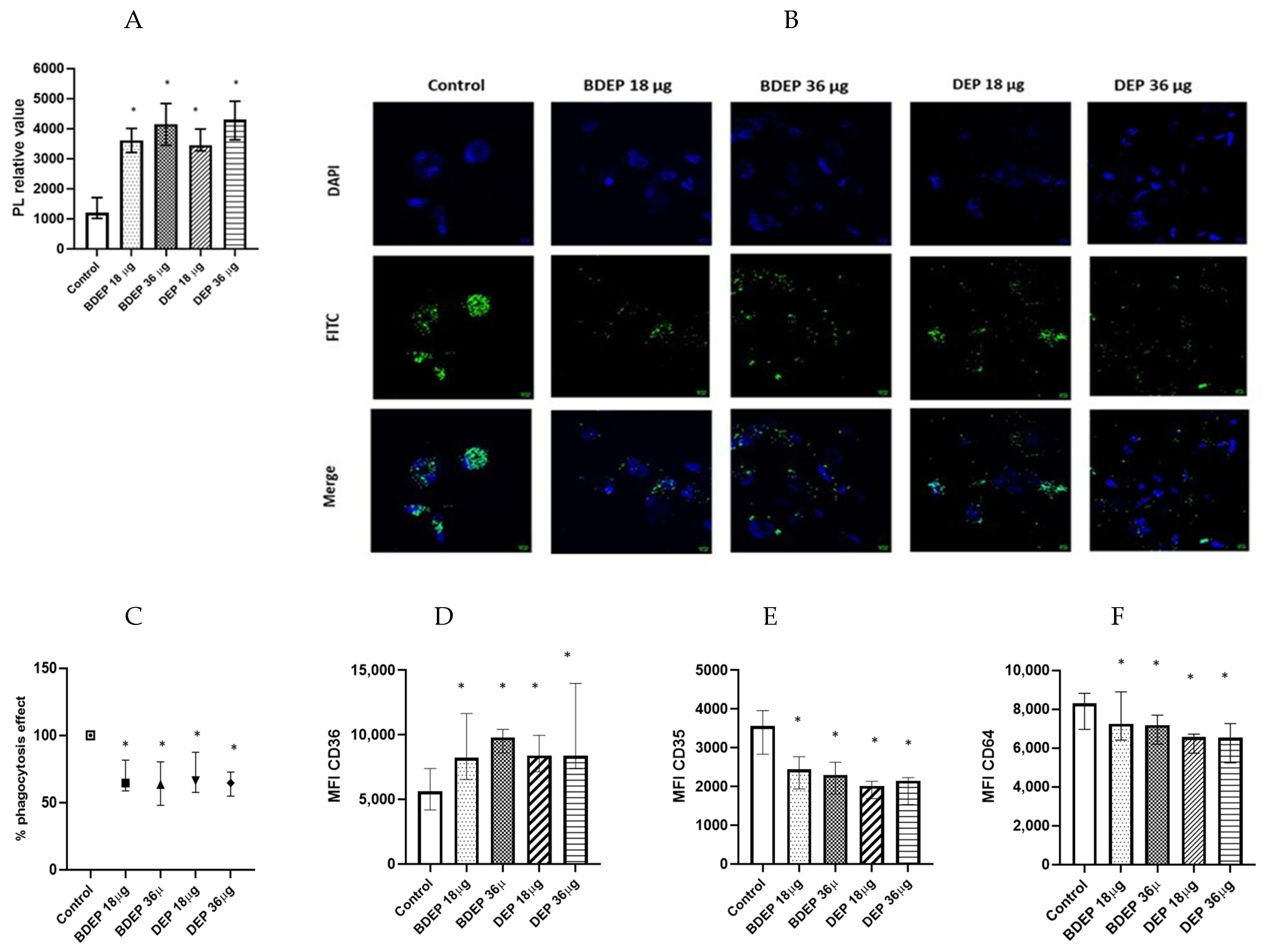
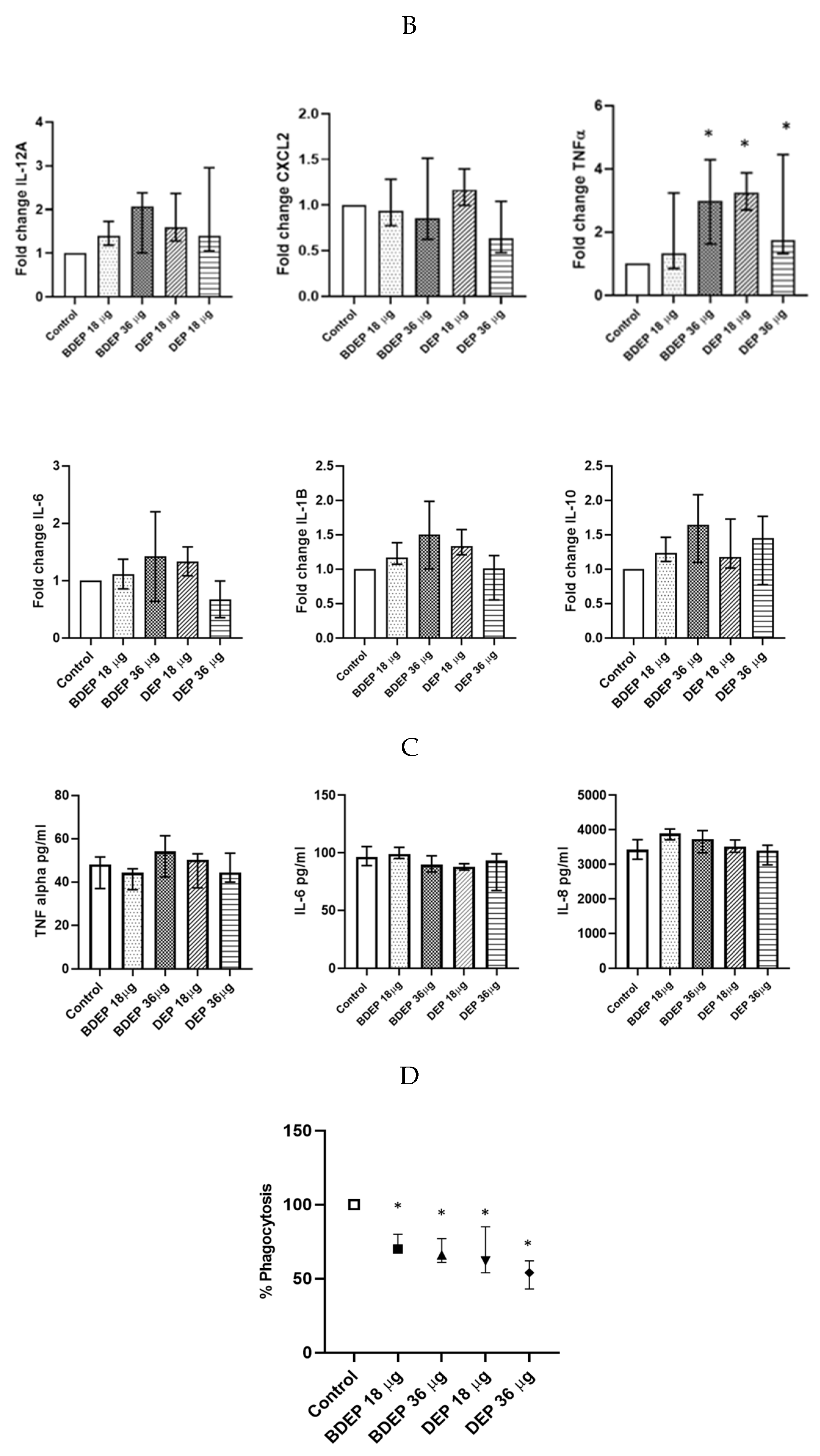
3.5. BDEP and DEP Increased Lipid Levels, but Reduced Phagocytic Activity in MQ-ALI
3.6. BDEP and DEP Induced Inflammation in PBEC-ALI Monocultures
3.7. BDEP and DEP Affect COX-2/PGE2 Pathway
3.8. BDEP and DEP Induced COX-2 Mediated DNA Damage
3.9. The Effect of DEP and BDEP Exposure in Co-Culture of PBEC and MQ
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, X.-Q.; Mei, X.-D.; Feng, D. Air pollution and chronic airway diseases: What should people know and do? J. Thorac. Dis. 2016, 8, E31–E40. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.who.int/news/item/22-09-2021-new-who-global-air-quality-guidelines-aim-to-save-millions-of-lives-from-air-pollution (accessed on 22 September 2021).
