Quantification of Morpholine in Peel and Pulp of Apples and Oranges by Gas Chromatography−Mass Spectrometry




Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Sample Collection
2.3. Optimization of Sample Preparation Method for Morpholine Analysis in Fruit Peel and Pulp
2.4. Method Validation (Method Detection Limit, Method Quantification Limit, Linearity, Accuracy, Precision, Cross-Lab Validation, and Measurement Uncertainty)
2.5. Morpholine Analysis by GC-MS
3. Results and Discussion
3.1. Method Validation for Morpholine Analysis
3.2. Applying the Method to Real Samples (Fruits and Vegetables)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuchowicz, E.; Rydzyński, K. Risk assessment of morpholine (tetrahydro-2H-1, 4-oxazine): A time for reevaluation of current occupational exposure standards? Appl. Occup. Environ. Hyg 1998, 13, 113–121. [Google Scholar] [CrossRef]
- Vakkalanka, M., Sr.; D’Souza, T.; Ray, S.; Yam, K.L.; Mir, N. 7—Emerging packaging technologies for fresh produce. In Emerging Food Packaging Technologies; Yam, K.L., Lee, D.S., Eds.; Woodhead Publishing: Cambridge, UK, 2012; pp. 109–133. [Google Scholar]
- Walker, M.J.; Gray, K.; Hopley, C.; Bell, D.; Colwell, P.; Maynard, P.; Burns, D.T. Forensically robust detection of the presence of morpholine in apples—Proof of principle. Food Anal. Methods 2012, 5, 874–880. [Google Scholar] [CrossRef]
- McGuire, R.G.; Hagenmaier, R.D. Shellac coatings for grapefruits that favor biological control of Penicillium digitatum by Candida oleophila. Biol. Control 1996, 7, 100–106. [Google Scholar] [CrossRef]
- El-Gamal, I.; Khidr, T.; Ghuiba, F. Nitrogen-based copolymers as wax dispersants for paraffinic gas oils. Fuel 1998, 77, 375–385. [Google Scholar] [CrossRef]
- Njombolwana, N.S.; Erasmus, A.; Van Zyl, J.G.; du Plooy, W.; Cronje, P.J.; Fourie, P.H. Effects of citrus wax coating and brush type on imazalil residue loading, green mould control and fruit quality retention of sweet oranges. Postharvest Biol. Tech. 2013, 86, 362–371. [Google Scholar] [CrossRef]
- Kumar, R.; Kapur, S. Morpholine: A glazing agent for fruits and vegetables coating/waxing. Int. J. Sci. Technol. Eng. 2016, 2, 694–697. [Google Scholar]
- USDA. Morpholine Was Included in EPA (Environment Protection Agencies, United State). Available online: https://www.ams.usda.gov/sites/default/files/media/Morph%20Technical%20Advisory%20Panel%20Report.pdf (accessed on 15 August 2019).
- Health Canada, Archived—A Summary of Health Hazard Assessment of Morpholine in Wax Coatings of Apples. Available online: https://www.canada.ca/en/health-canada/services/food-nutrition/food-safety/information-product/summary-health-hazard-assessment-morpholine-coatings-apples.html (accessed on 15 August 2019).
- Tannenbaum, S.R.; Archer, M.C.; Wishnok, J.S.; Bishop, W.W. Nitrosamine formation in human saliva. J. Natl. Cancer Inst. 1978, 60, 251–253. [Google Scholar] [CrossRef] [PubMed]
- European Union Food Additives Database. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32008R1333 (accessed on 15 August 2019).
- MFDS (Korea Ministry of Food and Drug Safety) Food Additives Code. Available online: https://www.mfds.go.kr/eng/brd/m_15/view.do?seq=72242&srchFr=&srchTo=&srchWord=code&srchTp=7&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&company_nm=&page=1 (accessed on 27 December 2019).
- MHLW (Ministry of Health, Labour and Welfare). Morpholine Salts of Fatty Acids. Available online: https://www.mhlw.go.jp/shingi/2005/10/dl/s1027-7g07b.pdf (accessed on 15 August 2019).
- MOH (Ministry of Health of the People’s Republic of China). Standards for Use of Food Additives. Available online: http://www.cirs-reach.com/China_Chemical_Regulation/GB_2760-2011_Food_Safety_National_Standards_for_the_Usage_of_Food_Additives.html (accessed on 15 August 2019).
- Korean Statistical Information Service. Available online: http://kosis.kr/index/index.do (accessed on 19 May 2020).
