

An Overview of Physiologically-Based Pharmacokinetic Models for Forensic Science

 ,
,  and
and
Abstract
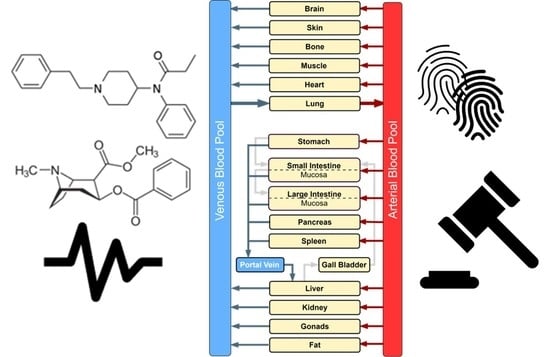
1. Introduction
2. Search Strategy
3. PBPK Model Development and Resources
- Step 1. Problem formulation: The purpose of why the model is built should be determined.
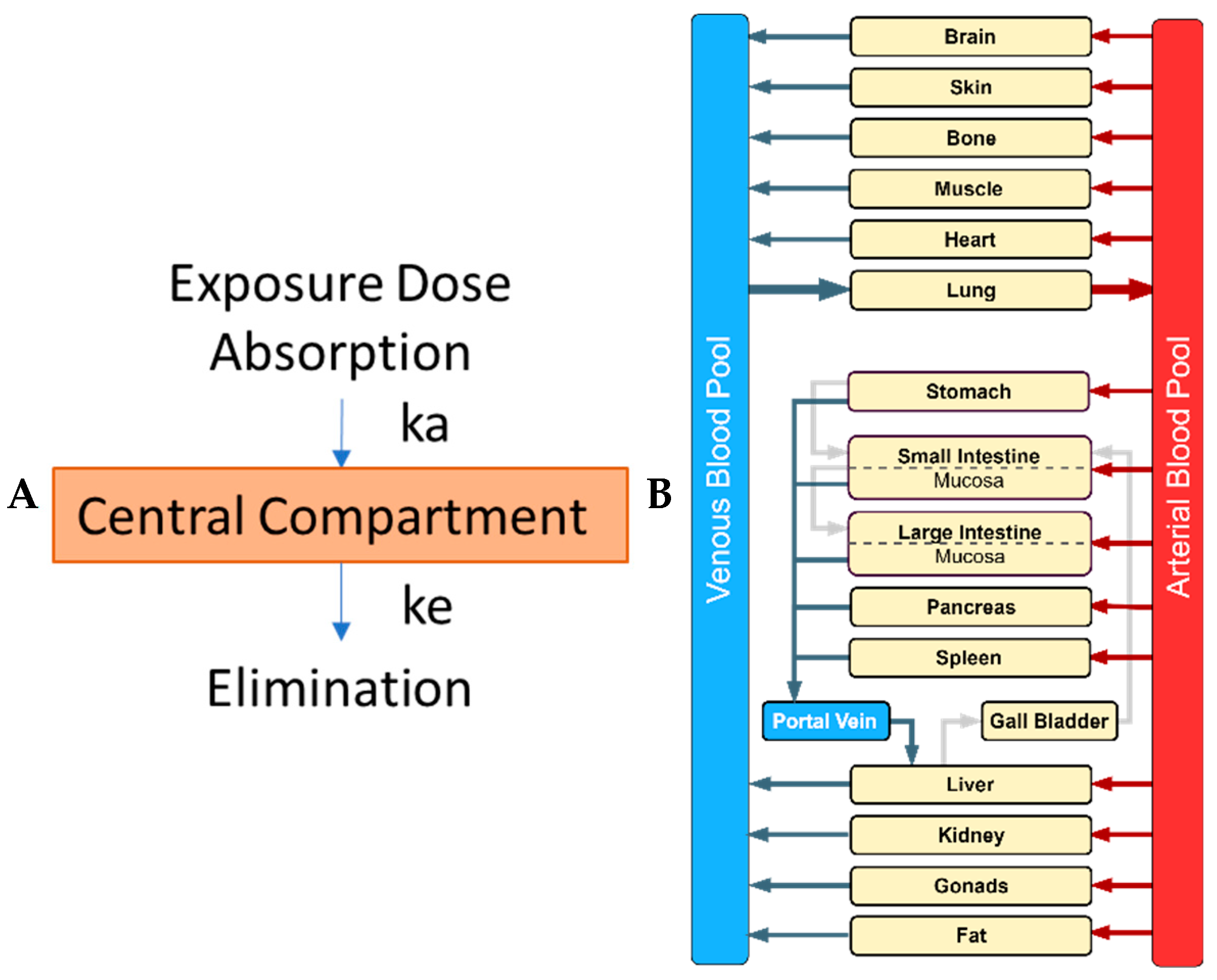
- Step 2. Model conceptualization: The structure of the PBPK model should be defined and informed by the problem formulation, knowledge of the underlying physiological and biokinetic mechanisms, and the availability of suitable data. The schematic model structure should be translated to mathematical equations to be implemented computationally.
- Step 3. Model parameterization: PBPK models are built using three sets of parameters: (i) physiological and anatomical parameters, with representative reference parameters obtained from the species under study (animal or human); (ii) biokinetic/ADME properties, such as clearance, which can be acquired using in vitro methods or by fitting the model to an in vivo data set; and (iii) physico-chemical parameters, such as lipophilicity, which are experimentally derived or obtained using in silico approaches such as quantitative structure activity relationship (QSAR) models. Several resources for model parameterization have been mapped [27].
- Step 4. Computer (software) implementation: This includes the choice of programming language to translate mathematical equations to computer code and the solver for execution of the model code. Currently, several open access and open-source modelling platforms, such as IndusChemFAte (Cefic LRI, http://cefic-lri.org/toolbox/induschemfate/), High-Throughput Toxicokinetics (httk)-r package (https://cran.r-project.org/web/packages/httk/index.html), MEGEN-RVis (https://megen.useconnect.co.uk/), PLETHEM (http://www.scitovation.com/plethem.html), MERLIN-EXPO (https://merlin-expo.eu/), and PK-Sim (www.systems-biology.com), and license-based platforms such as GastroPlus (www.simulations-plus.com) and SimCyp (https://www.certara.com), are available to individuals possessing varying degrees of expertise in PBPK modeling. These platforms provide different computational tools that allow non-programmers to develop and run model simulations with varying options to gain a better understanding and interpretation of model outputs. However, programmers or users with modeling skills can also use R, MATLAB, and Berkeley Madonna software to develop customized PBPK models.
- Step 6. Report and disseminate the model and simulations in a transparent and a “FAIR” way [28,29]. FAIR represents the Findability, Accessibility, Interoperability, and Reusability of scientific data. The FAIR guiding principles were first formally published in 2016 by Wilkinson et al. (2016) [30]. PBPK models can be reported using established templates [31] and or an international guidance document [21,22].