- Academy of Science Of South Africa; Brazilian Academy of Sciences; German National Academy of Sciences Leopoldina; U. S. National Academy of Medicine; U. S. National Academy of Sciences. Air Pollution and Health—A Science-Policy Initiative. Ann. Glob. Health 2019, 85, 140. [Google Scholar] [CrossRef] [PubMed]
- Doiron, D.; De Hoogh, K.; Probst-Hensch, N.; Fortier, I.; Cai, Y.; De Matteis, S.; Hansell, A.L. Air pollution, lung function and COPD: Results from the population-based UK Biobank study. Eur. Respir. J. 2019, 54, 1802140. [Google Scholar] [CrossRef] [PubMed]
- Sydbom, A.; Blomberg, A.; Parnia, S.; Stenfors, N.; Sandström, T.; Dahlén, S.-E. Health effects of diesel exhaust emissions. Eur. Respir. J. 2001, 17, 733–746. [Google Scholar] [CrossRef]
- Salvi, S.; Blomberg, A.; Rudell, B.; Kelly, F.; Sandström, T.; Holgate, S.T.; Frew, A. Acute Inflammatory Responses in the Airways and Peripheral Blood After Short-Term Exposure to Diesel Exhaust in Healthy Human Volunteers. Am. J. Respir. Crit. Care Med. 1999, 159, 702–709. [Google Scholar] [CrossRef]
- Behndig, A.F.; Shanmuganathan, K.; Whitmarsh, L.; Stenfors, N.; Brown, J.L.; Frew, A.J.; Kelly, F.J.; Mudway, I.S.; Sandström, T.; Wilson, S.J. Effects of controlled diesel exhaust exposure on apoptosis and proliferation markers in bronchial epithelium—An in vivo bronchoscopy study on asthmatics, rhinitics and healthy subjects. BMC Pulm. Med. 2015, 15, 99. [Google Scholar] [CrossRef]
- Pourazar, J.; Blomberg, A.; Kelly, F.J.; Davies, D.E.; Wilson, S.J.; Holgate, S.T.; Sandström, T. Diesel exhaust increases EGFR and phosphorylated C-terminal Tyr 1173 in the bronchial epithelium. Part. Fibre Toxicol. 2008, 5, 8. [Google Scholar] [CrossRef]
- Mills, N.L.; Törnqvist, H.; Robinson, S.D.; Gonzalez, M.; Darnley, K.; MacNee, W.; Boon, N.A.; Donaldson, K.; Blomberg, A.; Sandstrom, T.; et al. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation 2005, 112, 3930–3936. [Google Scholar] [CrossRef]
- Cosselman, K.E.; Krishnan, R.M.; Oron, A.P.; Jansen, K.; Peretz, A.; Sullivan, J.H.; Larson, T.V.; Kaufman, J.D. Blood Pressure Response to Controlled Diesel Exhaust Exposure in Human Subjects. Hypertension 2012, 59, 943–948. [Google Scholar] [CrossRef]
- Lundbäck, M.; Mills, N.L.; Lucking, A.; Barath, S.; Donaldson, K.; Newby, D.E.; Sandström, T.; Blomberg, A. Experimental exposure to diesel exhaust increases arterial stiffness in man. Part. Fibre Toxicol. 2009, 6, 7. [Google Scholar] [CrossRef]
- Wauters, A.; Vicenzi, M.; De Becker, B.; Riga, J.P.; Esmaeilzadeh, F.; Faoro, V.; Vachiéry, J.L.; van de Borne, P.; Argacha, J.F. At high cardiac output, diesel exhaust exposure increases pulmonary vascular resistance and decreases distensibility of pulmonary resistive vessels. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H2137–H2144. [Google Scholar] [CrossRef]
- Lucking, A.J.; Lundback, M.; Mills, N.L.; Faratian, D.; Barath, S.L.; Pourazar, J.; Cassee, F.R.; Donaldson, K.; Boon, N.A.; Badimon, J.J.; et al. Diesel exhaust inhalation increases thrombus formation in man. Eur. Heart J. 2008, 29, 3043–3051. [Google Scholar] [CrossRef]
