Oregano Phytocomplex Induces Programmed Cell Death in Melanoma Lines via Mitochondria and DNA Damage

 , ,
, ,  ,
, 
Abstract

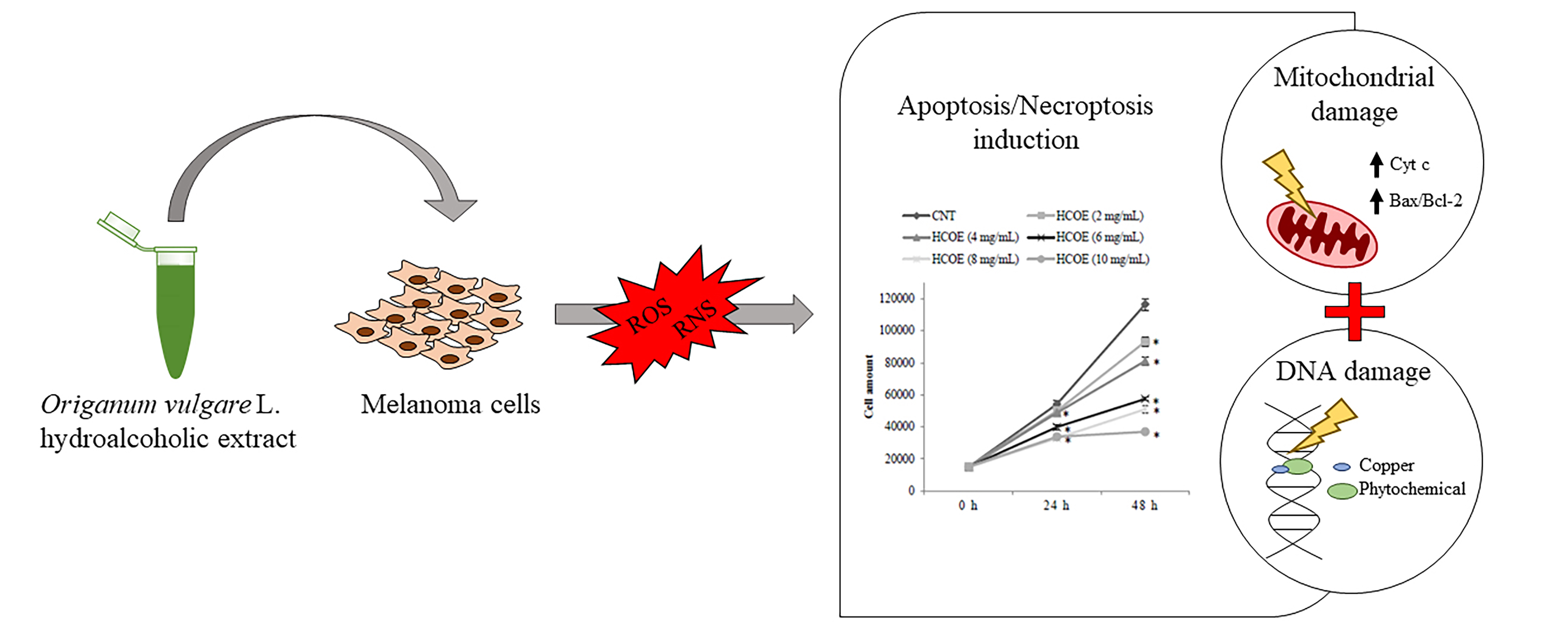
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Total Phenol and Flavonoid Content
2.3. High-Performance Liquid Chromatography-Diode Array Detector (HPLC-DAD) and Gas Chromatography-Mass Spectrometry (GC-MS) Analyses
2.4. Cell Cultures and Plant Treatments
2.5. Cell Proliferation, Selectivity Index, and Cell Cycle Analysis
2.6. Mutagen and Mutagen-Protective Activity
2.7. Protein Analysis
2.8. Real-Time-PCR (RT-PCR) Assay
2.9. Reactive Species Level and Mitochondrial Mass and Membrane Potential Measurement
2.10. Immunofluorescence Microscopy
2.11. Statistical Analysis
3. Results
3.1. Chemical Characterization of the O. vulgare L. Phytocomplex
3.2. O. vulgare L. Extract Reduces B16-F10 Cell Growth Not Affecting C2C12 Cell Viability
3.3. Oregano Treatment Shows Antiproliferative Activity Also on A375 Human Melanoma Cells
3.4. Oregano Extract Has Neither Mutagenic Nor Mutagen-Protective Effects
3.5. Oregano Phytocomplex Impairs MITF Pathway and Accumulates Reactive Species in A375 Cells
3.6. Apoptosis/Necroptosis via Mitochondrial Pathway Is Induced in A375 Cells by HCOE
3.7. HCOE Triggers DNA Breakages Mediated by Metal Ions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clardy, J.; Walsh, C. Lessons from natural molecules. Nature 2004, 432, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Thomford, N.; Senthebane, D.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, M.; Rahman, P.K. Medicinal plants: Their use in anticancer treatment. Int. J. Pharm. Sci. Res. 2015, 6, 4103–4112. [Google Scholar] [PubMed]
- Singh, S.; Sharma, B.; Kanwar, S.S.; Kumar, A. Lead phytochemicals for anticancer drug development. Front. Plant Sci. 2016, 7, 1667. [Google Scholar] [CrossRef]
- Gullett, N.P.; Ruhul Amin, A.R.M.; Bayraktar, S.; Pezzuto, J.M.; Shin, D.M.; Khuri, F.R.; Aggarwal, B.B.; Surh, Y.J.; Kucuk, O. Cancer prevention with natural compounds. Semin. Oncol. 2010, 37, 258–281. [Google Scholar] [CrossRef] [PubMed]
- Lesgards, J.F.; Baldovini, N.; Vidal, N.; Pietri, S. Anticancer activities of essential oils constituents and synergy with conventional therapies: A review. Phytother. Res. 2014, 28, 1423–1446. [Google Scholar] [CrossRef]
- Bharti, A.C.; Aggarwal, B.B. Nuclear factor-kappa B and cancer: Its role in prevention and therapy. Biochem. Pharmacol. 2002, 64, 883–888. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Holbeck, S.; Sausville, E.A. Natural products as leads to cell cycle pathway targets in cancer chemotherapy. Curr. Cancer Drug Targets 2002, 2, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Hollósy, F.; Kéri, G. Plant-derived protein tyrosine kinase inhibitors as anticancer agents. Curr. Med. Chem. Anticancer Agents 2004, 4, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.H. Naturally occurring NF-kappaB inhibitors. Mini Rev. Med. Chem. 2006, 6, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Inhibition of PI3K/Akt/mTOR signaling by natural products. Anticancer Agents Med. Chem. 2013, 13, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Kaur, S.; Kumar, M.; Kaur, S. Role of phytochemicals in MAPK signaling pathway-mediated apoptosis: A possible strategy in cancer chemoprevention. Stud. Nat. Prod. Chem. 2016, 50, 159–178. [Google Scholar]
- Low, H.B.; Zhang, Y. Regulatory roles of MAPK phosphatases in cancer. Immune Netw. 2016, 16, 85–98. [Google Scholar] [CrossRef]