4. PBPK Model Applications in Forensic Science
5. Illegally Used Drugs
5.1. Acute, Short-Term, & Long-Term Exposure
5.1.1. Opioids
5.1.2. Psychostimulants
5.1.3. Psychedelics
Psilocybin and Psilocin
Mitragynine
5.1.4. Multidrug Combinations
5.1.5. Alcohol
6. Environmental Chemicals
6.1. Acute, Short-Term, and Accidental Environmental Chemical Exposure
6.2. Long-Term Environmental Chemical Exposure
6.2.1. Cyanide and Human Continuous Cyanide Inhalation Predictor (HCCIP)
6.2.2. Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS)
6.2.3. Trichloroethylene (TCE)
7. PBPK Modelling in Post-Mortem Investigations
7.1. Considerations of Organ-Specific Toxicity (Cardiotoxicity and Drug Induced Liver Injury) in Forensic Cases
7.2. Post-Mortem Considerations
7.3. PBPK Model Gaps, Uncertainties, and Future Needs
8. Discussion and Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug or Chemical Names | Cas Number | Tissues (Sample Type) | Sample Type (Sample Number) | Ethnicity | Sex | Age | References |
|---|---|---|---|---|---|---|---|
| Alprazolam | 28981-97-7 | Heart, subclavian blood, urine, bile, vitreous humor, liver, kidney | Autopsy | White | Female | 44 | Jenkins et al. 1997 [146] |
| Amiodarone | 1951-25-3 | Liver | Autopsy and biopsy | N/A | Both | 17–78 | Adams et al. 1985 [147] |
| Amitriptyline | 549-18-8 | Blood, pericardial fluid (PF), psoas muscle (PM), vitreous humor, vastus lateralis muscle (VM) | Autopsy (n = 9) | N/A | N/A | N/A | Åse Marit Leere Øiestad et al. 2018 [148] |
| Amphetamine | 300-62-9 | Blood, urine, liver | Autopsy | N/A | Male | 30 | Adjutantis et al. 1975 [149] |
| Buprenorphine | 52485-79-7 | Blood, urine, bile, liver, brain, kidney, myocardium, hair | Autopsy (n = 20) | N/A | Both | 14–48 | Tracqui et al. 1998 [150] |
| Carbamazepine | 298-46-4 | Blood, liver, kidney | Autopsy (n = 16) | N/A | Both | 7–70 | Klys et al. 2003 [151] |
| Chloroquine | 54-05-07 | Blood, liver, kidney, brain | Autopsy (n = 27) | N/A | Both | 11 months–61 | Di Maio et al. 1974 [155] |
| Citalopram | 59729-33-8 | Blood, bile, urine, liver, kidney | Autopsy (n = 13) | N/A | N/A | N/A | Levine et al. 2001 [156] |
| Clozapine | 5786-21-0 | Peripheral blood, heart blood, cerebrospinal fluid, vitreous humor, bile | Autopsy (n = 1) | N/A | Female | 15 | Keller et al. 1997 [157] |
| Carfentanil | 59708-52-0 | Blood, vitreous humor, urine | Autopsy (n = 290) | N/A | Both | 16–69 | Chatterton et al. 2020 [158] |
| Codeine | 76-57-3 | Blood, urine, bile, brain | Autopsy (n = 14) | N/A | Male | 23–45 | Gambaro et al. 2014 [159] |
| Digoxin | 20830-75-5 | Heart, skeletal muscle, liver, kidney | Autopsy | N/A | Both | Infants and children (2 days–7 years) Adult (59–80) | Kim et al. 1975 [160] Karjalainen et al. 1974 [161] |
| Diltiazem | 42399-41-7 | Blood, urine, bile | Autopsy (n = 1) | N/A | Female | 78 | Engelhart et al. 1997 [162] |
| Diphenhydramine | 58-73-1 | Blood (n = 44), liver (n = 33) | Autopsy | N/A | N/A | N/A | Levine et al.1996 [163] |
| Fentanyl | 437-68-7 | Central blood, femoral blood, brain, muscle, liver | Autopsy | N/A | Both | 20–60 | Chatterton et al. 2018 [164] |
| Fluoxetine | 56296-78-7 | Blood, urine, vitreous humor, bile, liver, lung, kidney, spleen, muscle, brain, heart | Autopsy (n = 10) | N/A | N/A | N/A | Johnson et al. 1992 [165] |
| Lidocaine | 137-58-6 | Blood, brain, heart, kidney, lung, spleen, skeletal muscle, adipose | Autopsy (n = 1) | N/A | N/A | 64 | Poklis et al. 1984 [166] |
| Methadone | 76-99-3 | Blood, bile, urine, liver, kidney, lung, brain | Autopsy (n = 6) | White, Black African | Both | 18–46 | Garriott et al. 1973 [167] |
| Mirtazapine | 85650-52-8 | Heart blood, peripheral blood, urine, liver, kidney, bile | Autopsy (n = 8) | N/A | Both | N/A | Moore et al. 1999 [168] |
| Morphine | 57-27-2 | Blood (n = 21), cerebrospinal fluid (n = 11), urine (n = 6) | Autopsy | N/A | Both | 18–40 (1 unknown) | Bogusz et al. 1997 [169] |
| Olanzapine | 132539-06-1 | Peripheral blood, central blood, liver, vitreous humor | Autopsy (n = 28) | N/A | Both | 26–68 | Vance et al. 2009 [170] |
| Oxycodone | 76-42-6 | Blood, liver, urine, bile, vitreous humor | Autopsy (n = 36) | N/A | Both | 21–62 | Anderson et al. 2002 [171] |
| PFOS | Mixture (1763-23-1?) | Liver, kidney, adipose tissue, brain, basal ganglia, hypophysis, thyroid, gonads, pancreas, lung, skeletal muscle | Autopsy | N/A | Both | 12–83 | Maestri et al. 2006 [172] |
| PFOA | Mixture (335-67-1?) | Liver, kidney, adipose tissue, brain, basal ganglia, hypophysis, thyroid, gonads, pancreas, lung, skeletal muscle | Autopsy | NA | Both | 12–83 | Maestri et al. 2006 [172] |
| PFAS | mixture | Liver, Kidney, Lung, Brain, Bone (20 samples each) | Autopsy | Hispanic | N/A | 28–83 | Perez et al. 2013 [136] |
| Quetiapine | 111974-69-7 | Central blood, femoral blood, brain, muscle, liver | Autopsy | N/A | Both | 20–60 | Breivik et al. 2020 [173] |
| THC | 1972-08-3 | Blood, urine, liver, lung, kidney, spleen, muscle, brain, heart, bile | Autopsy | N/A | Male | 22–69 | Saenz et al. 2017 [174] |
| Tramadol | 123154-38-1 | Blood, urine, liver, kidney, bile | Autopsy (n = 8) | N/A | Both | 28–67 | Decker et al. 2007 [175] |
| Venlafaxine | 93413-69-5 | Blood, pericardial fluid (PF), psoas muscle (PM), vitreous humor, vastus lateralis muscle (VM) | Autopsy (n = 6) | N/A | N/A | N/A | Åse Marit Leere Øiestad et al. 2018 [151] |
| Zolpidem | 82626-48-0 | Blood, vitreous humor, bile, urine, gastric contents, liver | Autopsy (n = 2) | White, Black | Female | 36, 58 | Gock et al. 1999 [176] |
| Zopiclone | 43200-80-2 | Blood, liver | Autopsy (n = 2) | White, Indian | Both | 24–82 | Boniface et al. 1996 [177] |
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.-F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Barton, H.A.; Chiu, W.A.; Setzer, R.W.; Andersen, M.E.; Bailer, A.J.; Bois, F.Y.; De Woskin, R.S.; Hays, S.; Johanson, G.; Jones, N.; et al. Characterizing uncertainty and variability in physiologically based pharmacokinetic models: State of the science and needs for research and implementation. Toxicol. Sci. 2007, 99, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Krauss, M.; Tappe, K.; Schuppert, A.; Kuepfer, L.; Goerlitz, L. Bayesian Population Physiologically-Based Pharmacokinetic (PBPK) Approach for a Physiologically Realistic Characterization of Interindividual Variability in Clinically Relevant Populations. PLoS ONE 2015, 10, e0139423. [Google Scholar] [CrossRef] [PubMed]
- Bois, F.Y.; Jamei, M.; Clewell, H.J. PBPK modelling of inter-individual variability in the pharmacokinetics of environmental chemicals. Toxicology 2010, 278, 256–267. [Google Scholar] [CrossRef]
- Thompson, C.V.; Firman, J.W.; Goldsmith, M.R.; Grulke, C.M.; Tan, Y.-M.; Paini, A.; Penson, P.E.; Sayre, R.R.; Webb, S.; Madden, J.C. A Systematic Review of Published Physiologically-based Kinetic Models and an Assessment of their Chemical Space Coverage. Altern. Lab. Anim. 2021, 49, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Polasek, T.M.; Shakib, S.; Rostami-Hodjegan, A. Precision dosing in clinical medicine: Present and future. Expert Rev. Clin. Pharmacol. 2018, 11, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Paini, A.; Leonard, J.A.; Joossens, E.; Bessems, J.; Desalegn, A.; Dorne, J.; Gosling, J.; Heringa, M.; Klaric, M.; Kliment, T.; et al. Next generation physiologically based kinetic (NG-PBK) models in support of regulatory decision making. Comput. Toxicol. 2019, 9, 61–72. [Google Scholar] [CrossRef]
