Austrian Raw-Milk Hard-Cheese Ripening Involves Successional Dynamics of Non-Inoculated Bacteria and Fungi

 ,
,  , , , , and
, , , , and
Abstract
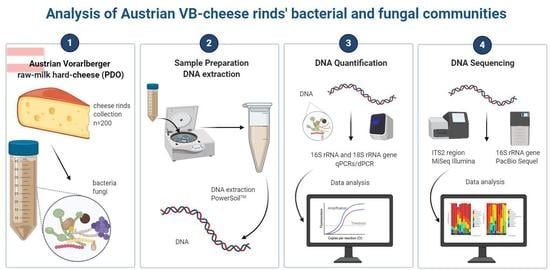
1. Introduction
2. Materials and Methods
2.1. VB-Cheese Production
2.2. Cheese Rind Sampling
2.3. DNA Extraction
2.4. qPCR and dPCR Analysis of 18S rRNA Gene
2.5. qPCR Statistics
2.6. Gene-Targeted Sequencing
2.7. Bioinformatic Data Analysis
3. Results
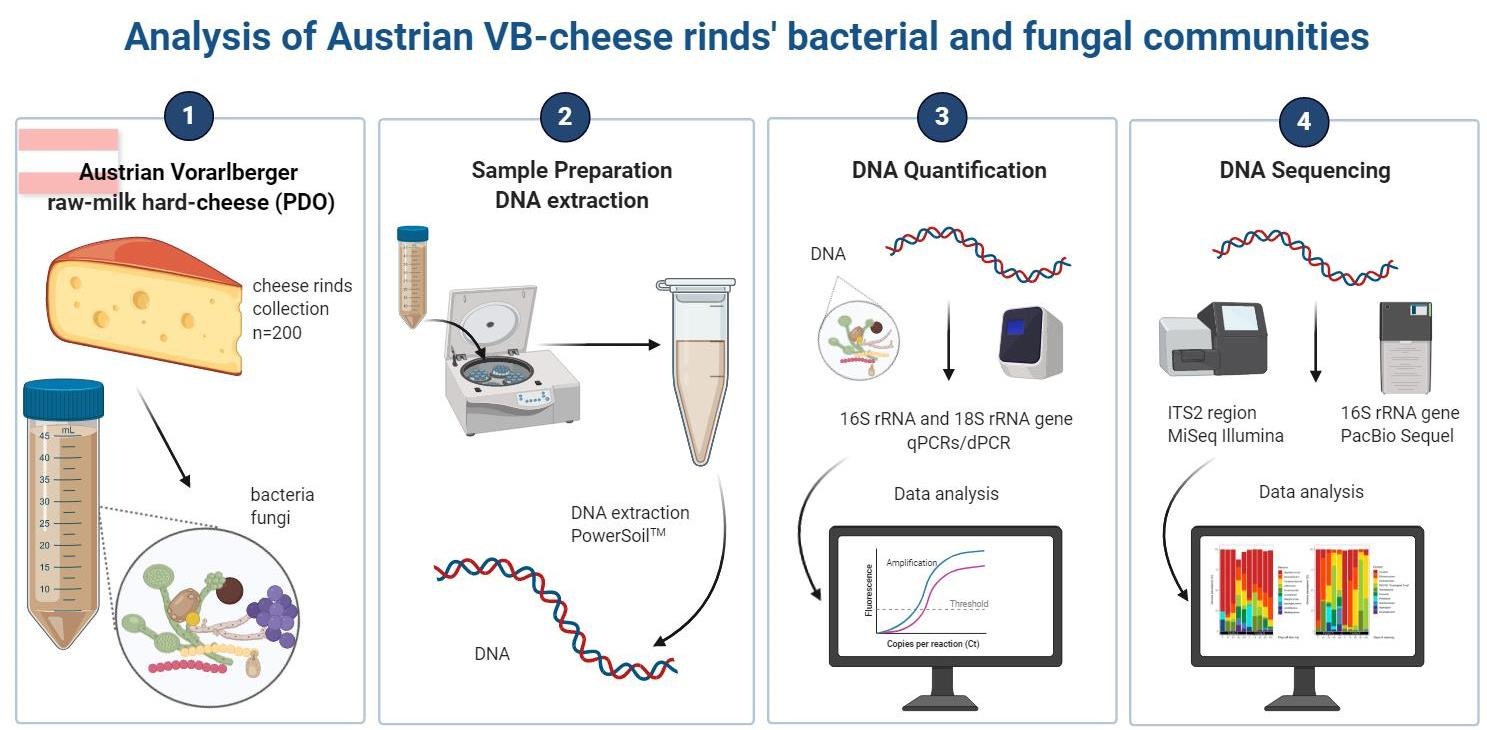
3.1. Quantification of Bacteria and Fungi in VB Rinds throughout Ripening Using qPCR and dPCR
3.2. Investigation of Microbial Communities in VB Rinds throughout Ripening by Gene-Targeted Sequencing
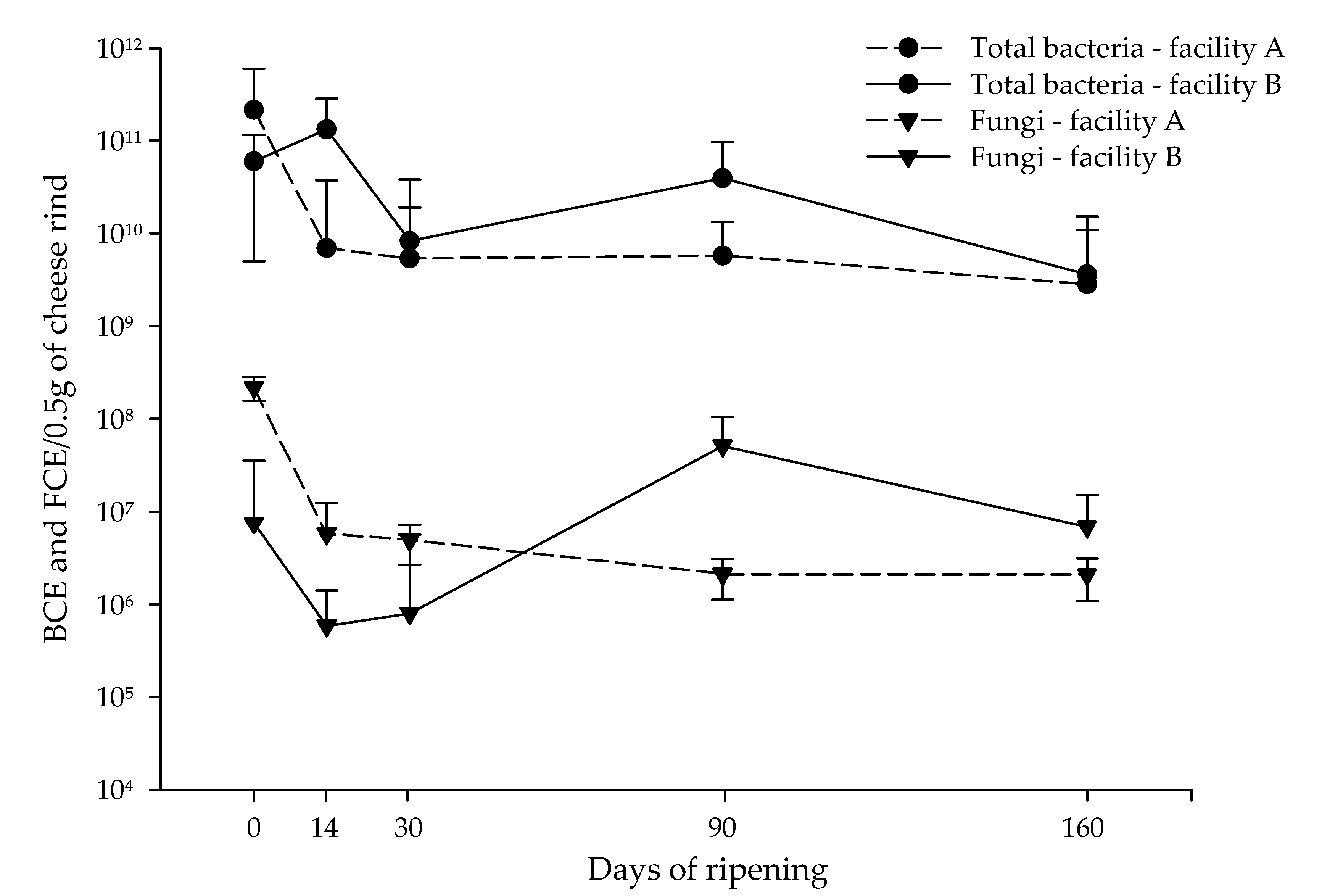
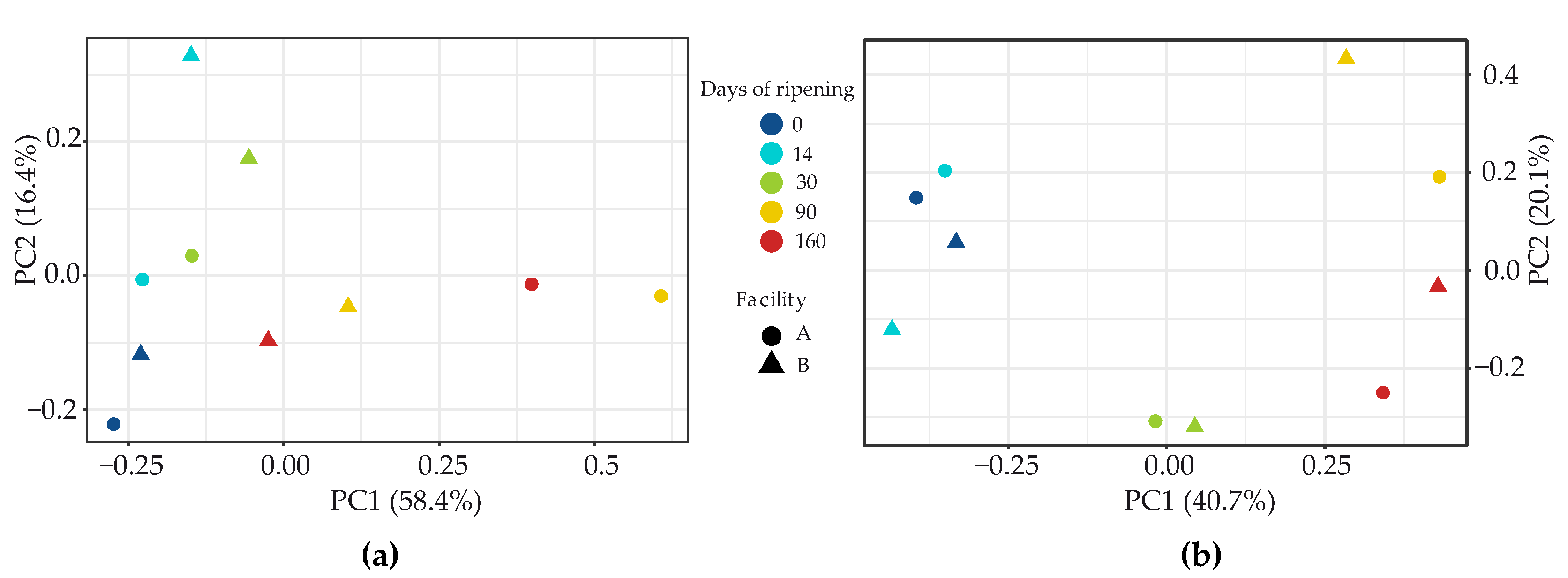
3.2.1. Bacteria
3.2.2. Fungi
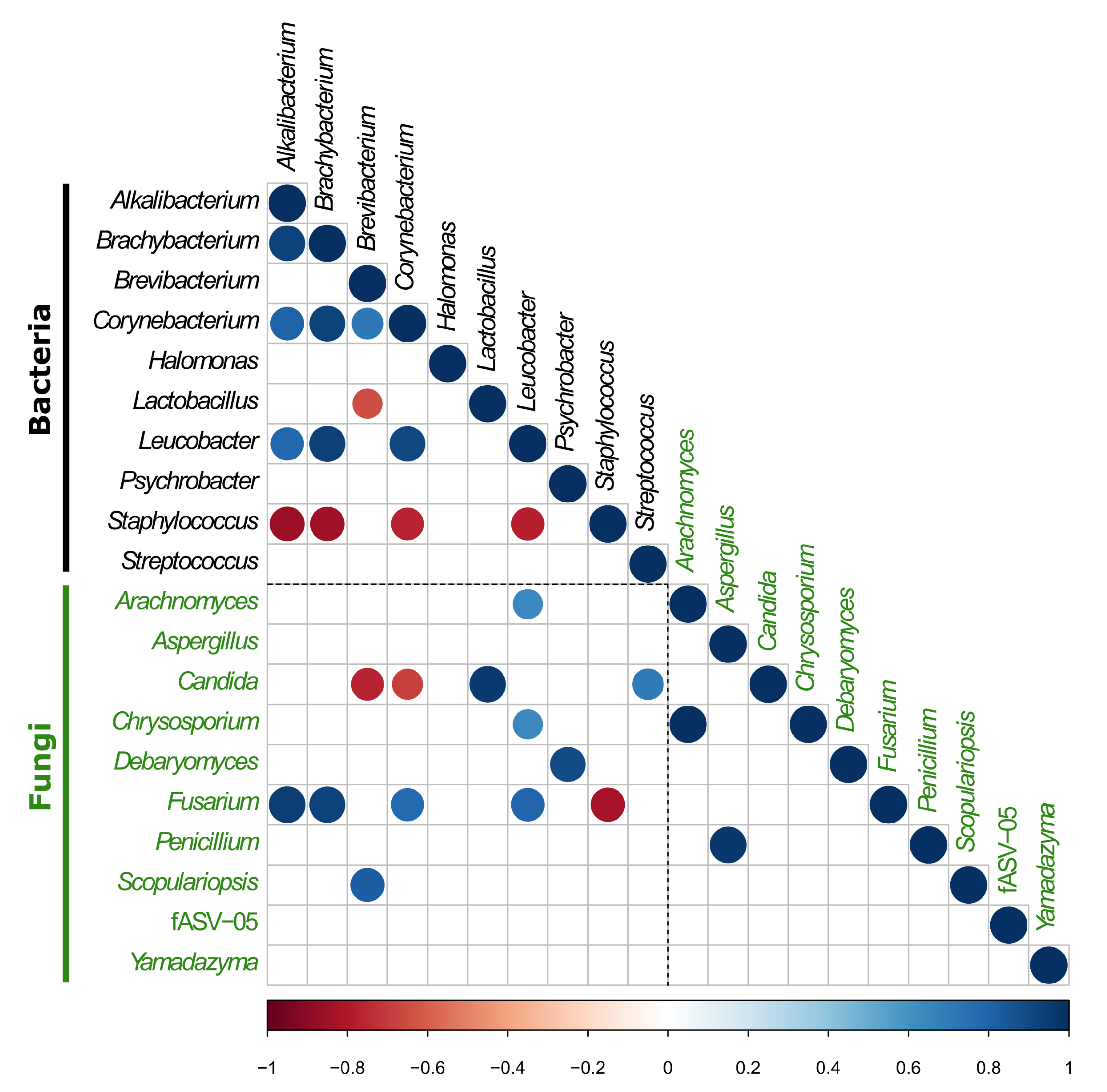
3.2.3. Investigation of the Potential Correlation between Bacterial and Fungal Communities in VB Rinds throughout Ripening
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salque, M.; Bogucki, P.I.; Pyzel, J.; Sobkowiak-Tabaka, I.; Grygiel, R.; Szmyt, M.; Evershed, R.P. Earliest evidence for cheese making in the sixth millennium bc in northern Europe. Nature 2012, 493, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Irlinger, F.; Layec, S.; Hélinck, S.; Dugat-Bony, E. Cheese rind microbial communities: Diversity, composition and origin. FEMS Microbiol. Lett. 2014, 362, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boldyreva, E.M.; Hill, A.; Griffiths, M.; Marcone, M. The quality and safety of washed-rind cheeses with a focus on antilisterial protection. Int. Dairy J. 2016, 55, 26–37. [Google Scholar] [CrossRef]