- Nosrati, N.; Bakovic, M.; Paliyath, G. Molecular mechanisms and pathways as targets for cancer prevention and progression with dietary compounds. Int. J. Mol. Sci. 2017, 18, 2050. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, V.; Murahari, M.; Khan, T.; Chaubey, P.; Sangave, P. Phytochemicals and PI3K inhibitors in cancer-An insight. Front. Pharmacol. 2017, 8, 916. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Hadi, S.M.; Asad, S.F.; Singh, S.; Ahmad, A. Putative mechanism for anticancer and apoptosis-inducing properties of plant-derived polyphenolic compounds. IUBMB Life 2000, 50, 167–171. [Google Scholar] [PubMed]
- Elbling, L.; Weiss, R.M.; Teufelhofer, O.; Uhl, M.; Knasmueller, S.; Schulte-Hermann, R.; Berger, W.; Micksche, M. Green tea extract and (-)-epigallocatechin-3-gallate, the major tea catechin, exert oxidant but lack antioxidant activities. FASEB J. 2005, 19, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of nitric oxide synthases in parkinson’s disease: A review on the antioxidant and anti-inflammatory activity of polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef]
- Martin-Cordero, C.; Jose Leon-Gonzalez, A.; Manuel Calderon-Montano, J.; Burgos-Moron, E.; Lopez-Lazaro, M. Pro-oxidant natural products as anticancer agents. Curr. Drug Targets 2012, 13, 1006–1028. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.Y.; Zubair, H.; Faisal, M.; Ullah, M.F.; Farhan, M.; Sarkar, F.H.; Ahmad, A.; Hadi, S.M. Plant polyphenol induced cell death in human cancer cells involves mobilization of intracellular copper ions and reactive oxygen species generation: A mechanism for cancer chemopreventive action. Mol. Nutr. Food Res. 2014, 58, 437–446. [Google Scholar] [CrossRef]
- Nanni, V.; Canuti, L.; Gismondi, A.; Canini, A. Hydroalcoholic extract of Spartium junceum L. flowers inhibits growth and melanogenesis in B16-F10 cells by inducing senescence. Phytomedecine 2018, 46, 1–10. [Google Scholar] [CrossRef]
- Kubatka, P.; Kello, M.; Kajo, K.; Kruzliak, P.; Výbohová, D.; Mojžiš, J.; Adamkov, M.; Fialová, S.; Veizerová, L.; Zulli, A.; et al. Oregano demonstrates distinct tumor-suppressive effects in the breast carcinoma model. Eur. J. Nutr. 2017, 56, 1303–1316. [Google Scholar] [CrossRef]
- Pezzani, R.; Vitalini, S.; Iriti, M. Bioactivities of Origanum vulgare L.: An update. Phytochem. Rev. 2017, 16, 1253–1268. [Google Scholar] [CrossRef]
- Marrelli, M.; Statti, G.A.; Conforti, F. Origanum spp.: An update of their chemical and biological profiles. Phytochem. Rev. 2018, 17, 873. [Google Scholar] [CrossRef]
- Kintzios, S.E. In Handbook of Herbs and Spices; Woodhead Publishing Ltd.: Abington Hall, Abington, MA, USA; Cambridge, UK, 2012; Volume 2, pp. 417–436. [Google Scholar]
- Leyva-López, N.; Gutiérrez-Grijalva, E.P.; Vazquez-Olivo, G.; Heredia, J.B. Essential oils of oregano: Biological activity beyond their antimicrobial properties. Molecules 2017, 22, 989. [Google Scholar]
- Oniga, I.; Pușcaș, C.; Silaghi-Dumitrescu, R.; Olah, N.K.; Sevastre, B.; Marica, R.; Marcus, I.; Sevastre-Berghian, A.C.; Benedec, D.; Pop, C.E.; et al. Origanum vulgare ssp. vulgare: Chemical composition and biological studies. Molecules 2018, 23, 2077. [Google Scholar] [CrossRef] [PubMed]
- Impei, S.; Gismondi, A.; Canuti, L.; Canini, A. Metabolic and biological profile of autochthonous Vitis vinifera L. ecotypes. Food Funct. 2015, 6, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Yang, M.H.; Wen, H.M.; Chern, J.C. Estimation of total flavonoid content in propolis by two complementary colorimetric methods. J. Food Drug Anal. 2002, 10, 3. [Google Scholar]
- Fidler, I.J. Selection of successive tumor lines for metastasis. Nat. New Biol. 1973, 242, 148–149. [Google Scholar] [CrossRef]