- Baldwin, E.A. Edible coatings for fresh fruits and vegetables: Past, present, and future. In Edible Coatings and Films to Improve Food Quality; CRC Press: Boca Raton, FL, USA, 1994; pp. 25–82. [Google Scholar]
- Chen, D.; Miao, H.; Zou, J.; Cao, P.; Ma, N.; Zhao, Y.; Wu, Y. Novel dispersive micro-solid-phase extraction combined with ultrahigh-performance liquid chromatography–high-resolution mass spectrometry to determine morpholine residues in citrus and apples. J. Agric. Food Chem. 2015, 63, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Hengel, M.J.; Jordan, R.; Maguire, W. Development and validation of a standardized method for the determination of morpholine residues in fruit commodities by liquid chromatography–mass spectrometry. J. Agric. Food Chem. 2014, 62, 3697–3701. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Han, C.; Qian, Y.; Ding, H.; Chen, X.; Xi, J. Determination of neonicotinoid pesticides residues in agricultural samples by solid-phase extraction combined with liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Kanrar, B.; Mandal, S.; Bhattacharyya, A. Validation and uncertainty analysis of a multiresidue method for 42 pesticides in made tea, tea infusion and spent leaves using ethyl acetate extraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Yan, H.; Fu, S.; Dai, H. Simultaneous determination of multiresidual phenyl acetanilide pesticides in different food commodities by solid-phase cleanup and gas chromatography-mass spectrometry. J. Sep. Sci. 2013, 36, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Gros, P.; Matignon, F.; Plonevez, S. Determination of morpholine in fruits using LC− MS/MS. Ann. Falsif. Expert. Chim. Toxicol. 2011, 974, 17–22. [Google Scholar]
- Sen, N.P.; Baddoo, P.A. An investigation on the possible presence of morpholine and N-nitrosomorpholine in wax-coated apples. J. Food Saf. 1988, 9, 183–191. [Google Scholar] [CrossRef]
- Hotchkiss, J.H.; Vecchio, A.J. Analysis of direct contact paper and paperboard food packaging for N-nitrosomorpholine and morpholine. J. Food Sci. 1983, 48, 240–242. [Google Scholar] [CrossRef]
- Fu, C.G.; Xu, H.-D. High-performance liquid chromatography with post-column chemiluminescence detection for simultaneous determination of trace N-nitrosamines and corresponding secondary amines in groundwater. Analyst 1995, 120, 1147–1151. [Google Scholar] [CrossRef]
- Ikeda, K.; Migliorese, K.G.; Curtis, H. Analysis of nitrosamines in cosmetics. J. Soc. Cosmet. Chem 1990, 41, 283–333. [Google Scholar]
- Gao, Y.; Cao, Y.; Chen, S.-L.; Wang, Y.; Zhu, R.; Sun, W.; Zhang, Q.; Fan, Y. Differences in product distribution measured with flame ionization detector gas chromatography and thermal conductivity detector gas chromatography during the dimethyl ether-to-olefins and methanol-to-olefins processes. Energ. Fuel 2017, 31, 13266–13272. [Google Scholar] [CrossRef]
- Cao, M.; Zhang, P.; Feng, Y.; Zhang, H.; Zhu, H.; Lian, K.; Kang, W.J. Development of a method for rapid determination of morpholine in juices and drugs by gas chromatography-mass spectrometry. J. Anal. Methods Chem. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y. Chapter 6—Drying of Citrus Peel and Processing of Foods and Feeds. In Comprehensive Utilization of Citrus By-Products; Shan, Y., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 75–83. [Google Scholar] [CrossRef]
- ICH. Validation of Analytical Procedures: Text and Methodology. Available online: https://www.ema.europa.eu/en/ich-q2-r1-validation-analytical-procedures-text-methodology. (accessed on 15 August 2019).
- KRISS (Korea Research Institute of Standards and Science). ISO/IEC Guide 98-3:2008 Guide to the Expression of Uncertainty in Measurement. Available online: https://www.kriss.re.kr/w/fileDownload.do?fileSeq=1008 (accessed on 15 August 2019).
- Eurachem. Quantifying Uncertainty in Analytical Measurement. Available online: https://www.eurachem.org/images/stories/Guides/pdf/QUAM2012_P1.pdf (accessed on 15 August 2019).
- ISO2008. ISO/IEC Guide 98-3: 2008 (E) Uncertainty of Measurement-Part 3: Guide to the Expression of Uncertainty in Measurement. Available online: http://www.iec.ch/dyn/www/f?p=103%3A391%3A0%3A%3A%3A%3AP391_PUB_ID%2CP391_LANG%3A11961 (accessed on 15 August 2019).