- Lautz, L.S.; Nebbia, C.; Hoeks, S.; Oldenkamp, R.; Hendriks, A.; Ragas, A.; Dorne, J. An open source physiologically based kinetic model for the chicken (Gallus gallus domesticus): Calibration and validation for the prediction residues in tissues and eggs. Environ. Int. 2020, 136, 105488. [Google Scholar] [CrossRef] [PubMed]
- Lautz, L.S.; Hoeks, S.; Oldenkamp, R.; Hendriks, A.J.; Dorne, J.; Ragas, A.M.J. Generic physiologically based kinetic modelling for farm animals: Part II. Predicting tissue concentrations of chemicals in swine, cattle and sheep. Toxicol. Lett. 2020, 318, 50–56. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.S.; Elwell-Cuddy, T.; Baynes, R.E.; Tell, L.A.; Davis, J.L.; Maunsell, F.P.; Riviere, J.E.; Lin, Z. Physiological parameter values for physiologically based pharmacokinetic models in food-producing animals. Part III: Sheep and goat. J. Vet. Pharmacol. Ther. 2021, 44, 456–477. [Google Scholar] [CrossRef]
- Li, M.; Gehring, R.; Riviere, J.E.; Lin, Z. Development and application of a population physiologically based pharmacokinetic model for penicillin G in swine and cattle for food safety assessment. Food Chem. Toxicol. 2017, 107, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, M.; Wang, Y.S.; Tell, L.A.; Baynes, R.E.; Davis, J.L.; Vickroy, T.W.; Riviere, J.E. Physiological parameter values for physiologically based pharmacokinetic models in food-producing animals. Part I: Cattle and swine. J. Vet. Pharmacol. Ther. 2020, 43, 385–420. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.W.; McKim, J.M.; Lien, G.J.; Hoffman, A.D.; Bertelsen, S.L.; Gallinat, C.A. Physiologically-based toxicokinetic modeling of three waterborne chloroethanes in channel catfish, Ictalurus punctatus. Aquat. Toxicol. 1993, 27, 83–111. [Google Scholar] [CrossRef]
- Nichols, J.W.; McKim, J.M.; Andersen, M.E.; Gargas, M.L.; Clewell, H.J.; Erickson, R.J. A physiologically based toxicokinetic model for the uptake and disposition of waterborne organic chemicals in fish. Toxicol. Appl. Pharmacol. 1990, 106, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Lien, G.J.; McKim, J.M.; Hoffman, A.D.; Jenson, C.T. A physiologically based toxicokinetic model for lake trout (Salvelinus namaycush). Aquat. Toxicol. 2001, 51, 335–350. [Google Scholar] [CrossRef]
- Brinkmann, M.; Eichbaum, K.; Kammann, U.; Hudjetz, S.; Cofalla, C.; Buchinger, S.; Reifferscheid, G.; Schüttrumpf, H.; Preuss, T.; Hollert, H. Physiologically-based toxicokinetic models help identifying the key factors affecting contaminant uptake during flood events. Aquat. Toxicol. 2014, 152, 38–46. [Google Scholar] [CrossRef]
- Salmina, E.S.; Wondrousch, D.; Kühne, R.; Potemkin, V.A.; Schüürmann, G. Variation in predicted internal concentrations in relation to PBPK model complexity for rainbow trout. Sci. Total Environ. 2016, 550, 586–597. [Google Scholar] [CrossRef]
- Baier, V.; Paini, A.; Schaller, S.; Scanes, C.G.; Bone, A.J.; Ebeling, M.; Preuss, T.G.; Witt, J.; Heckmann, D. A generic avian physiologically-based kinetic (PBK) model and its application in three bird species. Environ. Int. 2022, 169, 107547. [Google Scholar] [CrossRef] [PubMed]
- DOJ. Forensic Science. 2021. Available online: https://www.justice.gov/olp/forensic-science (accessed on 27 September 2022).
- Bravo-Gómez, M.E.; Camacho-García, L.N.; Castillo-Alanís, L.A.; Mendoza-Meléndez, M.; Quijano-Mateos, A. Revisiting a physiologically based pharmacokinetic model for cocaine with a forensic scope. Toxicol. Res. 2019, 8, 432–446. [Google Scholar] [CrossRef]
- WHO. Characterization and Application of Physiologically Based Phamacokinetic Models in Risk Assessment. 2010. Available online: https://apps.who.int/iris/handle/10665/44495 (accessed on 27 September 2022).
- Guidance Document on the Characterisation, Validation and Reporting of PBK Models for Regulatory Purposes. 2021. Available online: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV-CBC MONO(2021)1%20&doclanguage=en (accessed on 27 September 2022).
- Paini, A.; Worth, A.; Kulkarni, S.; Ebbrell, D.; Madden, J. Assessment of the predictive capacity of a physiologically based kinetic model using a read-across approach. Comput. Toxicol. 2021, 18, 100159. [Google Scholar] [CrossRef]
- Physiologically Based Pharmacokinetic Analyses—Format and Content Guidance for Industry. 2019. Available online: http://www.fda.gov/regulatory-information/search-fda-guidance-documents/physiologically-based-pharmacokinetic-analyses-format-and-content-guidance-industry (accessed on 27 September 2022).
- Guideline on the Qualification and Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation_en.pdf (accessed on 27 September 2022).
- EPA. Approaches for the Application of Physiologically Based Pharmacokinetic (PBPK) Models and Supporting Data in Risk Assessment (Final Report); United States Environmental Protection Agency: Washington, DC, USA, 2006. [Google Scholar]
- Madden, J.C.; Pawar, G.; Cronin, M.T.D.; Webb, S.; Tan, Y.-M.; Paini, A. In silico resources to assist in the development and evaluation of physiologically-based kinetic models. Comput. Toxicol. 2019, 11, 33–49. [Google Scholar] [CrossRef]
- Mons, B.; Schultes, E.; Liu, F.; Jacobsen, A. The FAIR Principles: First Generation Implementation Choices and Challenges. Data Intell. 2020, 2, 1–9. [Google Scholar] [CrossRef]
- Briggs, K.; Bosc, N.; Camara, T.; Diaz, C.; Drew, P.; Drewe, W.; Kors, J.; van Mulligen, E.; Pastor, M.; Pognan, F.; et al. Guidelines for FAIR sharing of preclinical safety and off-target pharmacology data. Altex 2021, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Tan, Y.M.; Chan, M.; Chukwudebe, A.; Domoradzki, J.; Fisher, J.; Hack, C.E.; Hinderliter, P.; Hirasawa, K.; Leonard, J.; Lumen, A.; et al. PBPK model reporting template for chemical risk assessment applications. Regul. Toxicol. Pharmacol. 2020, 115, 104691. [Google Scholar] [CrossRef]
- Prescription Drug Use in the Past 30 Days, by Sex, Race and Hispanic Origin, and Age: United States, Selected Years 1988–1994 through 2015–2018; Centers for Disease Control: Atlanta, GA, USA, 2019.
- Mattson, C.L.; Tanz, L.J.; Quinn, K.; Kariisa, M.; Patel, P.; Davis, N.L. Trends and Geographic Patterns in Drug and Synthetic Opioid Overdose Deaths—United States, 2013–2019. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 202–207. [Google Scholar] [CrossRef]
- Eaton, D.L.; Gilbert, S.G. Principles of Toxicology. In Casarett and Doull’s Toxicology: The Basic Science of Poisons; McGraw-Hill Education: New York, NY, USA, 2015. [Google Scholar]
- What Is the U.S. Opioid Epidemic? Available online: https://www.hhs.gov/opioids/about-the-epidemic/index.html (accessed on 27 September 2022).