- Gismondi, A.; Lentini, A.; Tabolacci, C.; Provenzano, B.; Beninati, S. Transglutaminase-dependent antiproliferative and differentiative properties of nimesulide on B16-F10 mouse melanoma cells. Amino Acids 2010, 38, 257–262. [Google Scholar] [CrossRef]
- Gismondi, A.; Canuti, L.; Impei, S.; Di Marco, G.; Kenzo, M.; Colizzi, V.; Canini, A. Antioxidant extracts of African medicinal plants induce cell cycle arrest and differentiation in B16F10 melanoma cells. Int. J. Oncol. 2013, 43, 956–964. [Google Scholar] [CrossRef]
- Rossi, D.; Guerrini, A.; Maietti, S.; Bruni, R.; Paganetto, G.; Poli, F.; Scalvenzi, L.; Radice, M.; Saro, K.; Sacchetti, G. Chemical fingerprinting and bioactivity of Amazonian Ecuador Croton lechleri Müll. Arg. (Euphorbiaceae) stem bark essential oil: A new functional food ingredient? Food Chem. 2011, 126, 837–848. [Google Scholar] [CrossRef]
- Rossi, D.; Guerrini, A.; Paganetto, G.; Bernacchia, G.; Conforti, F.; Statti, G.; Maietti, S.; Poppi, I.; Tacchini, M.; Sacchetti, G. Croton lechleri Müll. Arg. (Euphorbiaceae) stem bark essential oil as possible mutagen-protective food ingredient against heterocyclic amines from cooked food. Food Chem. 2013, 139, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Mansky, K.C.; Sulzbacher, S.; Purdom, G.; Nelsen, L.; Hume, D.A.; Rehli, M.; Ostrowski, M.C. The microphthalmia transcription factor and the related helix-loop-helix zipper factors TFE-3 and TFE-C collaborate to activate the tartrate-resistant acid phosphatase promoter. J. Leukoc. Biol. 2002, 71, 304–310. [Google Scholar]
- Vetrini, F.; Auricchio, A.; Du, J.; Angeletti, B.; Fisher, D.E.; Ballabio, A.; Marigo, V. The microphthalmia transcription factor (Mitf) controls expression of the ocular albinism type 1 gene: Link between melanin synthesis and melanosome biogenesis. Mol. Cell. Biol. 2004, 24, 6550–6559. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, Y.; Liang, P.; Guo, X.; Ying, Y.; Shu, X.S.; Gao, M., Jr.; Cheng, Y. OSR1 is a novel epigenetic silenced tumor suppressor regulating invasion and proliferation in renal cell carcinoma. Oncotarget 2017, 8, 30008–30018. [Google Scholar] [CrossRef]
- Lin, Y.T.; Lin, C.C.; Wang, H.C.; Hsu, Y.C. Induction of mitotic delay in pharyngeal and nasopharyngeal carcinoma cells using an aqueous extract of Ajuga bracteosa. Int. J. Med. Sci. 2017, 14, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.J.; Chabot, B. Control of hnRNP A1 alternative splicing: An intron element represses use of the common 3′ splice site. Mol. Cell. Biol. 2000, 20, 7353–7362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gismondi, A.; Canuti, L.; Grispo, M.; Canini, A. Biochemical composition and antioxidant properties of Lavandula angustifolia Miller essential oil are shielded by propolis against UV radiations. Photochem. Photobiol. 2014, 90, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Pendergrass, W.; Wolf, N.; Pool, M. Efficacy of MitoTracker GreenTM and CMXRosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytom. A 2004, 61, 162–169. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; Mcphail, A.T. Plant antitumor agents.VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.M.; Zhu, J.; McQueney, P.A.; Bollag, D.; Lazarides, E. Taxol-induced mitotic block triggers rapid onset of a p53-independent apoptotic pathway. Mol. Med. 1995, 5, 506–526. [Google Scholar] [CrossRef]
- Nurse, P. Universal control mechanism regulating onset of M-phase. Nature 1990, 344, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O. New roles for p21 and p27 cell-cycle inhibitors: A function for each cell compartment? Trends Cell Biol. 2003, 13, 65–70. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef]
