Detection of Additives and Chemical Contaminants in Turmeric Powder Using FT-IR Spectroscopy

Abstract
:1. Introduction
- (1)
- Identify the vibrational modes of Sudan Red G, white turmeric, and yellow turmeric.
- (2)
- Prepare yellow turmeric—Sudan Red and yellow turmeric—white turmeric mixture samples at different concentrations and acquire IR spectra.
- (3)
- Develop PLSR models to estimate concentrations of Sudan Red and white turmeric in the mixed samples.
2. Materials and Methods
2.1. Sample Preparation
2.2. Spectral Acquisition
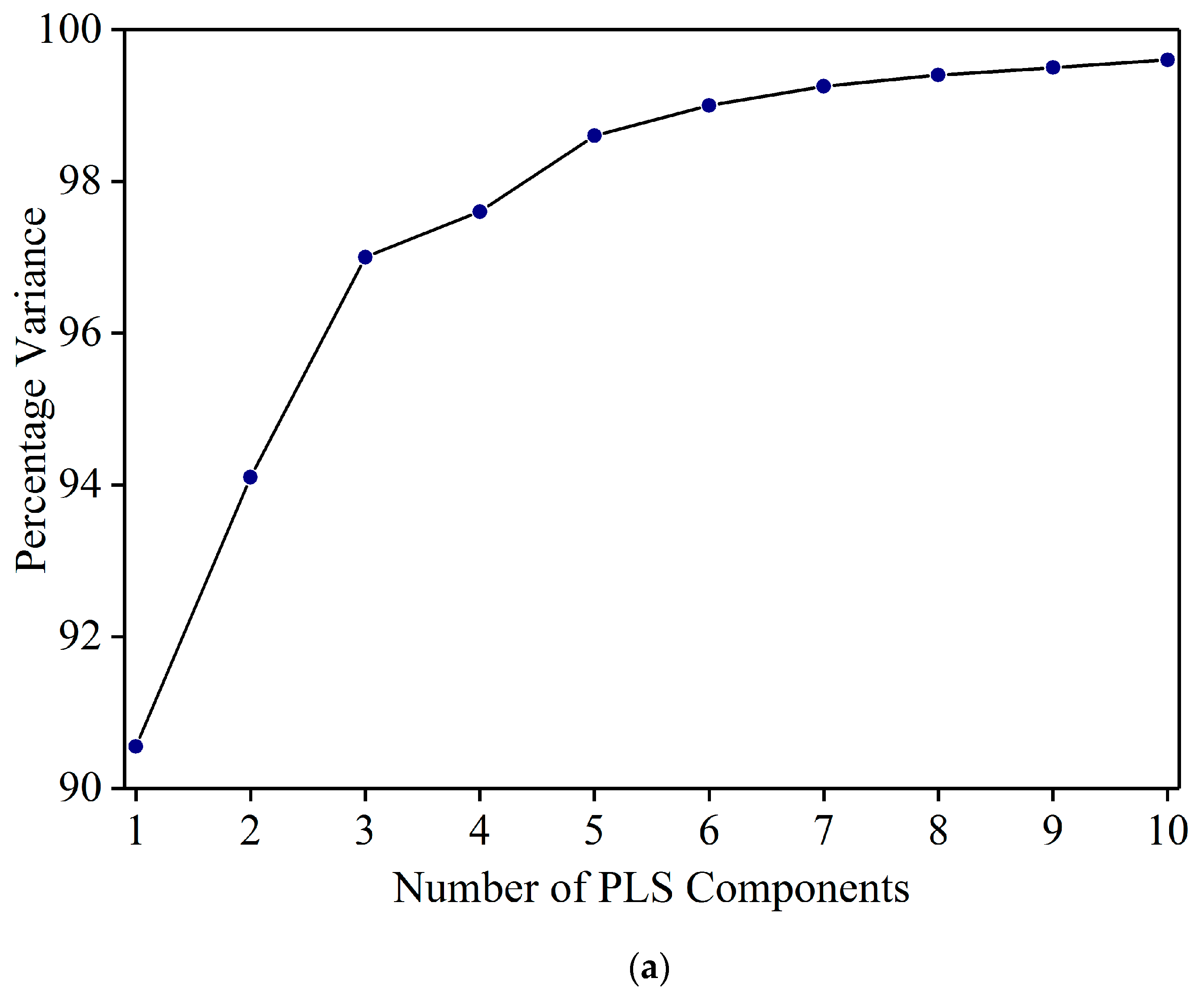
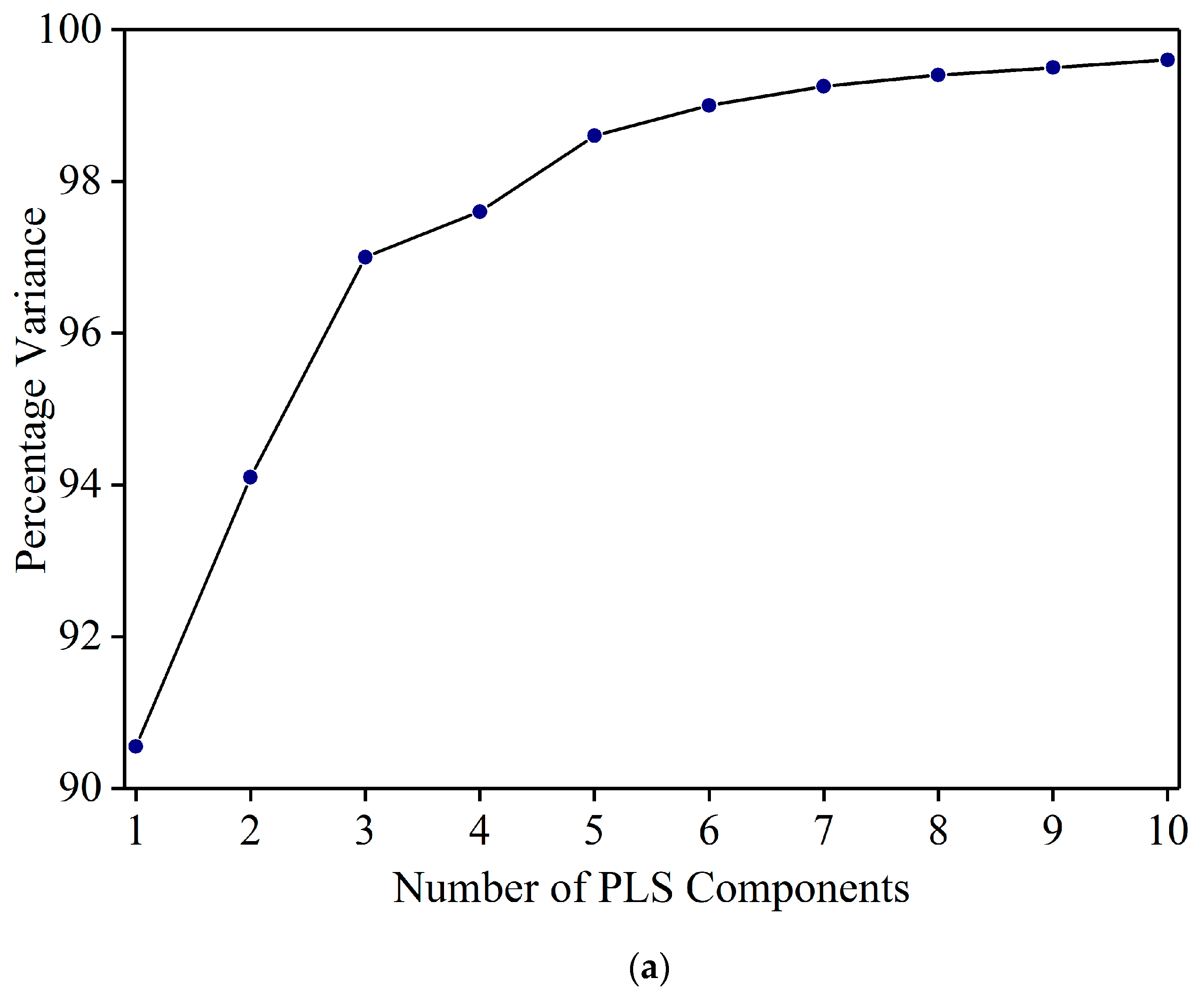
2.3. Data Analysis
3. Results and Discussion