- Schaefer, N.; Moj, D.; Lehr, T.; Schmidt, P.H.; Ramsthaler, F. The feasibility of physiologically based pharmacokinetic modeling in forensic medicine illustrated by the example of morphine. Int. J. Leg. Med. 2018, 132, 415–424. [Google Scholar] [CrossRef]
- German, C.; Pilvankar, M.; Przekwas, A. Computational framework for predictive PBPK-PD-Tox simulations of opioids and antidotes. J. Pharmacokinet. Pharmacodyn. 2019, 46, 513–529. [Google Scholar] [CrossRef]
- Sjöstedt, N.; Neuhoff, S.; Brouwer, K.L.R. Physiologically-Based Pharmacokinetic Model of Morphine and Morphine-3-Glucuronide in Nonalcoholic Steatohepatitis. Clin. Pharmacol. Ther. 2021, 109, 676–687. [Google Scholar] [CrossRef]
- Imaoka, T.; Huang, W.; Shum, S.; Hailey, D.W.; Chang, S.-Y.; Chapron, A.; Yeung, C.K.; Himmelfarb, J.; Isoherranen, N.; Kelly, E.J. Bridging the gap between in silico and in vivo by modeling opioid disposition in a kidney proximal tubule microphysiological system. Sci. Rep. 2021, 11, 21356. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Välitalo, P.A.; Wong, Y.C.; Huntjens, D.R.; Proost, J.H.; Vermeulen, A.; Krauwinkel, W.; Beukers, M.W.; Kokki, H.; Kokki, M.; et al. Prediction of human CNS pharmacokinetics using a physiologically-based pharmacokinetic modeling approach. Eur. J. Pharm. Sci. 2018, 112, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Uchaipichat, V.; Rowland, A.; Miners, J.O. Inhibitory effects of non-steroidal anti-inflammatory drugs on human liver microsomal morphine glucuronidation: Implications for drug-drug interaction liability. Drug Metab. Pharmacokinet. 2022, 42, 100442. [Google Scholar] [CrossRef] [PubMed]
- Hahn, D.; Emoto, C.; Euteneuer, J.C.; Mizuno, T.; Vinks, A.A.; Fukuda, T. Influence of OCT1 Ontogeny and Genetic Variation on Morphine Disposition in Critically Ill Neonates: Lessons from PBPK Modeling and Clinical Study. Clin. Pharmacol. Ther. 2019, 105, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Emoto, C.; Johnson, T.N.; Neuhoff, S.; Hahn, D.; Vinks, A.A.; Fukuda, T. PBPK Model of Morphine Incorporating Developmental Changes in Hepatic OCT1 and UGT2B7 Proteins to Explain the Variability in Clearances in Neonates and Small Infants. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Emoto, C.; Fukuda, T.; Johnson, T.N.; Neuhoff, S.; Sadhasivam, S.; Vinks, A.A. Characterization of Contributing Factors to Variability in Morphine Clearance Through PBPK Modeling Implemented with OCT1 Transporter. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Schlender, J.F.; Meyer, M.; Thelen, K.; Krauss, M.; Willmann, S.; Eissing, T.; Jaehde, U. Development of a Whole-Body Physiologically Based Pharmacokinetic Approach to Assess the Pharmacokinetics of Drugs in Elderly Individuals. Clin. Pharmacokinet. 2016, 55, 1573–1589. [Google Scholar] [CrossRef]
- Prasad, B.; Bhatt, D.K.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.E.C.A.; Mathias, A.; et al. Abundance of Phase 1 and 2 Drug-Metabolizing Enzymes in Alcoholic and Hepatitis C Cirrhotic Livers: A Quantitative Targeted Proteomics Study. Drug Metab. Dispos. 2018, 46, 943–952. [Google Scholar] [CrossRef]
- Strougo, A.; Eissing, T.; Yassen, A.; Willmann, S.; Danhof, M.; Freijer, J. First dose in children: Physiological insights into pharmacokinetic scaling approaches and their implications in paediatric drug development. J. Pharmacokinet. Pharmacodyn. 2012, 39, 195–203. [Google Scholar] [CrossRef][Green Version]
- Pang, K.S.; Yang, Q.J.; Noh, K. Unequivocal evidence supporting the segregated flow intestinal model that discriminates intestine versus liver first-pass removal with PBPK modeling. Biopharm. Drug Dispos. 2017, 38, 231–250. [Google Scholar] [CrossRef]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-gp DDI Prediction: A Modeling Network of Rifampicin, Itraconazole, Clarithromycin, Midazolam, Alfentanil, and Digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef]
- Abduljalil, K.; Pan, X.; Pansari, A.; Jamei, M.; Johnson, T.N. Preterm Physiologically Based Pharmacokinetic Model. Part II: Applications of the Model to Predict Drug Pharmacokinetics in the Preterm Population. Clin. Pharmacokinet. 2020, 59, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Edginton, A.N.; Willmann, S. Physiology-based simulations of a pathological condition: Prediction of pharmacokinetics in patients with liver cirrhosis. Clin. Pharmacokinet. 2008, 47, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, G.; Parrott, N.; Meille, C.; Iliadis, A.; Lavé, T. Physiologically based pharmacokinetic modeling of CYP3A4 induction by rifampicin in human: Influence of time between substrate and inducer administration. Eur. J. Pharm. Sci. 2014, 56, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Abbiati, R.A.; Manca, D. Innovations and Improvements in Pharmacokinetic Models Based on Physiology. Curr. Drug Deliv. 2017, 14, 190–202. [Google Scholar] [CrossRef][Green Version]
- Cascone, S.; Lamberti, G.; Piazza, O.; Abbiati, R.A.; Manca, D. A physiologically-based model to predict individual pharmacokinetics and pharmacodynamics of remifentanil. Eur. J. Pharm. Sci. 2018, 111, 20–28. [Google Scholar] [CrossRef]
- Sánchez Restrepo, F.; Hernández Valdivieso, A.M. Global sensitivity analysis in physiologically-based pharmacokinetic/pharmacodynamic models of inhaled and opioids anesthetics and its application to generate virtual populations. J. Pharmacokinet. Pharmacodyn. 2022, 49, 411–428. [Google Scholar] [CrossRef]
- Zhou, W.; Johnson, T.N.; Bui, K.H.; Cheung, S.A.; Li, J.; Xu, H.; Al-Huniti, N.; Zhou, D. Predictive Performance of Physiologically Based Pharmacokinetic (PBPK) Modeling of Drugs Extensively Metabolized by Major Cytochrome P450s in Children. Clin. Pharmacol. Ther. 2018, 104, 188–200. [Google Scholar] [CrossRef]
- T’Jollyn, H.; Vermeulen, A.; Van Bocxlaer, J. PBPK and its Virtual Populations: The Impact of Physiology on Pediatric Pharmacokinetic Predictions of Tramadol. AAPS J. 2018, 21, 8. [Google Scholar] [CrossRef]
- Xu, M.; Zheng, L.; Zeng, J.; Xu, W.; Jiang, X.; Wang, L. Physiologically based pharmacokinetic modeling of tramadol to inform dose adjustment and drug-drug interactions according to CYP2D6 phenotypes. Pharmacotherapy 2021, 41, 277–290. [Google Scholar] [CrossRef]
- T’Jollyn, H.; Snoeys, J.; Colin, P.; Van Bocxlaer, J.; Annaert, P.; Cuyckens, F.; Vermeulen, A.; Van Peer, A.; Allegaert, K.; Mannens, G.; et al. Physiology-based IVIVE predictions of tramadol from in vitro metabolism data. Pharm. Res. 2015, 32, 260–274. [Google Scholar] [CrossRef]
- T’Jollyn, H.; Snoeys, J.; Vermeulen, A.; Michelet, R.; Cuyckens, F.; Mannens, G.; Van Peer, A.; Annaert, P.; Allegaert, K.; Van Bocxlaer, J.; et al. Physiologically Based Pharmacokinetic Predictions of Tramadol Exposure Throughout Pediatric Life: An Analysis of the Different Clearance Contributors with Emphasis on CYP2D6 Maturation. AAPS J. 2015, 17, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; Cristofoletti, R.; Cicali, B.; Michaud, V.; Dow, P.; Turgeon, J.; Schmidt, S. Physiologically Based Pharmacokinetic Modeling to Assess the Impact of CYP2D6-Mediated Drug-Drug Interactions on Tramadol and O-Desmethyltramadol Exposures via Allosteric and Competitive Inhibition. J. Clin. Pharmacol. 2022, 62, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Shum, S.; Shen, D.D.; Isoherranen, N. Predicting Maternal-Fetal Disposition of Fentanyl Following Intravenous and Epidural Administration Using Physiologically Based Pharmacokinetic Modeling. Drug Metab. Dispos. 2021, 49, 1003–1015. [Google Scholar] [CrossRef]