- Tishler, R.B.; Lamppu, D.M.; Park, S.; Price, B.D. Microtubule-active drugs taxol, vinblastine, and nocodazole increase the levels of transcriptionally active p53. Cancer Res. 1995, 55, 6021–6025. [Google Scholar] [PubMed]
- Maron, D.M.; Ames, B.N. Revised methods for the Salmonella mutagenicity test. Mutat. Res. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef]
- Slee, E.A.; Zhu, H.; Chow, S.C.; Macfarlane, M.; Nicholson, D.W.; Cohen, G.M. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J. 1996, 315, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Chen, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell. Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, D. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef]
- Said, A.M.; Fazal, F.; Rahman, A.; Hadi, S.M.; Parish, J.H. Activities of flavonoids for the cleavage of DNA in the presence of Cu(II): Correlation with the generation of active oxygen species. Carcinogenesis 1992, 13, 605–608. [Google Scholar]
- The American Cancer Society. What are the Key Statistics about Melanoma Skin Cancer? 2017. Available online: http://www.cancer.org/cancer/skincancer-melanoma/detailedguide/melanoma-skin-cancer-key-statistics (accessed on 8 January 2020).
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Chan, M.; Harland, M.; Hayward, N.K.; Demenais, F.; Bishop, D.T.; Azizi, E.; Bergman, W.; Bianchi-Scarra, G.; Bruno, W.; et al. Features associated with germline CDKN2A mutations: A GenoMEL study of melanoma-prone families from three continents. J. Med. Genet. 2007, 44, 99–106. [Google Scholar] [CrossRef]
- Liu, Y.; Sheikh, M.S. Melanoma: Molecular pathogenesis and therapeutic management. Mol. Cell. Pharmacol. 2014, 6, 228. [Google Scholar]
- Gismondi, A.; Di Marco, G.; Canuti, L.; Canini, A. Antiradical activity of phenolic metabolites extracted from grapes of white and red Vitis vinifera L. cultivars. Vitis 2017, 56, 19–26. [Google Scholar]
- Jensen, J.D.; Wing, G.J.; Delvalle, R.P. Nutrition and melanoma prevention. Clin. Dermatol. 2010, 28, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Chinembiri, T.N.; du Plessis, L.H.; Gerber, M.; Hamman, J.H.; du Plessis, J. Review of natural compounds for potential skin cancer treatment. Molecules 2014, 19, 11679–11721. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.X.; Young, L.C. Nutrition: The future of melanoma prevention? J. Am. Acad. Dermatol. 2014, 71, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Działo, M.; Mierziak, J.; Korzun, U.; Preisner, M.; Szopa, J.; Kulma, A. The potential of plant phenolics in prevention and therapy of skin disorders. Int. J. Mol. Sci. 2016, 17, 160. [Google Scholar] [CrossRef] [PubMed]
- Ettorre, A.; Frosali, S.; Andreassi, M.; Di Stefano, A. Lycopene phytocomplex, but not pure lycopene, is able to trigger apoptosis and improve the efficacy of photodynamic therapy in HL60 human leukemia cells. Exp. Biol. Med. 2010, 235, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Mariño, G.; Lissa, D.; Vacchelli, E.; Malik, S.A.; Niso-Santano, M.; Zamzami, N.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Pro-autophagic polyphenols reduce the acetylation of cytoplasmic proteins. Cell Cycle 2012, 11, 3851–3860. [Google Scholar] [CrossRef] [PubMed]
- Hasa, D.; Perissutti, B.; Dall’Acqua, S.; Chierotti, M.R.; Gobetto, R.; Grabnar, I.; Cepek, C.; Voinovich, D. Rationale of using Vinca minor Linne dry extract phytocomplex as a vincamine’s oral bioavailability enhancer. Eur. J. Pharm. Biopharm. 2013, 84, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Kokkini, S.; Karousou, R.; Dardioti, A.; Krigas, N.; Lanaras, T. Autumn essential oils of greek oregano. Phytochemistry 1997, 44, 883–886. [Google Scholar] [CrossRef]