- Kim, D.I.; Song, M.-K.; Lee, K. Diesel Exhaust Particulates Enhances Susceptibility of LPS-Induced Acute Lung Injury through Upregulation of the IL-17 Cytokine-Derived TGF-β1/Collagen I Expression and Activation of NLRP3 Inflammasome Signaling in Mice. Biomolecules 2021, 11, 67. [Google Scholar] [CrossRef]
- Carlsten, C.; Blomberg, A.; Pui, M.; Sandstrom, T.; Wong, S.W.; Alexis, N.; Hirota, J. Diesel exhaust augments allergen-induced lower airway inflammation in allergic individuals: A controlled human exposure study. Thorax 2016, 71, 35–44, Erratum in: Thorax 2016, 71, 385. [Google Scholar] [CrossRef]
- Ryu, M.H.; Afshar, T.; Li, H.; Wooding, D.J.; Orach, J.; Zhou, J.S.; Murphy, S.; Lau, K.S.K.; Schwartz, C.; Yuen, A.C.Y.; et al. Impact of Exposure to Diesel Exhaust on Inflammation Markers and Proteases in Former Smokers with Chronic Obstructive Pulmonary Disease: A Randomized, Double-blinded, Crossover Study. Am. J. Respir. Crit. Care Med. 2022, 205, 1046–1052. [Google Scholar] [CrossRef]
- Ji, J.; Upadhyay, S.; Xiong, X.; Malmlöf, M.; Sandström, T.; Gerde, P.; Palmberg, L. Multi-cellular human bronchial models exposed to diesel exhaust particles: Assessment of inflammation, oxidative stress and macrophage polarization. Part. Fibre Toxicol. 2018, 15, 19. [Google Scholar] [CrossRef]
- Ahn, E.-K.; Yoon, H.-K.; Jee, B.K.; Ko, H.-J.; Lee, K.-H.; Kim, H.J.; Lim, Y. COX-2 expression and inflammatory effects by diesel exhaust particles in vitro and in vivo. Toxicol. Lett. 2008, 176, 178–187. [Google Scholar] [CrossRef]
- Rudra-Ganguly, N.; Reddy, S.T.; Korge, P.; Herschman, H.R. Diesel Exhaust Particle Extracts and Associated Polycyclic Aromatic Hydrocarbons Inhibit Cox-2-dependent Prostaglandin Synthesis in Murine Macrophages and Fibroblasts. J. Biol. Chem. 2002, 277, 39259–39265. [Google Scholar] [CrossRef]
- Cao, D.; Bromberg, P.A.; Samet, J.M. COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1. Am. J. Respir. Cell Mol. Biol. 2007, 37, 232–239. [Google Scholar] [CrossRef]
- Dagouassat, M.; Gagliolo, J.M.; Chrusciel, S.; Bourin, M.C.; Duprez, C.; Caramelle, P.; Boyer, L.; Hue, S.; Stern, J.B.; Validire, P.; et al. The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am. J. Respir. Crit. Care Med. 2013, 187, 703–714. [Google Scholar] [CrossRef]
- Zaslona, Z.; Serezani, C.H.; Okunishi, K.; Aronoff, D.; Peters-Golden, M. Prostaglandin E2 restrains macrophage maturation via E prostanoid receptor 2/protein kinase A signaling. Blood 2012, 119, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Gómez-Abellán, V.; Arizcun, M.; Mulero, V.; Sepulcre, M.P. Prostaglandin E2 promotes M2 polarization of macrophages via a cAMP/CREB signaling pathway and deactivates granulocytes in teleost fish. Fish Shellfish. Immunol. 2016, 55, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, N.K.; Li, M.; Poynter, M.E.; Palmer, B.C.; Parker, E.; Kasumba, J.; Holmén, B.A. Soy Biodiesel and Petrodiesel Emissions Differ in Size, Chemical Composition and Stimulation of Inflammatory Responses in Cells and Animals. Environ. Sci. Technol. 2013, 47, 12496–12504. [Google Scholar] [CrossRef] [PubMed]