- Huang, W.; Isoherranen, N. Sampling Site Has a Critical Impact on Physiologically Based Pharmacokinetic Modeling. J. Pharmacol. Exp. Ther. 2020, 372, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Liu, S.; Xue, Y.; He, X.; Man, V.H.; Xie, X.-Q.; Wang, J. Prediction of Drug-Drug Interactions Between Opioids and Overdosed Benzodiazepines Using Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation. Drugs R&D 2019, 19, 297–305. [Google Scholar] [CrossRef]
- Shen, M.; Chen, M.; Liang, T.; Wang, S.; Xue, Y.; Bertz, R.; Xie, X.-Q.; Feng, Z. Pain Chemogenomics Knowledgebase (Pain-CKB) for Systems Pharmacology Target Mapping and Physiologically Based Pharmacokinetic Modeling Investigation of Opioid Drug-Drug Interactions. ACS Chem. Neurosci. 2020, 11, 3245–3258. [Google Scholar] [CrossRef]
- Shankaran, H.; Adeshina, F.; Teeguarden, J.G. Physiologically-based pharmacokinetic model for Fentanyl in support of the development of Provisional Advisory Levels. Toxicol. Appl. Pharmacol. 2013, 273, 464–476. [Google Scholar] [CrossRef]
- Björkman, S. Reduction and lumping of physiologically based pharmacokinetic models: Prediction of the disposition of fentanyl and pethidine in humans by successively simplified models. J. Pharmacokinet. Pharmacodyn. 2003, 30, 285–307. [Google Scholar] [CrossRef]
- Rytkönen, J.; Ranta, V.P.; Kokki, M.; Hautajärvi, H.; Rinne, V.; Heikkinen, A.T. Physiologically based pharmacokinetic modelling of oxycodone drug-drug interactions. Biopharm. Drug Dispos. 2020, 41, 72–88. [Google Scholar] [CrossRef]
- Zheng, L.; Xu, M.; Tang, S.W.; Song, H.X.; Jiang, X.H.; Wang, L. Physiologically Based Pharmacokinetic Modeling of Oxycodone in Children to Support Pediatric Dosing Optimization. Pharm. Res. 2019, 36, 171. [Google Scholar] [CrossRef]
- Zhang, H.; Kalluri, H.V.; Bastian, J.R.; Chen, H.; Alshabi, A.; Caritis, S.N. Gestational changes in buprenorphine exposure: A physiologically-based pharmacokinetic analysis. Br. J. Clin. Pharmacol. 2018, 84, 2075–2087. [Google Scholar] [CrossRef] [PubMed]
- Kharidia, J.; Howgate, E.M.; Laffont, C.M.; Liu, Y.; Young, M.A. Evaluation of Drug-Drug Interaction Liability for Buprenorphine Extended-Release Monthly Injection Administered by Subcutaneous Route. Clin. Pharmacol. Drug Dev. 2021, 10, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.L.; Silvola, R.M.; Haas, D.M.; Quinney, S.K. Physiologically based pharmacokinetic modelling in pregnancy: Model reproducibility and external validation. Br. J. Clin. Pharmacol. 2022, 88, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, H.V.; Zhang, H.; Caritis, S.N.; Venkataramanan, R. A physiologically based pharmacokinetic modelling approach to predict buprenorphine pharmacokinetics following intravenous and sublingual administration. Br. J. Clin. Pharmacol. 2017, 83, 2458–2473. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gobburu, J.V.S. A Physiologically Based Pharmacokinetic Modeling Approach to Predict Drug-Drug Interactions of Buprenorphine After Subcutaneous Administration of CAM2038 with Perpetrators of CYP3A4. J. Pharm. Sci. 2018, 107, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Van Hoogdalem, M.W.; Johnson, T.N.; McPhail, B.T.; Kamatkar, S.; Wexelblatt, S.L.; Ward, L.P.; Christians, U.; Akinbi, H.T.; Vinks, A.A.; Mizuno, T. Physiologically-Based Pharmacokinetic Modeling to Investigate the Effect of Maturation on Buprenorphine Pharmacokinetics in Newborns with Neonatal Opioid Withdrawal Syndrome. Clin. Pharmacol. Ther. 2022, 111, 496–508. [Google Scholar] [CrossRef]
- Cao, Y.; Jusko, W.J. Applications of minimal physiologically-based pharmacokinetic models. J. Pharmacokinet. Pharmacodyn. 2012, 39, 711–723. [Google Scholar] [CrossRef]
- Noh, K.; Chen, S.; Yang, Q.J.; Pang, K.S. Physiologically based pharmacokinetic modeling revealed minimal codeine intestinal metabolism in first-pass removal in rats. Biopharm. Drug Dispos. 2017, 38, 50–74. [Google Scholar] [CrossRef]
- Badhan, R.K.S.; Gittins, R. Precision dosing of methadone during pregnancy: A pharmacokinetics virtual clinical trials study. J. Subst. Abuse Treat. 2021, 130, 108521. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Bouwmeester, H.; Rietjens, I.; Strikwold, M. Integrating in vitro data and physiologically based kinetic modeling-facilitated reverse dosimetry to predict human cardiotoxicity of methadone. Arch. Toxicol. 2020, 94, 2809–2827. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Dong, Y.; Bouwmeester, H.; Rietjens, I.; Strikwold, M. In vitro-in silico-based prediction of inter-individual and inter-ethnic variations in the dose-dependent cardiotoxicity of R- and S-methadone in humans. Arch. Toxicol. 2022, 96, 2361–2380. [Google Scholar] [CrossRef] [PubMed]
- Badhan, R.K.S.; Gittins, R.; Al Zabit, D. The optimization of methadone dosing whilst treating with rifampicin: A pharmacokinetic modeling study. Drug Alcohol Depend. 2019, 200, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.B.; Nallani, S.C.; Zhao, P.; Rostami-Hodjegan, A.; Unadkat, J.D. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br. J. Clin. Pharmacol. 2014, 77, 554–570. [Google Scholar] [CrossRef] [PubMed]
- McPhail, B.T.; Emoto, C.; Fukuda, T.; Butler, D.; Wiles, J.R.; Akinbi, H.; Vinks, A.A. Utilizing Pediatric Physiologically Based Pharmacokinetic Models to Examine Factors That Contribute to Methadone Pharmacokinetic Variability in Neonatal Abstinence Syndrome Patients. J. Clin. Pharmacol. 2020, 60, 453–465. [Google Scholar] [CrossRef]
- Yang, F.; Tong, X.; McCarver, D.G.; Hines, R.N.; Beard, D.A. Population-based analysis of methadone distribution and metabolism using an age-dependent physiologically based pharmacokinetic model. J. Pharmacokinet. Pharmacodyn. 2006, 33, 485–518. [Google Scholar] [CrossRef]
- Michael, G.; Feasel, R.J.L.; Kristovich, R.L.; Wohlfarth, A.; Huestis, M.A. Translational Human Health Assessment of Carfentanil Using an Experimentally Refined PBPK Model; Center ECB: Gunpowder, MD, USA, 2018; Available online: https://apps.dtic.mil/sti/pdfs/AD1060142.pdf (accessed on 27 September 2022).
- Cannaert, A.; Ambach, L.; Blanckaert, P.; Stove, C.P. Activity-Based Detection and Bioanalytical Confirmation of a Fatal Carfentanil Intoxication. Front. Pharmacol. 2018, 9, 486. [Google Scholar] [CrossRef]
- Nichols, D.E. Psychedelics. Pharmacol. Rev. 2016, 68, 264–355. [Google Scholar] [CrossRef]
- Marek, G.J.; Carpenter, L.L.; McDougle, C.J.; Price, L.H. Synergistic Action of 5-HT2A Antagonists and Selective Serotonin Reuptake Inhibitors in Neuropsychiatric Disorders. Neuropsychopharmacology 2003, 28, 402–412. [Google Scholar] [CrossRef]
- Musikaphongsaku, P.; Ya, K.; Subsoontorn, P.; Lohitnavy, M. Development of a physiologically based pharmacokinetic (PBPK) model of psilocybin and psilocin from magic mushroom in rats and humans. F1000Research 2021, 10, 209. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, S.; Lin, W.; Wu, N.; Wang, Y.; Chen, M.; Xie, X.-Q.; Feng, Z. Computational Systems Pharmacology-Target Mapping for Fentanyl-Laced Cocaine Overdose. ACS Chem. Neurosci. 2019, 10, 3486–3499. [Google Scholar] [CrossRef]
- Key Substance Use and Mental Health Indicators in the United States: Results from the 2017 National Survey on Drug Use and Health. 2018. Available online: https://www.samhsa.gov/data/sites/default/files/cbhsq-reports/NSDUHFFR2017/NSDUHFFR2017.pdf (accessed on 27 September 2022).