- Ohnishi, T.; Kubota, M.; Okada, A.; Tonami, K. Residual survey investigation and removal efficiency by washing with kitchen detergent of food additive morpholine. Hokuriku Koshu Eisei Gakkaishi 1983, 10, 63–67. [Google Scholar]


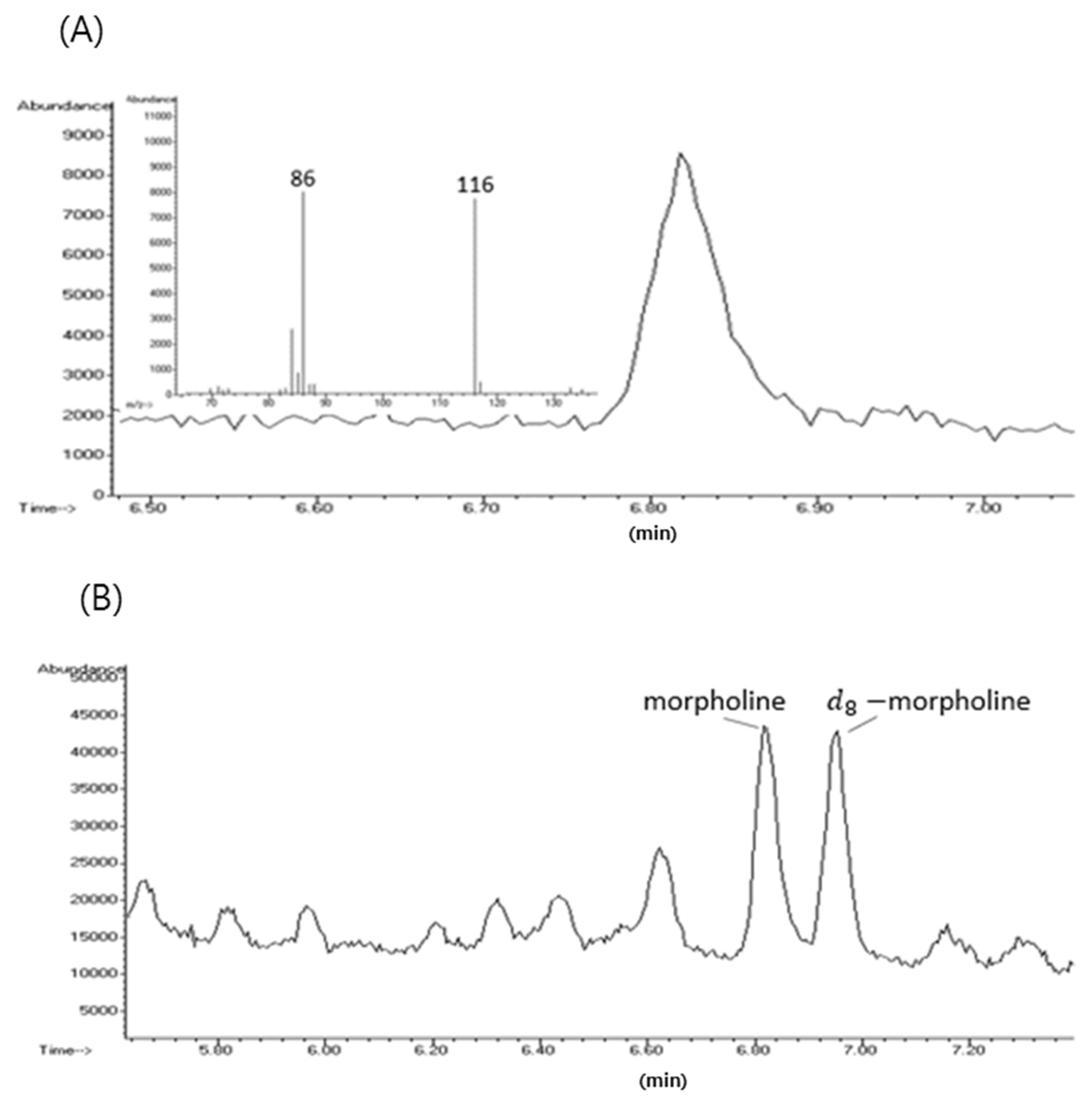{kind=link}
| Matrix | Standard Curve a | Linearity (R2) | Calibration Range (µg/kg) | MDL b (µg/kg) | MQL c (µg/kg) |
|---|---|---|---|---|---|
| Apple peel | y = 1.0280x − 0.0081 | 0.9999 | 10–400 | 3.3 | 10.1 |
| Apple pulp | y = 1.0136x − 0.0041 | 1.0000 | 10–400 | 1.3 | 4.1 |
| Orange peel | y = 1.0173x − 0.0035 | 1.0000 | 10–400 | 2.3 | 8.5 |
| Orange pulp | y = 1.0126x − 0.0051 | 0.9999 | 10–400 | 2.9 | 8.7 |
| Intra-Day (n = 5) | ||||||||
| Morpholine Concentration (µg/kg) | Apple Peel | Apple Pulp | Orange Peel | Orange Pulp | ||||
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |
| 10 | 104.0 | 4.1 | 102.2 | 0.9 | 104.4 | 0.9 | 102.0 | 9.4 |
| 25 | 101.3 | 4.1 | 102.0 | 1.4 | 100.5 | 1.4 | 99.8 | 1.5 |
| 50 | 99.7 | 2.5 | 98.9 | 3.7 | 98.0 | 3.7 | 97.8 | 1.6 |
| 100 | 101.0 | 2.2 | 98.7 | 3.0 | 100.7 | 3.0 | 101.2 | 1.6 |
| 200 | 98.7 | 1.6 | 100.8 | 2.7 | 99.8 | 2.7 | 99.9 | 3.4 |
| 400 | 100.3 | 2.9 | 99.9 | 0.8 | 100.0 | 0.8 | 102.2 | 2.4 |
| Inter-Day (3 Days) | ||||||||
| Morpholine Concentration (µg/kg) | Apple Peel | Apple Pulp | Orange Peel | Orange Pulp | ||||
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |
| 25 | 101.3 | 2.8 | 99.8 | 1.9 | 98.5 | 1.9 | 103.2 | 1.9 |
| 100 | 99.8 | 2.2 | 99.7 | 2.0 | 100.5 | 2.0 | 100.0 | 1.5 |
| 400 | 99.4 | 2.2 | 100.4 | 1.8 | 99.6 | 1.8 | 99.1 | 2.3 |
| Theoretical Morpholine Concentration (μg/kg DW) | Observed Concentration (μg/kg DW) | Recovery% | RSD% | ||
|---|---|---|---|---|---|
| Lab A | Lab B | Lab C | |||
| Apple peel | |||||
| 25 | 25 | 27 | 25 | 102.7% | 6.9 |
| 100 | 106 | 104 | 103 | 104.3% | 4.0 |
| 400 | 416 | 433 | 418 | 104.1% | 3.5 |
| Apple pulp | |||||
| 25 | 26 | 24 | 26 | 101.3% | 5.7 |
| 100 | 101 | 112 | 104 | 105.7% | 5.4 |