- Russo, M.; Galletti, G.C.; Bocchini, P.; Carnacini, A. Essential oil chemical composition of wild populations of Italian oregano spice (Origanum vulgare ssp. hirtum (Link) Ietswaart): A preliminary evaluation of their use in chemotaxonomy by cluster analysis. 1. Inflorescences. J. Agric. Food Chem. 1998, 46, 3741–3746. [Google Scholar] [CrossRef]
- De Martino, L.; De Feo, V.; Formisano, C.; Mignola, E.; Senatore, F. Chemical composition and antimicrobial activity of the essential oils from three chemotypes of Origanum vulgare L. ssp. hirtum (Link) Ietswaart growing wild in Campania (Southern Italy). Molecules 2009, 14, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.S.; Vattem, D.A.; Lin, Y.T.; Shetty, K. Phenolic antioxidants from clonal oregano (Origanum vulgare) with antimicrobial activity against Helicobacter pylori. Process Biochem. 2005, 40, 809–816. [Google Scholar] [CrossRef]
- Skendi, A.; Irakli, M.; Chatzopoulou, P. Analysis of phenolic compounds in Greek plants of Lamiaceae family by HPLC. J. Appl. Res. Med. Aromat. Plants 2017, 6, 62–69. [Google Scholar] [CrossRef]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar] [PubMed]
- EFSA Scientific Committee. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA J. 2011, 9, 2379. [Google Scholar]
- Nurmi, A.; Mursu, J.; Nurmi, T.; Nyyssönen, K.; Alfthan, G.; Hiltunen, R.; Kaikkonen, J.; Salonen, J.T.; Voutilainen, S. Consumption of juice fortified with oregano extract markedly increases excretion of phenolic acids but lacks short- and long-term effects on lipid peroxidation in healthy nonsmoking men. J. Agric. Food Chem. 2006, 54, 5790–5796. [Google Scholar] [CrossRef] [PubMed]
- Llana-Ruiz-Cabello, M.; Puerto, M.; Maisanaba, S.; Guzmán-Guillén, R.; Pichardo, S.; Cameán, A.M. Use of micronucleus and comet assay to evaluate evaluate the genotoxicity of oregano essential oil (Origanum vulgare L. virens) in rats orally exposed for 90 days. J. Toxicol. Environ. Health A 2018, 81, 525–533. [Google Scholar] [CrossRef]
- Vaughn, C.J. Drugs and Lactation Database: LactMed. J. Electron. Resour. Med. Libr. 2012, 9, 272–277. [Google Scholar] [CrossRef]
- EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS). Scientific Opinion on the use of oregano and lemon balm extracts as a food additive on request of the European Commission. EFSA J. 2010, 8, 1514. [Google Scholar]
- EFSA Scientific Committee. Technical report on the outcome of the consultation with Member States and EFSA on the basic substance application for Origanum vulgare L. essential oil for use in plant protection as fungicide, bactericide and insecticide. EFSA Support. Publ. 2016, 13, 1054. [Google Scholar]
- Liang, C.H.; Chou, T.H.; Ding, H.Y. Inhibition of melanogensis by a novel origanoside from Origanum vulgare. J. Dermatol. Sci. 2010, 57, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Arunasree, K.M. Anti-proliferative effects of carvacrol on a human metastatic breast cancer cell line, MDA-MB 231. Phytomedicine 2010, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Khan, I.; Farooqui, A.; Ansari, I. Carvacrol induces reactive oxygen species (ROS)-mediated apoptosis along with cell cycle arrest at G0/G1 in human prostate cancer cells. Nutr. Cancer 2017, 69, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Varoni, E.M.; Iriti, M.; Martorell, M.; Setzer, W.N.; del Mar Contreras, M.; Salehi, B.; Soltani-Nejad, A.; Rajabi, S.; Tajbakhsh, M.; et al. Carvacrol and human health: A comprehensive review. Phytother. Res. 2018, 32, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Rodolfo, C.; Rocco, M.; Cattaneo, L.; Tartaglia, M.; Sassi, M.; Aducci, P.; Scaloni, A.; Camoni, L.; Marra, M. Ophiobolin a induces autophagy and activates the mitochondrial pathway of apoptosis in human melanoma cells. PLoS ONE 2016, 11, e0167672. [Google Scholar] [CrossRef]