- Irlinger, F.; Mounier, J. Microbial interactions in cheese: Implications for cheese quality and safety. Curr. Opin. Biotechnol. 2009, 20, 142–148. [Google Scholar] [CrossRef]
- Monnet, C.; Landaud, S.; Bonnarme, P.; Swennen, D. Growth and adaptation of microorganisms on the cheese surface. FEMS Microbiol. Lett. 2015, 362, 1–9. [Google Scholar] [CrossRef]
- Calasso, M.; Ercolini, D.; Mancini, L.; Stellato, G.; Minervini, F.; Cagno, R.D.; Angelis, M.D.; Gobbetti, M. Relationships among house, rind and core microbiotas during manufacture of traditional Italian cheeses at the same dairy plant. Food Microbiol. 2016, 54, 115–126. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Mills, D.A. Facility-Specific “House” Microbiome Drives Microbial Landscapes of Artisan Cheesemaking Plants. Appl. Environ. Microbiol. 2013, 79, 5214–5223. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese Rind Communities Provide Tractable Systems for In Situ and In Vitro Studies of Microbial Diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef]
- Dugat-Bony, E.; Straub, C.; Teissandier, A.; Onésime, D.; Loux, V.; Monnet, C.; Irlinger, F.; Landaud, S.; Leclercq-Perlat, M.N.; Bento, P.; et al. Overview of a Surface-Ripened Cheese Community Functioning by Meta-Omics Analyses. PLoS ONE 2015, 10, e0124360. [Google Scholar] [CrossRef]