- Landwehr, K.R.; Hillas, J.; Mead-Hunter, R.; O’leary, R.A.; Kicic, A.; Mullins, B.J.; Larcombe, A.N.; AusREC; WAERP. Soy Biodiesel Exhaust is More Toxic than Mineral Diesel Exhaust in Primary Human Airway Epithelial Cells. Environ. Sci. Technol. 2019, 53, 11437–11446. [Google Scholar] [CrossRef] [PubMed]
- Mullins, B.J.; Kicic, A.; Ling, K.M.; Mead-Hunter, R.; Larcombe, A.N. Biodiesel exhaust-induced cytotoxicity and proinflammatory mediator production in human airway epithelial cells. Environ. Toxicol. 2016, 31, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.; Stevanovic, S.; Banks, A.P.W.; Zare, A.; Rahman, M.; Bowman, R.V.; Fong, K.M.; Ristovski, Z.D.; Yang, I.A. The cytotoxic, inflammatory and oxidative potential of coconut oil-substituted diesel emissions on bronchial epithelial cells at an air-liquid interface. Environ. Sci. Pollut. Res. 2019, 26, 27783–27791. [Google Scholar] [CrossRef]
- Unosson, J.; Kabéle, M.; Boman, C.; Nyström, R.; Sadiktsis, I.; Westerholm, R.; Mudway, I.S.; Purdie, E.; Raftis, J.; Miller, M.R.; et al. Acute cardiovascular effects of controlled exposure to dilute Petrodiesel and biodiesel exhaust in healthy volunteers: A crossover study. Part. Fibre Toxicol. 2021, 18, 22. [Google Scholar] [CrossRef]
- Dayem, A.A.; Hossain, M.K.; Lee, S.B.; Kim, K.; Saha, S.K.; Yang, G.-M.; Choi, H.Y.; Cho, S.-G. The Role of Reactive Oxygen Species (ROS) in the Biological Activities of Metallic Nanoparticles. Int. J. Mol. Sci. 2017, 18, 120. [Google Scholar] [CrossRef]
- Wu, C.-W.; Biggar, K.K.; Zhang, J.; Tessier, S.N.; Pifferi, F.; Perret, M.; Storey, K.B. Induction of Antioxidant and Heat Shock Protein Responses During Torpor in the Gray Mouse Lemur, Microcebus murinus. Genom. Proteom. Bioinform. 2015, 13, 119–126. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Schwarze, P.E.; Totlandsdal, A.I.; Låg, M.; Refsnes, M.; Holme, J.A.; Øvrevik, J. Inflammation-Related Effects of Diesel Engine Exhaust Particles: Studies on Lung CellsIn Vitro. BioMed Res. Int. 2013, 2013, 685142. [Google Scholar] [CrossRef]
- Miyata, R.; van Eeden, S.F. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226. [Google Scholar] [CrossRef]
- Chaudhuri, N.; Jary, H.; Lea, S.; Khan, N.; Piddock, K.C.; Dockrell, D.H.; Donaldson, K.; Duffin, R.; Singh, D.; Parker, L.C.; et al. Diesel Exhaust Particle Exposure In Vitro Alters Monocyte Differentiation and Function. PLoS ONE 2012, 7, e51107. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Rahman, M.; Irmler, M.; Keshavan, S.; Introna, M.; Beckers, J.; Palmberg, L.; Johanson, G.; Ganguly, K.; Upadhyay, S. Differential Effect of SARS-CoV-2 Spike Glycoprotein 1 on Human Bronchial and Alveolar Lung Mucosa Models: Implications for Pathogenicity. Viruses 2021, 13, 2537. [Google Scholar] [CrossRef]