- Savini, I.; Arnone, R.; Catani, M.V.; Avigliano, L. Origanum vulgare induces apoptosis in human colon cancer caco2 cells. Nutr. Cancer 2009, 61, 381–389. [Google Scholar] [CrossRef]
- Rubin, B.; Mansi, J.; Monticelli, H.; Bertazza, L.; Redaelli, M.; Sensi, F.; Zorzan, M.; Scaroni, C.; Mian, C.; Iacobone, M.; et al. Crude extract of Origanum vulgare L. induced cell death and suppressed MAPK and PI3/Akt signaling pathways in SW13 and H295R cell lines. Nat. Prod. Res. 2018, 15, 1646–1649. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Salvesen, G. Regulated cell death: Signaling and mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 337–356. [Google Scholar] [CrossRef]
- Sawai, H.; Domae, N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem. Biophys. Res. Commun. 2011, 411, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Al Dhaheri, Y.; Eid, A.; AbuQamar, S.; Attoub, S.; Khasawneh, M.; Aiche, G.; Hisaindee, S.; Iratni, R. Mitotic arrest and apoptosis in breast cancer cells induced by Origanum majorana extract: Upregulation of TNF-α and downregulation of survivin and mutant p53. PLoS ONE 2013, 8, e56649. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Mironov, S.L.; Ivannikov, M.V.; Schmidt, J.; Richter, D.W. Mitochondrial organization and motility probed by two-photon microscopy in cultured mouse brainstem neurons. Exp. Cell Res. 2005, 303, 114–127. [Google Scholar] [CrossRef]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar]
- Hemann, M.T.; Lowe, S.W. The p53-Bcl-2 connection. Cell Death Differ. 2006, 13, 1256–1259. [Google Scholar] [CrossRef]
- Bourgarel-Rey, V.; Savry, A.; Hua, G.; Carré, M.; Bressin, C.; Chacon, C.; Imbert, J.; Braguer, D.; Barra, Y. Transcriptional down-regulation of Bcl-2 by vinorelbine: Identification of a novel binding site of p53 on Bcl-2 promoter. Biochem. Pharmacol. 2009, 78, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef]
- Kagawa, T.F.; Geierstanger, B.H.; Wang, A.H.J.; Ho, P.S. Covalent modification of guanine bases in double-stranded DNA. The 1.2-A Z-DNA structure of d(CGCGCG) in the presence of CuCl2. J. Biol. Chem. 1991, 266, 20175–20184. [Google Scholar] [PubMed]
- Ullah, M.F.; Ahmad, A.; Khan, H.Y.; Zubair, H.; Sarkar, F.H.; Hadi, S.M. The prooxidant action of dietary antioxidants leading to cellular DNA breakage and anticancer effects: Implications for chemotherapeutic action against cancer. Cell Biochem. Biophys. 2013, 67, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Narciso, L.; Fortini, P.; Pajalunga, D.; Franchitto, A.; Liu, P.; Degan, P.; Frechet, M.; Demple, B.; Crescenzi, M.; Dogliotti, E. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc. Natl. Acad. Sci. USA 2007, 104, 17010–17015. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Feeney, L.; Revet, I. Phosphorylated H2Ax is not an unambiguous marker for DNA double strand breaks. Cell Cycle 2011, 10, 3223–3224. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Ferretti, C.; Pascucci, B.; Narciso, L.; Pajalunga, D.; Puggioni, E.M.R.; Castino, R.; Isidoro, C.; Crescenzi, M.; Dogliotti, E. DNA damage response by single-strand breaks in terminally differentiated muscle cells and the control of muscle integrity. Cell Death Differ. 2012, 19, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Minn, K.; van Deursen, J.; Chen, J. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003, 23, 2556–2563. [Google Scholar] [CrossRef]