- Afshari, R.; Pillidge, C.J.; Dias, D.A.; Osborn, A.M.; Gill, H. Cheesomics: The future pathway to understanding cheese flavour and quality. Crit. Rev. Food Sci. Nutr. 2018, 60, 33–47. [Google Scholar] [CrossRef]
- Coton, M.; Delbés-Paus, C.; Irlinger, F.; Desmasures, N.; Fleche, A.L.; Stahl, V.; Montel, M.C.; Coton, E. Diversity and assessment of potential risk factors of Gram-negative isolates associated with French cheeses. Food Microbiol. 2012, 29, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Gori, K.; Ryssel, M.; Arneborg, N.; Jespersen, L. Isolation and Identification of the Microbiota of Danish Farmhouse and Industrially Produced Surface-Ripened Cheeses. Microb. Ecol. 2012, 65, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Monnet, C.; Dugat-Bony, E.; Swennen, D.; Beckerich, J.M.; Irlinger, F.; Fraud, S.; Bonnarme, P. Investigation of the Activity of the Microorganisms in a Reblochon-Style Cheese by Metatranscriptomic Analysis. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Schornsteiner, E.; Mann, E.; Bereuter, O.; Wagner, M.; Schmitz-Esser, S. Cultivation-independent analysis of microbial communities on Austrian raw milk hard cheese rinds. Int. J. Food Microbiol. 2014, 180, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Quijada, N.M.; Mann, E.; Wagner, M.; Rodríguez-Lázaro, D.; Hernández, M.; Schmitz-Esser, S. Autochthonous facility-specific microbiota dominates washed-rind Austrian hard cheese surfaces and its production environment. Int. J. Food Microbiol. 