- Rahman, M.; Irmler, M.; Introna, M.; Beckers, J.; Palmberg, L.; Johanson, G.; Upadhyay, S.; Ganguly, K. Insight into the pulmonary molecular toxicity of heated tobacco products using human bronchial and alveolar mucosa models at air-liquid interface. Sci. Rep. 2022, 12, 16396. [Google Scholar] [CrossRef]
- Jaggi, U.; Yang, M.; Matundan, H.H.; Hirose, S.; Shah, P.K.; Sharifi, B.G.; Ghiasi, H. Increased phagocytosis in the presence of enhanced M2-like macrophage responses correlates with increased primary and latent HSV-1 infection. PLoS Pathog. 2020, 16, e1008971. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Hodge, S.; Hodge, G.; Scicchitano, R.; Reynolds, P.N.; Holmes, M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell Biol. 2003, 81, 289–296, Erratum in: Immunol. Cell Biol. 2003, 81, 499. [Google Scholar] [CrossRef]
- Jubrail, J.; Kurian, N.; Niedergang, F. Macrophage phagocytosis cracking the defect code in COPD. Biomed. J. 2017, 40, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Rudell, B.; Blomberg, A.; Helleday, R.; Ledin, M.C.; Lundback, B.; Stjernberg, N.; Horstedt, P.; Sandstrom, T. Bronchoalveolar inflammation after exposure to diesel exhaust: Comparison between unfiltered and particle trap filtered exhaust. Occup. Environ. Med. 1999, 56, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Teng, O.; Ang, C.K.E.; Guan, X.L. Macrophage–Bacteria Interactions—A Lipid-Centric Relationship. Front. Immunol. 2017, 8, 1836. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.P.J.; Bitterle, E.; Beck-Speier, I.; Maier, K.L.; Frankenberger, M.; Heyder, J.; Ziegler-Heitbrock, L. Diesel exhaust particles increase LPS-stimulated COX-2 expression and PGE2 production in human monocytes. J. Leukoc. Biol. 2004, 75, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.-I.; Takano, H.; Yanagisawa, R.; Ichinose, T.; Sadakane, K.; Yoshino, S.; Yamaki, K.; Uchiyama, K.; Yoshikawa, T. Components of diesel exhaust particles differentially affect lung expression of cyclooxygenase-2 related to bacterial endotoxin. J. Appl. Toxicol. 2004, 24, 415–418. [Google Scholar] [CrossRef]
- Cao, Y.; Jantzen, K.; Gouveia, A.C.D.; Skovmand, A.; Roursgaard, M.; Loft, S.; Møller, P. Automobile diesel exhaust particles induce lipid droplet formation in macrophages in vitro. Environ. Toxicol. Pharmacol. 2015, 40, 164–171. [Google Scholar] [CrossRef]
- Yin, F.; Lawal, A.; Ricks, J.; Fox, J.R.; Larson, T.; Navab, M.; Fogelman, A.M.; Rosenfeld, M.E.; Araujo, J.A. Diesel Exhaust Induces Systemic Lipid Peroxidation and Development of Dysfunctional Pro-Oxidant and Pro-Inflammatory High-Density Lipoprotein. Arter. Thromb. Vasc. Biol. 2013, 33, 1153–1161. [Google Scholar] [CrossRef]
- Inoue, H.; Nanayama, T.; Hara, S.; Yokoyama, C.; Tanabe, T. The cyclic AMP response element plays an essential role in the expression of the human prostaglandin-endoperoxide synthase 2 gene in differentiated U937 monocytic cells. FEBS Lett. 1994, 350, 51–54. [Google Scholar] [CrossRef]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar] [CrossRef]
- Smith, W.L.; Dewitt, D.L. Prostaglandin endoperoxide H synthases-1 and -2. Adv. Immunol. 1996, 62, 167–215. [Google Scholar] [CrossRef]