- Product Information: VANTRELA(TM) ER Oral Extended-Release Tablets, Hydrocodone Bitartrate Oral Extended-Release Tablets. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/207975s000lbl.pdf (accessed on 27 September 2022).
- McCance-Katz, E.F. Drug interactions associated with methadone, buprenorphine, cocaine, and HIV medications: Implications for pregnant women. Life Sci. 2011, 88, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Deaths from Excessive Alcohol Use in the United States. Centers for Disease Control and Prevention. Updated July 6, 2022. 2022. Available online: https://www.cdc.gov/alcohol/features/excessive-alcohol-deaths.html (accessed on 27 September 2022).
- Crowell, S.R.; Smith, J.N.; Creim, J.A.; Faber, W.; Teeguarden, J.G. Physiologically based pharmacokinetic modeling of ethyl acetate and ethanol in rodents and humans. Regul. Toxicol. Pharmacol. 2015, 73, 452–462. [Google Scholar] [CrossRef]
- Dennison, J.E.; Bigelow, P.L.; Andersen, M.E. Occupational exposure limits in the context of solvent mixtures, consumption of ethanol, and target tissue dose. Toxicol. Ind. Health 2004, 20, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Delerme, C.; Artigou, C.; Bodin, L.; Tardif, R.; Charest-Tardif, G.; Verdier, C.; Sater, N.; Ould-Elhkim, M.; Desmares, C. Short Communication: Is Ethanol-Based Hand Sanitizer Involved in Acute Pancreatitis after Excessive Disinfection? An Evaluation with the Use of PBPK Model. J. Toxicol. 2012, 2012, 959070. [Google Scholar] [CrossRef] [PubMed]
- Jongeneelen, F.J.; Berge, W.F. A generic, cross-chemical predictive PBTK model with multiple entry routes running as application in MS Excel; design of the model and comparison of predictions with experimental results. Ann. Occup. Hyg. 2011, 55, 841–864. [Google Scholar] [CrossRef]
- Han, A.A.; Buerger, A.N.; Allen, H.; Vincent, M.; Thornton, S.A.; Unice, K.M.; Maier, A.; Quiñones-Rivera, A. Assessment of ethanol exposure from hand sanitizer use and potential for developmental toxicity in nursing infants. J. Appl. Toxicol. 2022, 42, 1424–1442. [Google Scholar] [CrossRef]
- Martin, S.A.; Oshiro, W.M.; Evansky, P.A.; Degn, L.L.; Ledbetter, A.D.; Ford, J.; Krantz, Q.T.; LeFew, W.R.; Beasley, T.E.; El-Masri, H.; et al. Use of novel inhalation kinetic studies to refine physiologically-based pharmacokinetic models for ethanol in non-pregnant and pregnant rats. Inhal. Toxicol. 2014, 26, 598–619. [Google Scholar] [CrossRef]
- Martin, S.A.; McLanahan, E.D.; Bushnell, P.J.; Hunter, E.S.; 3rd El-Masri, H. Species extrapolation of life-stage physiologically-based pharmacokinetic (PBPK) models to investigate the developmental toxicology of ethanol using in vitro to in vivo (IVIVE) methods. Toxicol. Sci. 2015, 143, 512–535. [Google Scholar] [CrossRef]
- Martin, P.R.; Singleton, C.K.; Hiller-Sturmhöfel, S. The role of thiamine deficiency in alcoholic brain disease. Alcohol. Res. Health 2003, 27, 134–142. [Google Scholar]
- More, S.L.; Thornton, S.A.; Maskrey, J.R.; Sharma, A.; De Gandiaga, E.; Cheng, T.J.; Fung, E.S.; Bernal, A.J.; Madl, A.K. PBPK modeling characterization of potential acute impairment effects from inhalation of ethanol during e-cigarette use. Inhal. Toxicol. 2020, 32, 14–23. [Google Scholar] [CrossRef]
- Dumas-Campagna, J.; Tardif, R.; Charest-Tardif, G.; Haddad, S. Ethanol toxicokinetics resulting from inhalation exposure in human volunteers and toxicokinetic modeling. Inhal. Toxicol. 2014, 26, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Plawecki, M.H.; Han, J.J.; Doerschuk, P.C.; Ramchandani, V.A.; O’Connor, S.J. Physiologically based pharmacokinetic (PBPK) models for ethanol. IEEE Trans. Biomed. Eng. 2008, 55, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Han, J.J.; Plawecki, M.H.; Doerschuk, P.C.; Ramchandani, V.A.; O’Connor, S. Ordinary differential equation models for ethanol pharmacokinetic based on anatomy and physiology. In Proceedings of the 2006 International Conference of the IEEE Engineering in Medicine and Biology Society, New York, NY, USA, 30 August–3 September 2006; IEEE: Piscataway, NJ, USA, 2006; pp. 5033–5036. [Google Scholar] [CrossRef]
- Ramchandani, V.A.; Plawecki, M.; Li, T.K.; O’Connor, S. Intravenous ethanol infusions can mimic the time course of breath alcohol concentrations following oral alcohol administration in healthy volunteers. Alcohol. Clin. Exp. Res. 2009, 33, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Plawecki, M.H.; Decarlo, R.; Ramchandani, V.A.; O’Connor, S. Improved Transformation of Morphometric Measurements for A Priori Parameter Estimation in a Physiologically-Based Pharmacokinetic Model of Ethanol. Biomed. Signal. Process. Control 2007, 2, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Plawecki, M.H.; DeCarlo, R.A.; Ramchandani, V.A.; O’Connor, S. Estimation of ethanol infusion profile to produce specified BrAC time course using physiologically-based pharmacokinetic (PBPK) models. Proceedngs of the 26th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, San Francisco, CA, USA, 1–5 September 2004; IEEE: Piscataway, NJ, USA, 2004; pp. 778–781. [Google Scholar] [CrossRef]
- Ramchandani, V.A.; Bolane, J.; Li, T.K.; O’Connor, S. A physiologically-based pharmacokinetic (PBPK) model for alcohol facilitates rapid BrAC clamping. Alcohol. Clin. Exp. Res. 1999, 23, 617–623. [Google Scholar] [CrossRef]
- O’Connor, S.; Ramchandani, V.A.; Li, T.K. PBPK modeling as a basis for achieving a steady BrAC of 60 +/− 5 mg% within ten minutes. Alcohol. Clin. Exp. Res. 2000, 24, 426–427. [Google Scholar] [CrossRef]
- Boyes, W.K.; Simmons, J.E.; Eklund, C.; Benignus, V.A.; Janssen, P.; Bushnell, P.J. Applications of dosimetry modeling to assessment of neurotoxic risk. Environ. Toxicol. Pharmacol. 2005, 19, 599–605. [Google Scholar] [CrossRef]
- Umulis, D.M.; Gürmen, N.M.; Singh, P.; Fogler, H.S. A physiologically based model for ethanol and acetaldehyde metabolism in human beings. Alcohol 2005, 35, 3–12. [Google Scholar] [CrossRef]
- Zhu, L.; Pei, W.; Thiele, I.; Mahadevan, R. Integration of a physiologically-based pharmacokinetic model with a whole-body, organ-resolved genome-scale model for characterization of ethanol and acetaldehyde metabolism. PLoS Comput. Biol. 2021, 17, e1009110. [Google Scholar] [CrossRef]