2018, 267, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Esser, S.; Dzieciol, M.; Nischler, E.; Schornsteiner, E.; Bereuter, O.; Mann, E.; Wagner, M. Abundance and potential contribution of Gram-negative cheese rind bacteria from Austrian artisanal hard cheeses. Int. J. Food Microbiol. 2018, 266, 95–103. [Google Scholar] [CrossRef]
- Anast, J.M.; Dzieciol, M.; Schultz, D.L.; Wagner, M.; Mann, E.; Schmitz-Esser, S. Brevibacterium from Austrian hard cheese harbor a putative histamine catabolism pathway and a plasmid for adaptation to the cheese environment. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Bleve, G.; Rizzotti, L.; Dellaglio, F.; Torriani, S. Development of Reverse Transcription (RT)-PCR and Real-Time RT-PCR Assays for Rapid Detection and Quantification of Viable Yeasts and Molds Contaminating Yogurts and Pasteurized Food Products. Appl. Environ. Microbiol. 2003, 69, 4116–4122. [Google Scholar] [CrossRef]
- Leclercq-Perlat, M.N.; Buono, F.; Lambert, D.; Latrille, E.; Spinnler, H.E.; Corrieu, G. Controlled production of Camembert-type cheeses. Part I: Microbiological and physicochemical evolutions. J. Dairy Res. 2004, 71, 346–354. [Google Scholar] [CrossRef]
- Lessard, M.H.; Bélanger, G.; St-Gelais, D.; Labrie, S. The Composition of Camembert Cheese-Ripening Cultures Modulates both Mycelial Growth and Appearance. Appl. Environ. Microbiol. 2012, 78, 1813–1819. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2018, 17, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Kachur, S.; Dwan, M.G.; Abraham, A.G.; Aziz, M.; Hsueh, P.R.; Huang, Y.T.; Busch, J.D.; Lamit, L.J.; Gehring, C.A.; et al. FungiQuant: A broad-coverage fungal quantitative real-time PCR assay. BMC Microbiol. 2012, 12, 255. [Google Scholar] [CrossRef]
- Kobayashi, T. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci. 2011, 68, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The Digital MIQE Guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- New England Biolabs. Restriction Enzymes for Droplet Digital PCR (ddPCR). 2020. Available online: international.neb.com (accessed on 7 November 2019).
- Whale, A.S.; Huggett, J.F.; Cowen, S.; Speirs, V.; Shaw, J.; Ellison, S.; Foy, C.A.; Scott, D.J. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 2012, 40, e82. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols; Elsevier: Amsterdam, The Netherlands, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing (v3.6.1); R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. GigaScience 2012, 1. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- UNITE Community. UNITE QIIME Release for Fungi; UNITE Community: London, UK, 2019. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric-Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- SIMPSON, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix, Version 0.84. 2017. Available online: https://github.com/taiyun/corrplot (accessed on 6 July 2020).