| 400 | 404 | 440 | 407 | 103.7% | 5.3 |
| Orange peel | |||||
| 25 | 25 | 25 | 25 | 100.0% | 3.4 |
| 100 | 96 | 107 | 102 | 101.7% | 5.6 |
| 400 | 420 | 427 | 406 | 103.9% | 3.5 |
| Orange pulp | |||||
| 25 | 26 | 27 | 25 | 104.0% | 6.0 |
| 100 | 98 | 104 | 100 | 100.7% | 6.2 |
| 400 | 421 | 420 | 414 | 103.4% | 3.3 |
| Matrix | U1 a | U2 b | U3 c | U4 d | U5 e | U f |
|---|---|---|---|---|---|---|
| Apple peel | 0.0084 | 0.0116 | 0.0058 | 0.0374 | 0.0530 | 13.15 |
| Apple pulp | 0.0084 | 0.0116 | 0.0058 | 0.0301 | 0.0050 | 6.79 |
| Orange peel | 0.0084 | 0.0116 | 0.0058 | 0.0250 | 0.0270 | 6.67 |
| Orange pulp | 0.0084 | 0.0116 | 0.0058 | 0.0035 | 0.0110 | 3.27 |
| n | Morpholine Range (mg/kg DW) | Average Morpholine (mg/kg DW) | |||
|---|---|---|---|---|---|
| Apple peel | |||||
| Korea | 8 | n.d. b | n.d. | ||
| USA | 4 | 1.15–11.19 | 4.4 | ± | 4.7 |
| China | 5 | n.d. | n.d. | ||
| Apple pulp | |||||
| Korea | 8 | n.d. | n.d. | ||
| USA | 2 | n.d. | n.d. | ||
| China | 5 | n.d. | n.d. | ||
| Orange peel | |||||
| Korea | 5 | 0.97–12.82 | 5.8 | ± | 5.4 |
| China | 2 | n.d. | n.d. | ||
| Orange pulp | |||||
| Korea | 5 | n.d. | n.d. | ||
| China | 2 | n.d. | n.d. | ||
| Mandarin peel | |||||
| Korea | 2 | 0.91–0.92 | 0.9 | ± | 0.1 |
| Mandarin pulp | |||||
| Korea | 2 | n.d. | n.d. | ||
| Cucumbera | 1 | n.d. | n.d. | ||
| Squasha | 1 | n.d. | n.d. | ||
| Paprikaa | 1 | n.d. | n.d. | ||
| Sample Information (Purchased Location) | Origin | Morpholine Content in Peel (mg/kg DW) | Morpholine Content in Pulp (mg/kg DW) | ||
|---|---|---|---|---|---|
| Apple1 (Korea) | Korea | n.d. a | n.d. | ||
| Apple2 (Korea) | Korea | n.d. | n.d. | ||
| Apple3 (Korea) | Korea | n.d. | n.d. | ||
| Apple4 (Korea) | Korea | n.d. | n.d. | ||
| Apple5 (Korea) | Korea | n.d. | n.d. | ||
| Apple6 (Korea) | Korea | n.d. | n.d. | ||
| Apple7 (Korea) | Korea | n.d. | n.d. | ||
| Apple8 (Korea) | Korea | n.d. | n.d. | ||
| Apple9 (China) | Dalian, China | n.d. | n.d. | ||
| Apple10 (China) | Dalian, China | n.d. | n.d. | ||