- Nikolic, D.; van Breemen, R. DNA Oxidation Induced by Cyclooxygenase-2. Chem. Res. Toxicol. 2001, 14, 351–354. [Google Scholar] [CrossRef]
- Gouveia-Figueira, S.; Karimpour, M.; Bosson, J.A.; Blomberg, A.; Unosson, J.; Pourazar, J.; Sandström, T.; Behndig, A.F.; Nording, M.L. Mass spectrometry profiling of oxylipins, endocannabinoids, and N-acylethanolamines in human lung lavage fluids reveals responsiveness of prostaglandin E2 and associated lipid metabolites to biodiesel exhaust exposure. Anal. Bioanal. Chem. 2017, 409, 2967–2980. [Google Scholar] [CrossRef]
- Löndahl, J.; Swietlicki, E.; Rissler, J.; Bengtsson, A.; Boman, C.; Blomberg, A.; Sandstrom, T. Experimental determination of the respiratory tract deposition of diesel combustion particles in patients with chronic obstructive pulmonary disease. Part. Fibre Toxicol. 2012, 9, 30. [Google Scholar] [CrossRef]
- Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [Google Scholar] [CrossRef]
- Karavalakis, G.; Deves, G.; Fontaras, G.; Stournas, S.; Samaras, Z.; Bakeas, E. The impact of soy-based biodiesel on PAH, nitro-PAH and oxy-PAH emissions from a passenger car operated over regulated and nonregulated driving cycles. Fuel 2010, 89, 3876–3883. [Google Scholar] [CrossRef]
- Nyström, R.; Sadiktsis, I.; Ahmed, T.; Westerholm, R.; Koegler, J.; Blombergd, A.; Sandström, T.; Bomana, C. Physical and chemical properties of RME biodiesel exhaust particles without engine modifications. Fuel 2016, 186, 261–269. [Google Scholar] [CrossRef]
- Turpin, B.J.; Saxena, P.; Andrews, A. Measuring and simulating particulate organics in the atmosphere: Problems and prospects. Atmos Environ. 2000, 34, 2983–3013. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.; Upadhyay, S.; Ganguly, K.; Introna, M.; Ji, J.; Boman, C.; Muala, A.; Blomberg, A.; Sandström, T.; Palmberg, L. Comparable Response Following Exposure to Biodiesel and Diesel Exhaust Particles in Advanced Multicellular Human Lung Models. Toxics 2023, 11, 532. https://doi.org/10.3390/toxics11060532
Rahman M, Upadhyay S, Ganguly K, Introna M, Ji J, Boman C, Muala A, Blomberg A, Sandström T, Palmberg L. Comparable Response Following Exposure to Biodiesel and Diesel Exhaust Particles in Advanced Multicellular Human Lung Models. Toxics. 2023; 11(6):532. https://doi.org/10.3390/toxics11060532
Chicago/Turabian StyleRahman, Mizanur, Swapna Upadhyay, Koustav Ganguly, Micol Introna, Jie Ji, Christoffer Boman, Ala Muala, Anders Blomberg, Thomas Sandström, and Lena Palmberg. 2023. "Comparable Response Following Exposure to Biodiesel and Diesel Exhaust Particles in Advanced Multicellular Human Lung Models" Toxics 11, no. 6: 532. https://doi.org/10.3390/toxics11060532
APA StyleRahman, M., Upadhyay, S., Ganguly, K., Introna, M., Ji, J., Boman, C., Muala, A., Blomberg, A., Sandström, T., & Palmberg, L. (2023). Comparable Response Following Exposure to Biodiesel and Diesel Exhaust Particles in Advanced Multicellular Human Lung Models. Toxics, 11(6), 532. https://doi.org/10.3390/toxics11060532