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. R Rackage Version 1.0.0. 2020. Available online: https://github.com/tidyverse/dplyr (accessed on 6 July 2020).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer International Publishing: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Joachimiak, M.P.; Weisman, J.L.; May, B.C. JColorGrid: Software for the visualization of biological measurements. BMC Bioinform. 2006, 7, 225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karst, S.; Albertsen, M.; Kirkegaard, R.; Dueholm, M.; Nielsen, P. Molecular methods. In Experimental Methods in Wastewater Treatment; van Loosdrecht, M., Halkjær Nielsen, P., Lopez-Vazquez, C., Brdjanovic, D., Eds.; Water Intelligence Online; IWA Publishing: London, UK, 2016; pp. 285–324. [Google Scholar] [CrossRef]
- Mve, M.J.B.B.; Cloutier, Y.; Lacombe, N.; Lavoie, J.; Debia, M.; Marchand, G. Comparison of methods to evaluate the fungal biomass in heating, ventilation, and air-conditioning (HVAC) dust. Environ. Monit. Assess. 2016, 189. [Google Scholar] [CrossRef]
- Maza-Márquez, P.; Vílchez-Vargas, R.; González-Martínez, A.; González-López, J.; Rodelas, B. Assessing the abundance of fungal populations in a full-scale membrane bioreactor (MBR) treating urban wastewater by using quantitative PCR (qPCR). J. Environ. Manag. 2018, 223, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Simon, U.K.; Weiss, M. Intragenomic Variation of Fungal Ribosomal Genes Is Higher than Previously Thought. Mol. Biol. Evol. 2008, 25, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Bonk, F.; Popp, D.; Harms, H.; Centler, F. PCR-based quantification of taxa-specific abundances in microbial communities: Quantifying and avoiding common pitfalls. J. Microbiol. Methods 2018, 153, 139–147. [Google Scholar] [CrossRef]
- Mounier, J.; Monnet, C.; Vallaeys, T.; Arditi, R.; Sarthou, A.S.; Helias, A.; Irlinger, F. Microbial Interactions within a Cheese Microbial Community. Appl. Environ. Microbiol. 2007, 74, 172–181. [Google Scholar] [CrossRef]
- Cogan, T.M.; Goerges, S.; Gelsomino, R.; Larpin, S.; Hohenegger, M.; Bora, N.; Jamet, E.; Rea, M.C.; Mounier, J.; Vancanneyt, M.; et al. Biodiversity of the Surface Microbial Consortia from Limburger, Reblochon, Livarot, Tilsit, and Gubbeen Cheeses. In Cheese and Microbes; American Society of Microbiology: Washington, DC, USA, 2014; pp. 219–250. [Google Scholar] [CrossRef]
- Montel, M.C.; Buchin, S.; Mallet, A.; Delbes-Paus, C.; Vuitton, D.A.; Desmasures, N.; Berthier, F. Traditional cheeses: Rich and diverse microbiota with associated benefits. Int. J. Food Microbiol. 2014, 177, 136–154. [Google Scholar] [CrossRef]
- Fröhlich-Wyder, M.T.; Arias-Roth, E.; Jakob, E. Cheese yeasts. Yeast 2019, 36, 129–141. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.K.; Fister, S.; Mester, P.; Schoder, D.; Rossmanith, P. Evaluation of the performance of quantitative detection of the Listeria monocytogenes prfA locus with droplet digital PCR. Anal. Bioanal. Chem. 2016, 408, 7583–7593. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a Droplet Digital Polymerase Chain Reaction Format for DNA Copy Number Quantification. Anal. Chem. 2011, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Ropars, J.; Cruaud, C.; Lacoste, S.; Dupont, J. A taxonomic and ecological overview of cheese fungi. Int. J. Food Microbiol. 2012, 155, 199–210. [Google Scholar] [CrossRef]
- Quigley, L.; O’Sullivan, O.; Stanton, C.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; Cotter, P.D. The complex microbiota of raw milk. FEMS Microbiol. Rev. 2013, 37, 664–698. [Google Scholar] [CrossRef]