| Apple11 (China) | Dalian, China | n.d. | n.d. | ||
| Apple12 (China) | Dalian, China | n.d. | n.d. | ||
| Apple13 (China) | Dalian, China | n.d. | n.d. | ||
| Apple14 (USA) | USA | 3.97 | ± | 0.10 | n.d. |
| Apple15 (USA) | USA | 11.19 | ± | 0.37 | n.d. |
| Apple16 (USA) | USA | 1.25 | ± | 0.25 | n.d. |
| Apple17 (USA) | USA | 1.15 | ± | 0.02 | n.d. |
| Orange1 (Korea) | California, USA | n.d. | n.d. | ||
| Orange2 (Korea) | Cobram, Australia | 3.68 | ± | 0.08 | n.d. |
| Orange3 (Korea) | Cobram, Australia | 12.82 | ± | 0.79 | n.d. |
| Orange4 (Korea) | Cobram, Australia | 0.97 | ± | 0.05 | n.d. |
| Orange5 (Korea) | California, USA | n.d. | n.d. | ||
| Orange6 (China) | California, USA | n.d. | n.d. | ||
| Orange7 (China) | California, USA | n.d. | n.d. | ||
| Mandarine1 (Korea) | Jeju, Korea | 0.92 | ± | 0.09 | n.d. |
| Mandarine2 (Korea) | Jeju, Korea | 0.91 | ± | 0.01 | n.d. |
| Cucumber (Korea) | Korea | n.d. | n.d. | ||
| Squash (Korea) | Korea | n.d. | n.d. | ||
| Paprika (Korea) | Korea | n.d. | n.d. | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, K.; Kim, I.; Lee, C.; Moon, J.-K.; Suh, H.-J.; Lee, J. Quantification of Morpholine in Peel and Pulp of Apples and Oranges by Gas Chromatography−Mass Spectrometry. Foods 2020, 9, 746. https://doi.org/10.3390/foods9060746
An K, Kim I, Lee C, Moon J-K, Suh H-J, Lee J. Quantification of Morpholine in Peel and Pulp of Apples and Oranges by Gas Chromatography−Mass Spectrometry. Foods. 2020; 9(6):746. https://doi.org/10.3390/foods9060746
Chicago/Turabian StyleAn, Kunho, Inhwan Kim, Chan Lee, Joon-Kwan Moon, Hee-Jae Suh, and Jihyun Lee. 2020. "Quantification of Morpholine in Peel and Pulp of Apples and Oranges by Gas Chromatography−Mass Spectrometry" Foods 9, no. 6: 746. https://doi.org/10.3390/foods9060746
APA StyleAn, K., Kim, I., Lee, C., Moon, J.-K., Suh, H.-J., & Lee, J. (2020). Quantification of Morpholine in Peel and Pulp of Apples and Oranges by Gas Chromatography−Mass Spectrometry. Foods, 9(6), 746. https://doi.org/10.3390/foods9060746