- Loizou, G.D.; Spendiff, M. A human PBPK model for ethanol describing inhibition of gastric motility. J. Mol. Histol. 2004, 35, 687–696. [Google Scholar] [CrossRef]
- Pastino, G.M.; Flynn, E.J.; Sultatos, L.G. Genetic polymorphisms in ethanol metabolism: Issues and goals for physiologically based pharmacokinetic modeling. Drug Chem. Toxicol. 2000, 23, 179–201. [Google Scholar] [CrossRef] [PubMed]
- Pastino, G.M.; Sultatos, L.G.; Flynn, E.J. Development and application of a physiologically based pharmacokinetic model for ethanol in the mouse. Alcohol Alcohol. 1996, 31, 365–374. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.J.; Rostami-Hodjegan, A.; Tucker, G.T.; Linkens, D.A. Analysis of solvent central nervous system toxicity and ethanol interactions using a human population physiologically based kinetic and dynamic model. Regul. Toxicol. Pharmacol. 2002, 35, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, P.; Ginsberg, G.; Hattis, D.; Johns, D.O.; Guyton, K.Z.; Sonawane, B. Genetic polymorphism in CYP2E1: Population distribution of CYP2E1 activity. J. Toxicol. Environ. Health B Crit Rev. 2009, 12, 362–388. [Google Scholar] [CrossRef] [PubMed]
- Loizou, G.D.; McNally, K.; Jones, K.; Cocker, J. The application of global sensitivity analysis in the development of a physiologically based pharmacokinetic model for m-xylene and ethanol co-exposure in humans. Front. Pharmacol. 2015, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Sadighi, A.; Leggio, L.; Akhlaghi, F. Development of a Physiologically Based Pharmacokinetic Model for Prediction of Ethanol Concentration-Time Profile in Different Organs. Alcohol Alcohol. 2021, 56, 401–414. [Google Scholar] [CrossRef]
- Kirman, C.R.; Sweeney, L.M.; Meek, M.E.; Gargas, M.L. Assessing the dose-dependency of allometric scaling performance using physiologically based pharmacokinetic modeling. Regul. Toxicol. Pharmacol. 2003, 38, 345–367. [Google Scholar] [CrossRef] [PubMed]
- Levitt, D.G. PKQuest_Java: Free, interactive physiologically based pharmacokinetic software package and tutorial. BMC Res. Notes 2009, 2, 158. [Google Scholar] [CrossRef]
- Levitt, D.G. PKQuest: Measurement of intestinal absorption and first pass metabolism—Application to human ethanol pharmacokinetics. BMC Clin. Pharmacol. 2002, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Levitt, D.G. Physiologically based pharmacokinetic modeling of arterial-antecubital vein concentration difference. BMC Clin. Pharmacol. 2004, 4, 2. [Google Scholar] [CrossRef]
- Liu, L.; Koo, Y.; Akwitti, C.; Russell, T.; Gay, E.; Laskowitz, D.T.; Yun, Y. Three-dimensional (3D) brain microphysiological system for organophosphates and neurochemical agent toxicity screening. PLoS ONE 2019, 14, e0224657. [Google Scholar] [CrossRef] [PubMed]
- Morzorati, S.L.; Ramchandani, V.A.; Li, T.K.; O’Connor, S. Maintaining steady state arterial alcohol levels in rats by using a physiologically based pharmacokinetic model. Alcohol 2002, 28, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Intertek. Industrial Forensic Science. 2022. Available online: www.intertek.com/forensics/industrial/ (accessed on 4 October 2022).
- Tyan, Y.C.; Yang, M.H.; Jong, S.B.; Wang, C.K.; Shiea, J. Melamine contamination. Anal. Bioanal. Chem. 2009, 395, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.B.; Phenrat, T.; Lohitnavy, M. Human continuous hydrogen cyanide inhalation predictor with a physiologically based pharmacokinetic (PBPK) model. Environ. Sci. Pollut. Res. Int. 2020, 27, 24650–24658. [Google Scholar] [CrossRef]
- Lai, I.K.; Dhakal, K.; Gadupudi, G.S.; Li, M.; Ludewig, G.; Robertson, L.W.; Olivier, A.K. N-acetylcysteine (NAC) diminishes the severity of PCB 126-induced fatty liver in male rodents. Toxicology 2012, 302, 25–33. [Google Scholar] [CrossRef]
- Guo, W.; Pan, B.; Sakkiah, S.; Yavas, G.; Ge, W.; Zou, W.; Tong, W.; Hong, H. Persistent Organic Pollutants in Food: Contamination Sources, Health Effects and Detection Methods. Int. J. Environ. Res. Public Health 2019, 16, 4361. [Google Scholar] [CrossRef]
- Loccisano, A.E.; Campbell, J.L., Jr.; Butenhoff, J.L.; Andersen, M.E.; Clewell, H.J., 3rd. Evaluation of placental and lactational pharmacokinetics of PFOA and PFOS in the pregnant, lactating, fetal and neonatal rat using a physiologically based pharmacokinetic model. Reprod. Toxicol. 2012, 33, 468–490. [Google Scholar] [CrossRef]
- Loccisano, A.E.; Campbell, J.L., Jr.; Andersen, M.E.; Clewell, H.J., 3rd. Evaluation and prediction of pharmacokinetics of PFOA and PFOS in the monkey and human using a PBPK model. Regul. Toxicol. Pharmacol. 2011, 59, 157–175. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Lin, Z. Bayesian evaluation of a physiologically based pharmacokinetic (PBPK) model for perfluorooctane sulfonate (PFOS) to characterize the interspecies uncertainty between mice, rats, monkeys, and humans: Development and performance verification. Environ. Int. 2019, 129, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Pérez, F.; Nadal, M.; Navarro-Ortega, A.; Fabrega, F.; Domingo, J.L.; Barcelo, D.; Farré, M. Accumulation of perfluoroalkyl substances in human tissues. Environ. Int. 2013, 59, 354–362. [Google Scholar] [CrossRef]
- Fàbrega, F.; Kumar, V.; Schuhmacher, M.; Domingo, J.L.; Nadal, M. PBPK modeling for PFOS and PFOA: Validation with human experimental data. Toxico.l Lett. 2014, 230, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Worley, R.R.; Yang, X.; Fisher, J. Physiologically based pharmacokinetic modeling of human exposure to perfluorooctanoic acid suggests historical non drinking-water exposures are important for predicting current serum concentrations. Toxicol. Appl. Pharmacol. 2017, 330, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Da Broi, U.; Colatutto, A.; Sala, P.; Desinan, L. Medico legal investigations into sudden sniffing deaths linked with trichloroethylene. J. Forensic Leg. Med. 2015, 34, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.C.; Fisher, J.W. Pharmacokinetic Modeling of Trichloroethylene and Trichloroacetic Acid in Humans. Risk Anal. 1993, 13, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Husband, H. Prevention of Organ-Specific Doxorubicin Induced Toxicity Using Physiologically-Based Pharmacokinetic Modeling and Therapeutic Drug Monitoring. 2021. Available online: https://digitalcommons.latech.edu/dissertations/906 (accessed on 27 September 2022).