- Dearfield, K.L.; Cimino, M.C.; Mccarroll, N.E.; Mauer, I.; Valcovic, L.R. Genotoxicity risk assessment: A proposed classification strategy. Mutat. Res. 2002, 521, 121–135. [Google Scholar] [CrossRef]
- Laughton, M.J.; Halliwell, B.; Evans, P.J.; Robin, J.; Hoult, S. Antioxidant and pro-oxidant actions of the plant phenolics quercetin, gossypol and myricetin. Effects on lipid peroxidation, hydroxyl radical generation and bleomycin-dependent damage to DNA. Biochem. Pharmacol. 1989, 38, 2859–2865. [Google Scholar] [CrossRef]
- Hadi, S.M.; Bhat, S.H.; Azmi, A.S.; Hanif, S.; Shamim, U.; Ullah, M.F. Oxidative breakage of cellular DNA by plant polyphenols: A putative mechanism for anticancer properties. Semin. Cancer Biol. 2007, 17, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Wang, Y.H.; Liou, C.C.; Lin, Y.C.; Huang, H.; Liu, Y.C. Induction of oxidative DNA damage by flavonoids of propolis: Its mechanism and implication about antioxidant capacity. Chem. Res. Toxicol. 2012, 25, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Swanson, S. The status of zinc, copper, and metallothionein in cancer patients. Prog. Clin. Biol. Res. 1988, 259, 161–175. [Google Scholar] [PubMed]
- Yoshida, D.; Ikeda, Y.; Nakazawa, S. Quantitative analysis of copper, zinc and copper/zinc ratio in selected human brain tumors. J. Neurooncol. 1993, 16, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.Y.; Zubair, H.; Ullah, M.F.; Ahmad, A.; Hadi, S.M. A prooxidant mechanism for the anticancer and chemopreventive properties of plant polyphenols. Curr. Drug Targets 2012, 13, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: “Copper That Cancer”. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Cancer’s copper connections. Science 2015, 349, 129. [Google Scholar] [CrossRef]
- Sergediene, E.; Jönsson, K.; Szymusiak, H.; Tyrakowska, B.; Rietjens, I.M.C.M.; Čenas, N. Prooxidant toxicity of polyphenolic antioxidants to HL-60 cells: Description of quantitative structure-activity relationships. FEBS Lett. 1999, 462, 392–396. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yoshida, T.; Horinaka, M.; Yasuda, T.; Goda, A.E.; Konishi, M.; Wakada, M.; Kataoka, K.; Yoshikawa, T.; Sakai, T. Baicalein overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance via two different cell-specific pathways in cancer cells but not in normal cells. Cancer Res. 2008, 68, 8918–8927. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Lu, N.; Dai, Q.; Rong, J.; Chen, Y.; Li, Z.; You, Q.; Guo, Q. Different apoptotic effects of wogonin via induction of H2O2 generation and Ca2+ overload in malignant hepatoma and normal hepatic cells. J. Cell. Biochem. 2010, 111, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Montaño, J.M.; Martínez-Sánchez, S.M.; Burgos-Morón, E.; Guillén-Mancina, E.; Jiménez-Alonso, J.J.; García, F.; Aparicio, A.; López-Lázaro, M. Screening for selective anticancer activity of plants from Grazalema Natural Park, Spain. Nat. Prod. Res. 2018, 33, 3454–3458. [Google Scholar] [CrossRef] [PubMed]
- Cheimonidi, C.; Samara, P.; Polychronopoulos, P.; Tsakiri, E.N.; Nikou, T.; Myrianthopoulos, V.; Sakellaropoulos, T.; Zoumpourlis, V.; Mikros, E.; Papassideri, I.; et al. Selective cytotoxicity of the herbal substance acteoside against tumor cells and its mechanistic insights. Redox. Biol. 2018, 16, 169–178. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | µg/100 mg DMW ± SD |
|---|---|
| Chrysin | 8.47 ± 0.06 |
| Rutin | 2.15 ± 0.05 |
| Myricetin | 0.03 ± 0.01 |
| Caffeic acid | 0.35 ± 0.02 |
| 1,1-Dimethylallyl caffeate | 1.28 ± 0.03 |