- Tilocca, B.; Costanzo, N.; Morittu, V.M.; Spina, A.A.; Soggiu, A.; Britti, D.; Roncada, P.; Piras, C. Milk microbiota: Characterization methods and role in cheese production. J. Proteom. 2020, 210, 103534. [Google Scholar] [CrossRef]
- Larpin, S.; Mondoloni, C.; Goerges, S.; Vernoux, J.P.; Guéguen, M.; Desmasures, N. Geotrichum candidumdominates in yeast population dynamics in Livarot, a French red-smear cheese. FEMS Yeast Res. 2006, 6, 1243–1253. [Google Scholar] [CrossRef]
- Mounier, J.; Monnet, C.; Jacques, N.; Antoinette, A.; Irlinger, F. Assessment of the microbial diversity at the surface of Livarot cheese using culture-dependent and independent approaches. Int. J. Food Microbiol. 2009, 133, 31–37. [Google Scholar] [CrossRef]
- Garnier, L.; Valence, F.; Mounier, J. Diversity and Control of Spoilage Fungi in Dairy Products: An Update. Microorganisms 2017, 5, 42. [Google Scholar] [CrossRef]
- Bednarski, W.; Adamczak, M.; Tomasik, J.; Płaszczyk, M. Application of oil refinery waste in the biosynthesis of glycolipids by yeast. Bioresour. Technol. 2004, 95, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Vega-Alvarado, L.; Gómez-Angulo, J.; Escalante-García, Z.; Grande, R.; Gschaedler-Mathis, A.; Amaya-Delgado, L.; Sanchez-Flores, A.; Arrizon, J. High-Quality Draft Genome Sequence of Candida apicola NRRL Y-50540. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Seiler, H.; Busse, M. The yeasts of cheese brines. Int. J. Food Microbiol. 1990, 11, 289–303. [Google Scholar] [CrossRef]
- Hou, L.; Guo, L.; Wang, C.; Wang, C. Genome sequence of Candida versatilis and comparative analysis with other yeast. J. Ind. Microbiol. Biotechnol. 2016, 43, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Irlinger, F.; Morvan, A.; Solh, N.E.; Bergere, J. Taxonomic Characterization of Coagulase-Negative Staphylococci in Ripening Flora from Traditional French Cheeses. Syst. Appl. Microbiol. 1997, 20, 319–328. [Google Scholar] [CrossRef]
- Mounier, J.; Goerges, S.; Gelsomino, R.; Vancanneyt, M.; Vandemeulebroecke, K.; Hoste, B.; Brennan, N.; Scherer, S.; Swings, J.; Fitzgerald, G.; et al. Sources of the adventitious microflora of a smear-ripened cheese. J. Appl. Microbiol. 2006, 101, 668–681. [Google Scholar] [CrossRef]
- Rea, M.; Görges, S.; Gelsomino, R.; Brennan, N.; Mounier, J.; Vancanneyt, M.; Scherer, S.; Swings, J.; Cogan, T. Stability of the Biodiversity of the Surface Consortia of Gubbeen, a Red-Smear Cheese. J. Dairy Sci. 2007, 90, 2200–2210. [Google Scholar] [CrossRef]
- Vermote, L.; Verce, M.; Vuyst, L.D.; Weckx, S. Amplicon and shotgun metagenomic sequencing indicates that microbial ecosystems present in cheese brines reflect environmental inoculation during the cheese production process. Int. Dairy J. 2018, 87, 44–53. [Google Scholar] [CrossRef]
- Mounier, J.; Gelsomino, R.; Goerges, S.; Vancanneyt, M.; Vandemeulebroecke, K.; Hoste, B.; Scherer, S.; Swings, J.; Fitzgerald, G.F.; Cogan, T.M. Surface Microflora of Four Smear-Ripened Cheeses. Appl. Environ. Microbiol. 2005, 71, 6489–6500. [Google Scholar] [CrossRef]
- Martínez-Cuesta, M.D.C.; Peláez, C.; Requena, T. Methionine Metabolism: Major Pathways and Enzymes Involved and Strategies for Control and Diversification of Volatile Sulfur Compounds in Cheese. Crit. Rev. Food Sci. Nutr. 2011, 53, 366–385. [Google Scholar] [CrossRef]
- Kastman, E.K.; Kamelamela, N.; Norville, J.W.; Cosetta, C.M.; Dutton, R.J.; Wolfe, B.E. Biotic Interactions Shape the Ecological Distributions of Staphylococcus Species. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mayr, R.; Fricker, M.; Maoz, A.; Scherer, S. Anti-listerial activity and biodiversity of cheese surface cultures: Influence of the ripening temperature regime. Eur. Food Res. Technol. 2004, 218, 242–247. [Google Scholar] [CrossRef]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the Evolutionary History of an Ancient Phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef] [PubMed]