- Stewart, D.J.; Grewaal, D.; Green, R.M.; Mikhael, N.; Goel, R.; Montpetit, V.A.; Redmond, M.D. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res. 1993, 13, 1945–1952. [Google Scholar]
- Subedi, N.; Yadav, B.N.; Jha, S.; Gurung, S.; Pradhan, A. An autopsy study of liver injuries in a tertiary referral centre of eastern Nepal. J. Clin. Diagn. Res. 2013, 7, 1686–1688. [Google Scholar] [CrossRef]
- Darwich, A.S.; Ogungbenro, K.; Vinks, A.A.; Powell, J.R.; Reny, J.-L.; Marsousi, N.; Daali, Y.; Fairman, D.; Cook, J.; Lesko, L.J.; et al. Why has model-informed precision dosing not yet become common clinical reality? lessons from the past and a roadmap for the future. Clin. Pharmacol. Ther. 2017, 101, 646–656. [Google Scholar] [CrossRef]
- Mantinieks, D.; Gerostamoulos, D.; Glowacki, L.; Di Rago, M.; Schumann, J.; Woodford, N.W.; Drummer, O.H. Postmortem Drug Redistribution: A Compilation of Postmortem/Antemortem Drug Concentration Ratios. J. Anal. Toxicol. 2020, 45, 368–377. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Levine, B.; Locke, J.L.; Smialek, J.E. A Fatality Due to Alprazolam Intoxication. J. Anal. Toxicol. 1997, 21, 218–220. [Google Scholar] [CrossRef][Green Version]
- Adams, P.C.; Holt, D.W.; Storey, G.C.; Morley, A.R.; Callaghan, J.; Campbell, R.W. Amiodarone and its desethyl metabolite: Tissue distribution and morphologic changes during long-term therapy. Circulation 1985, 72, 1064–1075. [Google Scholar] [CrossRef]
- Øiestad, Å.M.L.; Karinen, R.; Rogde, S.; Nilsen, S.; Eldor, K.-B.B.; Brochmann, G.-W.; Arnestad, M.; Øiestad, E.L.; Peres, M.D.; Kristoffersen, L.; et al. Comparative Study of Postmortem Concentrations of Antidepressants in Several Different Matrices. J. Anal. Toxicol. 2018, 42, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Adjutantis, G.; Coutselinis, A.; Dimopoulous, G. Fatal Intoxication with Amphetamines (A Case Report). Med. Sci. Law 1975, 15, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Tracqui, A.; Kintz, P.; Ludes, B. Buprenorphine-Related Deaths Among Drug Addicts in France: A Report on 20 Fatalities. J. Anal. Toxicol. 1998, 22, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Klys, M.; Bystrowska, B.; Bujak-Gizycka, B. Postmortem Toxicology of Carbamazepine. J. Anal. Toxicol. 2003, 27, 243–248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferner, R.E. Post-mortem clinical pharmacology. Br. J. Clin. Pharmacol. 2008, 66, 430–443. [Google Scholar] [CrossRef]
- Magliaro, C.; Ahluwalia, A. Biomedical research on substances of abuse: The Italian case of study. Altern. Lab. Anim. 2022, 50, 423–436. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Triyatni, M.; Mueller, M.B.; Klammers, F.; Leonard, B.; Turley, D.; Schmaler, J.; Ekiciler, A.; Molitor, B.; Walter, I.; et al. Simultaneous Assessment of Clearance, Metabolism, Induction and Drug-Drug Interaction Potential Using a Long-Term In Vitro Liver Model for a Novel Hepatitis B Virus Inhibitor. J. Pharmacol. Exp. Ther. 2018, 365, 237–248. [Google Scholar] [CrossRef]
- Di Maio, V.J.M.; Henry, L.D. Chloroquine Poisoning. South. Med. J. 1974, 67, 1031–1035. [Google Scholar] [CrossRef]
- Levine, B.; Zhang, X.; Smialek, J.E.; Kunsman, G.W.; Frontz, M.E. Citalopram Distribution in Postmortem Cases. J. Anal. Toxicol. 2001, 25, 641–644. [Google Scholar] [CrossRef][Green Version]
- Keller, T.; Miki, A.; Binda, S.; Dirnhofer, R. Fatal overdose of clozapine. Forensic Sci. Int. 1997, 86, 119–125. [Google Scholar] [CrossRef]
- Chatterton, C.N.; Handy, R.P.; Shoemaker, G.K.; Scott-Ham, M. The distribution and redistribution of carfentanil in post mortem samples. Forensic Sci. Int. 2020, 309, 110215. [Google Scholar] [CrossRef] [PubMed]
- Gambaro, V.; Argo, A.; Cippitelli, M.; Dell’Acqua, L.; Farè, F.; Froldi, R.; Guerrini, K.; Roda, G.; Rusconi, C.; Procaccianti, P. Unexpected Variation of the Codeine/Morphine Ratio Following Fatal Heroin Overdose. J. Anal. Toxicol. 2014, 38, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.W.; Krasula, R.W.; Soyka, L.F.; Hastreiter, A.R. Postmortem tissue digoxin concentrations in infants and children. Circulation 1975, 52, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, J.; Ojala, K.; Reissell, P. Tissue Concentrations of Digoxin in an Autopsy Material. Acta Pharmacol. Toxicol. 1974, 34, 385–390. [Google Scholar] [CrossRef]
- Engelhart, D.A.; Lavins, E.S.; Seligman, S.S.; Sutheimer, C.A. Diltiazem and pentoxifylline determination in postmortem specimens. J. Anal. Toxicol. 1997, 21, 576–579. [Google Scholar] [CrossRef][Green Version]
- Levine, B.; Klette, K.; Radentz, S.; Smith, M.L.; Smialek, J.E. Antihistamine concentrations in postmortem blood and liver specimens. Forensic Sci. Int. 1996, 81, 73–76. [Google Scholar] [CrossRef]
- Chatterton, C.N.; Scott-Ham, M. The distribution and redistribution of fentanyl & norfentanyl in post mortem samples. Forensic Sci. Int. 2018, 284, 146–152. [Google Scholar] [CrossRef]
- Johnson, R.D.; Lewis, R.J.; Angier, M.K. The Distribution of Fluoxetine in Human Fluids and Tissues. J. Anal. Toxicol. 2007, 31, 409–414. [Google Scholar] [CrossRef]
- Poklis, A.; Mackell, M.A.; Tucker, E.F. Tissue distribution of lidocaine after fatal accidental injection. J. Forensic Sci. 1984, 29, 1229–1236. [Google Scholar] [CrossRef]
- Garriott, J.C.; Sturner, W.Q.; Mason, M.F. Toxicologic findings in six fatalities involving methadone. Clin. Toxicol. 1973, 6, 163–173. [Google Scholar] [CrossRef]
- Moore, K.A.; Levine, B.; Smith, M.L.; Saki, S.; Schames, J.; Smialek, J.E. Tissue distribution of mirtazapine (Remeron) in postmortem cases. J. Anal. Toxicol. 1999, 23, 541–543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bogusz, M.J. Postmortem distribution pattern of morphine and morphine glucuronides in heroin overdose. Int. J. Legal. Med. 1997, 110, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; McIntyre, I.M. Postmortem tissue concentrations of olanzapine. J. Anal. Toxicol. 2009, 33, 15–26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anderson, D.T.; Fritz, K.L.; Muto, J.J. Oxycontin: The concept of a “ghost pill” and the postmortem tissue distribution of oxycodone in 36 cases. J. Anal. Toxicol. 2002, 26, 448–459. [Google Scholar] [CrossRef][Green Version]
- Maestri, L.; Negri, S.; Ferrari, M.; Ghittori, S.; Fabris, F.; Danesino, P.; Imbriani, M. Determination of perfluorooctanoic acid and perfluorooctanesulfonate in human tissues by liquid chromatography/single quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 2728–2734. [Google Scholar] [CrossRef]
- Breivik, H.; Frost, J.; Løkken, T.N.; Slørdal, L. Post mortem tissue distribution of quetiapine in forensic autopsies. Forensic Sci. Int. 2020, 315, 110413. [Google Scholar] [CrossRef]
- Saenz, S.R.; Lewis, R.J.; Angier, M.K.; Wagner, J.R. Postmortem Fluid and Tissue Concentrations of THC, 11-OH-THC and THC-COOH. J. Anal. Toxicol. 2017, 41, 508–516. [Google Scholar] [CrossRef]
- De Decker, K.; Cordonnier, J.; Jacobs, W.; Coucke, V.; Schepens, P.; Jorens, P.G. Fatal intoxication due to tramadol alone: Case report and review of the literature. Forensic Sci. Int. 2008, 175, 79–82. [Google Scholar] [CrossRef]
- Gock, S.B.; Wong, S.H.; Nuwayhid, N.; Venuti, S.E.; Kelley, P.D.; Teggatz, J.R.; Jentzen, J.M. Acute zolpidem overdose—Report of two cases. J. Anal. Toxicol. 1999, 23, 559–562. [Google Scholar] [CrossRef]
- Boniface, P.J.; Russell, S.G. Two cases of fatal zopiclone overdose. J. Anal. Toxicol. 1996, 20, 131–133. [Google Scholar] [CrossRef][Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fairman, K.; Choi, M.-K.; Gonnabathula, P.; Lumen, A.; Worth, A.; Paini, A.; Li, M. An Overview of Physiologically-Based Pharmacokinetic Models for Forensic Science. Toxics 2023, 11, 126. https://doi.org/10.3390/toxics11020126
Fairman K, Choi M-K, Gonnabathula P, Lumen A, Worth A, Paini A, Li M. An Overview of Physiologically-Based Pharmacokinetic Models for Forensic Science. Toxics. 2023; 11(2):126. https://doi.org/10.3390/toxics11020126
Chicago/Turabian StyleFairman, Kiara, Me-Kyoung Choi, Pavani Gonnabathula, Annie Lumen, Andrew Worth, Alicia Paini, and Miao Li. 2023. "An Overview of Physiologically-Based Pharmacokinetic Models for Forensic Science" Toxics 11, no. 2: 126. https://doi.org/10.3390/toxics11020126
APA StyleFairman, K., Choi, M.-K., Gonnabathula, P., Lumen, A., Worth, A., Paini, A., & Li, M. (2023). An Overview of Physiologically-Based Pharmacokinetic Models for Forensic Science. Toxics, 11(2), 126. https://doi.org/10.3390/toxics11020126