| Caffeic acid phenethyl ester | 0.76 ± 0.03 |
| Gallic acid | 0.18 ± 0.01 |
| Kaempferol | 0.04 ± 0.01 |
| p-Coumaric acid | 0.30 ± 0.01 |
| Genistein | 1.02 ± 0.02 |
| Quercetin-3-o-arabinoside | 2.37 ± 0.04 |
| Chlorogenic acid | 1.03 ± 0.04 |
| Apigenin | 0.26 ± 0.01 |
| GC-MS Detected Compound | % |
|---|---|
| m-Cymol | 0.57 |
| p-Mentha-1,3,8-triene | 0.31 |
| p-Cymene-2,5-dione | 0.87 |
| Thymol | 16.64 |
| Carvacrol | 34.82 |
| Caryophyllene oxide | 0.90 |
| t-Butylhydroquinone | 1.86 |
| Isopropyl laurate | 0.94 |
| Palmitic acid | 2.50 |
| Ethyl palmitate | 1.33 |
| Phytol | 0.84 |
| Retinoic acid | 0.73 |
| Methyl linolenate | 7.96 |
| Ethyl linolenate | 7.35 |
| 3,3-Dimethylbutanoic acid | 0.03 |
| p-Mentha-1,4-diene | 0.13 |
| alpha-Aminoisobutanoic acid | 0.03 |
| p-Mentha-6,8-dien-2-ol | 0.94 |
| Myrtenyl acetate | 0.54 |
| 2,6-Dimethyl-1,3,5,7-octatetraene | 0.29 |
| 3,5-Dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one | 0.33 |
| Thujone | 0.52 |
| Octyl acetate | 0.19 |
| 2,3-Dimethyl-2-pentanol | 0.11 |
| Dimethyl malonate | 0.04 |
| Ehtyl acetimidate | 0.09 |
| Dimethylhexynediol | 0.20 |
| Linalool oxide | 0.89 |
| trans-2-Hexenyl caproate | 0.29 |
| 2-Ethyl-3-hydroxyhexyl 2-methylpropanoate | 0.09 |
| 1-Tetradecanol | 0.10 |
| Hydroxydehydrostevic acid | 0.11 |
| 2,2,4-Trimethyl-1,3-pentanediol diisobutyrate | 1.22 |
| Caprylic ether | 0.07 |
| alpha-Methylglucoside | 7.95 |
| Hexadecane | 0.92 |
| Retinyl acetate | 0.84 |
| Stearic acid | 0.99 |
| Butyl citrate | 3.58 |
| Jasmone | 0.44 |
| Oleic Acid | 0.33 |
| Oleic acid amide | 0.72 |
| 3-Hexadecanol | 0.40 |
| Cell Line | Treatment | Time | ||
|---|---|---|---|---|
| 24 h | 48 h | |||
| C2C12 | CNT (PBS) | 6.07 ± 0.80 | 4.61 ± 0.11 | |
| HCOE | (2 mg/mL) | 13.33 ± 0.41 | 11.67 ± 2.60 | |
| (4 mg/mL) | 13.16 ± 0.32 | 11.33 ± 3.43 | ||
| (6 mg/mL) | 18.86 ± 0.28 | 16.35 ± 0.80 | ||
| (8 mg/mL) | 20.25 ± 0.51 * | 19.78 ± 1.57 * | ||
| (10 mg/mL) | 20.67 ± 0.17 * | 21.25 ± 2.36 * | ||
| B16-F10 | CNT (PBS) | 1.86 ± 0.13 | 5.31 ± 0.26 | |
| HCOE | (2 mg/mL) | 14.90 ± 1.16 | 14.84 ± 1.17 | |
| (4 mg/mL) | 12.98 ± 2.38 | 26.62 ± 0.95 * | ||
| (6 mg/mL) | 24.36 ± 0.84 * | 33.29 ± 1.49 * | ||
| (8 mg/mL) | 37.30 ± 2.90 * | 39.45 ± 3.44 * | ||
| (10 mg/mL) | 42.00 ± 3.73 * | 44.75 ± 1.86 * | ||
| A375 | CNT (PBS) | 5.38 ± 1.09 | 5.00 ± 1.71 | |
| HCOE | (2 mg/mL) | 17.78 ± 1.38 | 13.10 ± 1.92 | |
| (4 mg/mL) | 16.82 ± 0.75 | 25.38 ± 2.96 * | ||
| (6 mg/mL) | 29.44 ± 1.17 * | 34.71 ± 1.86 * | ||
| (8 mg/mL) | 31.21 ± 3.81 * | 48.26 ± 0.77 * | ||
| (10 mg/mL) | 47.14 ± 1.73 * | 55.13 ± 1.47 * | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanni, V.; Di Marco, G.; Sacchetti, G.; Canini, A.; Gismondi, A. Oregano Phytocomplex Induces Programmed Cell Death in Melanoma Lines via Mitochondria and DNA Damage. Foods 2020, 9, 1486. https://doi.org/10.3390/foods9101486
Nanni V, Di Marco G, Sacchetti G, Canini A, Gismondi A. Oregano Phytocomplex Induces Programmed Cell Death in Melanoma Lines via Mitochondria and DNA Damage. Foods. 2020; 9(10):1486. https://doi.org/10.3390/foods9101486
Chicago/Turabian StyleNanni, Valentina, Gabriele Di Marco, Gianni Sacchetti, Antonella Canini, and Angelo Gismondi. 2020. "Oregano Phytocomplex Induces Programmed Cell Death in Melanoma Lines via Mitochondria and DNA Damage" Foods 9, no. 10: 1486. https://doi.org/10.3390/foods9101486
APA StyleNanni, V., Di Marco, G., Sacchetti, G., Canini, A., & Gismondi, A. (2020). Oregano Phytocomplex Induces Programmed Cell Death in Melanoma Lines via Mitochondria and DNA Damage. Foods, 9(10), 1486. https://doi.org/10.3390/foods9101486