- Callon, C.; Retureau, E.; Didienne, R.; Montel, M.C. Microbial biodiversity in cheese consortia and comparative Listeria growth on surfaces of uncooked pressed cheeses. Int. J. Food Microbiol. 2014, 174, 98–109. [Google Scholar] [CrossRef]
- Bachmann, H.; Bobst, C.; Bütikofer, U.; Casey, M.; Torre, M.D.; Fröhlich-Wyder, M.; Fürst, M. Occurrence and significance of Fusarium domesticum alias Anticollanti on smear-ripened cheeses. LWT Food Sci. Technol. 2005, 38, 399–407. [Google Scholar] [CrossRef]
- Gallardo, G.; Ruiz-Moyano, S.; Hernández, A.; Benito, M.; Córdoba, M.; Pérez-Nevado, F.; Martín, A. Application of ISSR-PCR for rapid strain typing of Debaryomyces hansenii isolated from dry-cured Iberian ham. Food Microbiol. 2014, 42, 205–211. [Google Scholar] [CrossRef]
- Stellato, G.; Filippis, F.D.; Storia, A.L.; Ercolini, D. Coexistence of Lactic Acid Bacteria and Potential Spoilage Microbiota in a Dairy Processing Environment. Appl. Environ. Microbiol. 2015, 81, 7893–7904. [Google Scholar] [CrossRef]
- Haastrup, M.K.; Johansen, P.; Malskær, A.H.; Castro-Mejía, J.L.; Kot, W.; Krych, L.; Arneborg, N.; Jespersen, L. Cheese brines from Danish dairies reveal a complex microbiota comprising several halotolerant bacteria and yeasts. Int. J. Food Microbiol. 2018, 285, 173–187. [Google Scholar] [CrossRef]
- Stielow, J.; Lévesque, C.; Seifert, K.; Meyer, W.; Irinyi, L.; Smits, D.; Renfurm, R.; Verkley, G.; Groenewald, M.; Chaduli, D.; et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia Mol. Phylogeny Evol. Fungi 2015, 35, 242–263. [Google Scholar] [CrossRef]
- Větrovský, T.; Kolařík, M.; Žifčáková, L.; Zelenka, T.; Baldrian, P. The rpb2 gene represents a viable alternative molecular marker for the analysis of environmental fungal communities. Mol. Ecol. Resour. 2015, 16, 388–401. [Google Scholar] [CrossRef]
- Xu, J. Fungal DNA barcoding. Genome 2016, 59, 913–932. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Facility | Days of | Temperature | Humidity | Cheese Treatment | Brine | Dry |
|---|---|---|---|---|---|---|
| Ripening | (C) | (%) | with Brine | Concentration (%) | Salting * | |
| A | 0, 14, 30, 90 | 13.5 | 96 | daily | 20 | no |
| 160 | 10 | 95–96 | once a week | 10 | ||
| B | 0, 14, 30, 90 | 13 | 93–94 | 2–3 times a week | 10 | yes |
| 160 | 13 | 93–94 | once a week | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quijada, N.M.; Schmitz-Esser, S.; Zwirzitz, B.; Guse, C.; Strachan, C.R.; Wagner, M.; Wetzels, S.U.; Selberherr, E.; Dzieciol, M. Austrian Raw-Milk Hard-Cheese Ripening Involves Successional Dynamics of Non-Inoculated Bacteria and Fungi. Foods 2020, 9, 1851. https://doi.org/10.3390/foods9121851
Quijada NM, Schmitz-Esser S, Zwirzitz B, Guse C, Strachan CR, Wagner M, Wetzels SU, Selberherr E, Dzieciol M. Austrian Raw-Milk Hard-Cheese Ripening Involves Successional Dynamics of Non-Inoculated Bacteria and Fungi. Foods. 2020; 9(12):1851. https://doi.org/10.3390/foods9121851
Chicago/Turabian StyleQuijada, Narciso M., Stephan Schmitz-Esser, Benjamin Zwirzitz, Christian Guse, Cameron R. Strachan, Martin Wagner, Stefanie U. Wetzels, Evelyne Selberherr, and Monika Dzieciol. 2020. "Austrian Raw-Milk Hard-Cheese Ripening Involves Successional Dynamics of Non-Inoculated Bacteria and Fungi" Foods 9, no. 12: 1851. https://doi.org/10.3390/foods9121851
APA StyleQuijada, N. M., Schmitz-Esser, S., Zwirzitz, B., Guse, C., Strachan, C. R., Wagner, M., Wetzels, S. U., Selberherr, E., & Dzieciol, M. (2020). Austrian Raw-Milk Hard-Cheese Ripening Involves Successional Dynamics of Non-Inoculated Bacteria and Fungi. Foods, 9(12), 1851. https://doi.org/10.3390/foods9121851