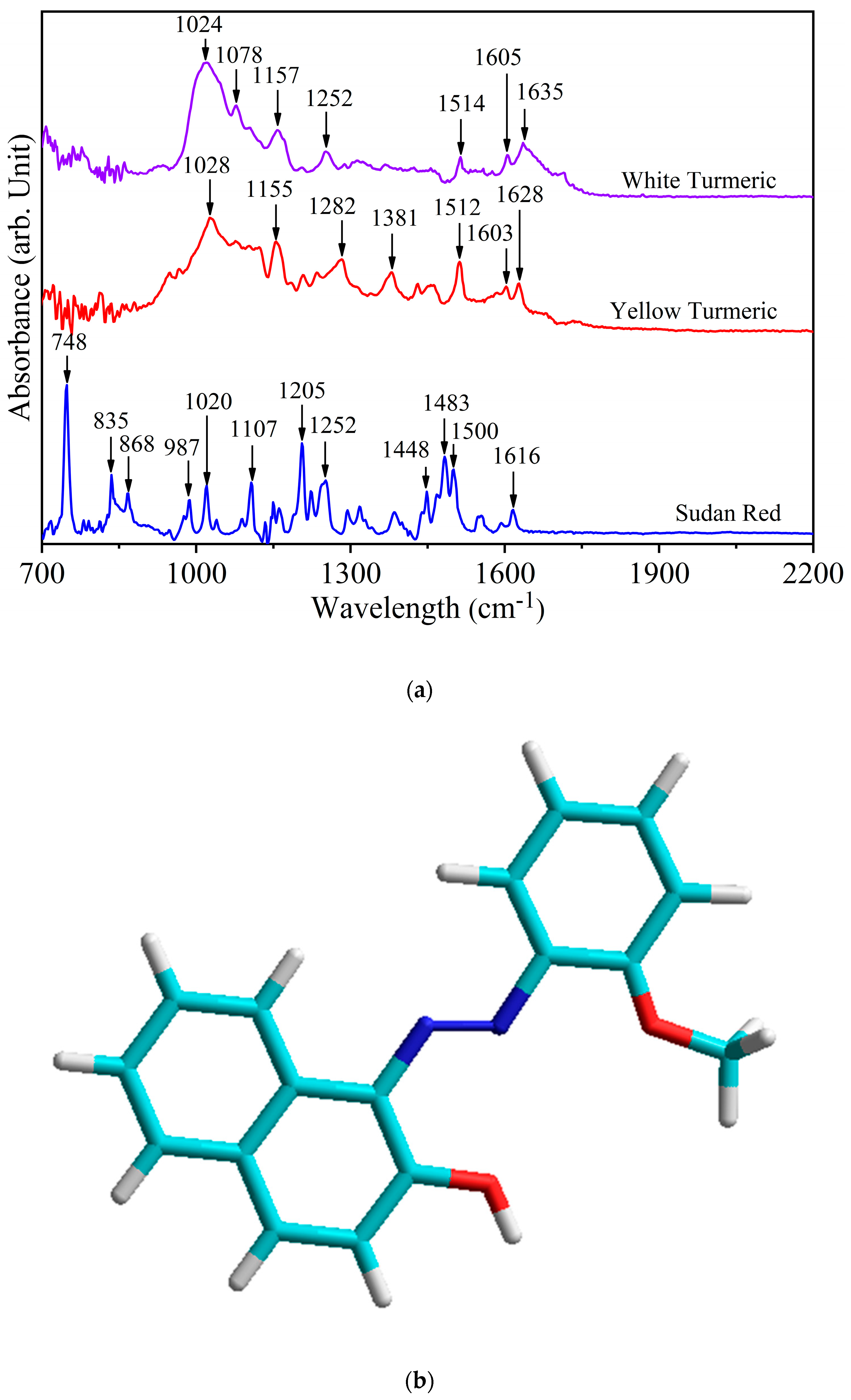
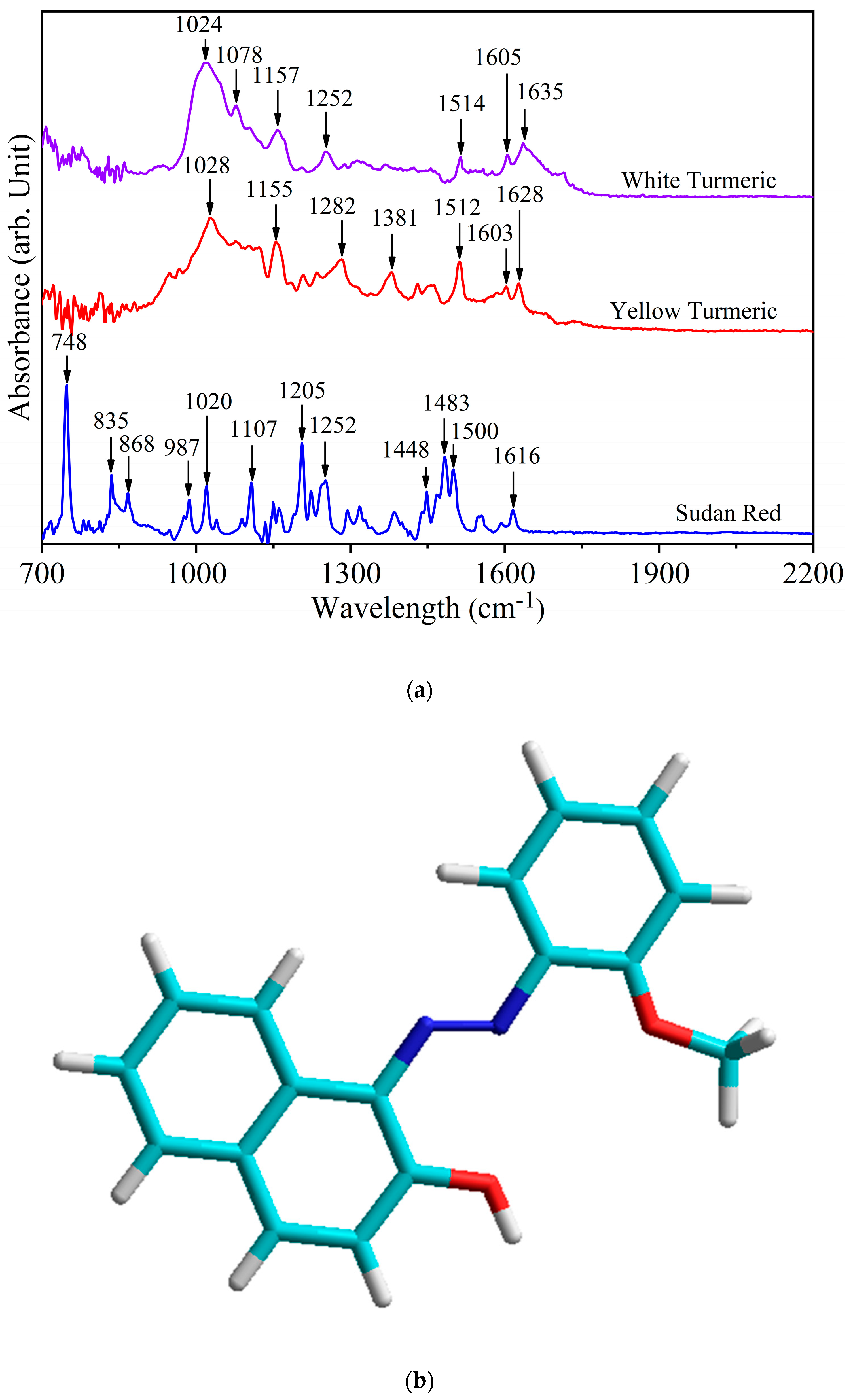
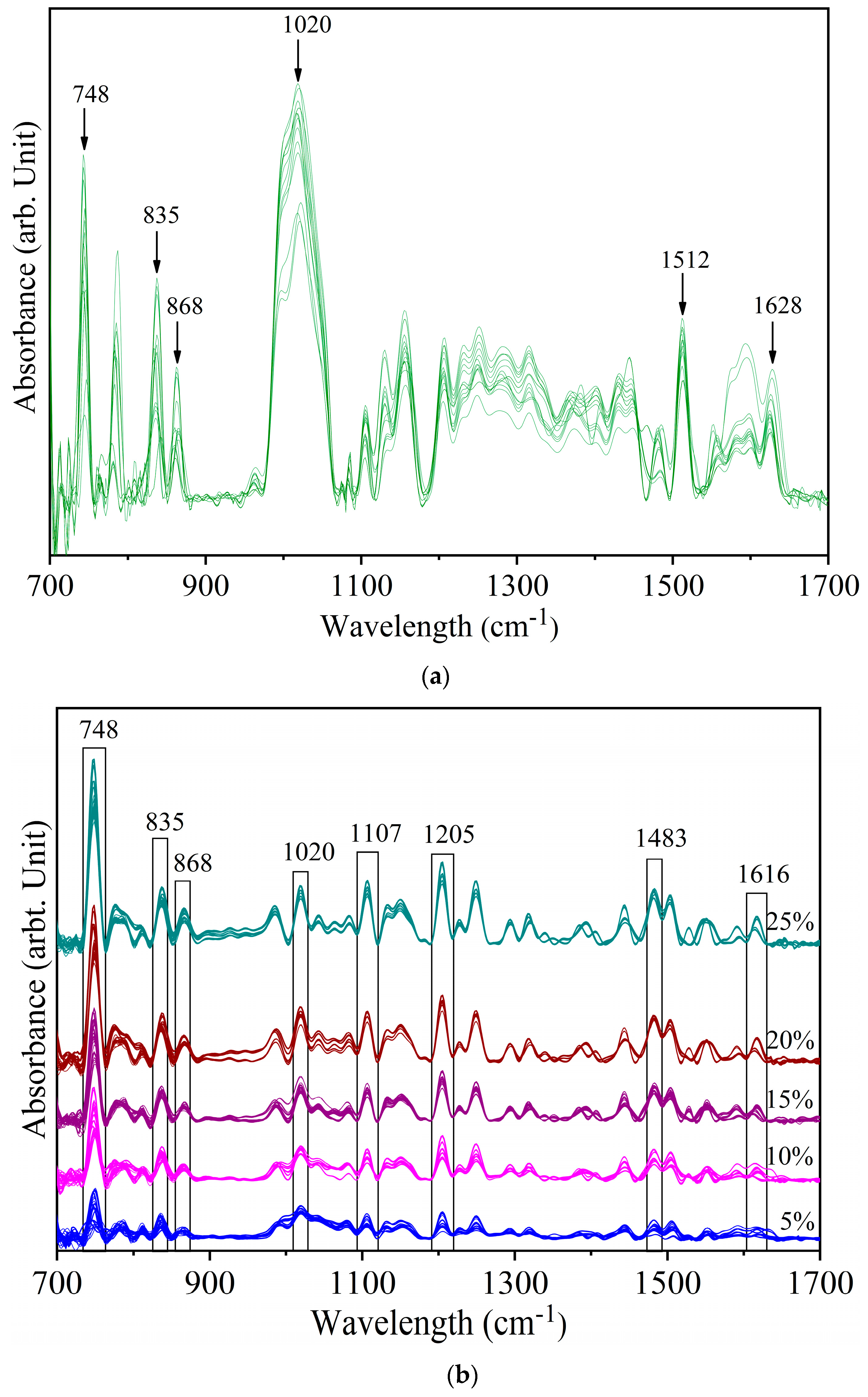
3.1. Assignment of Spectral Bands
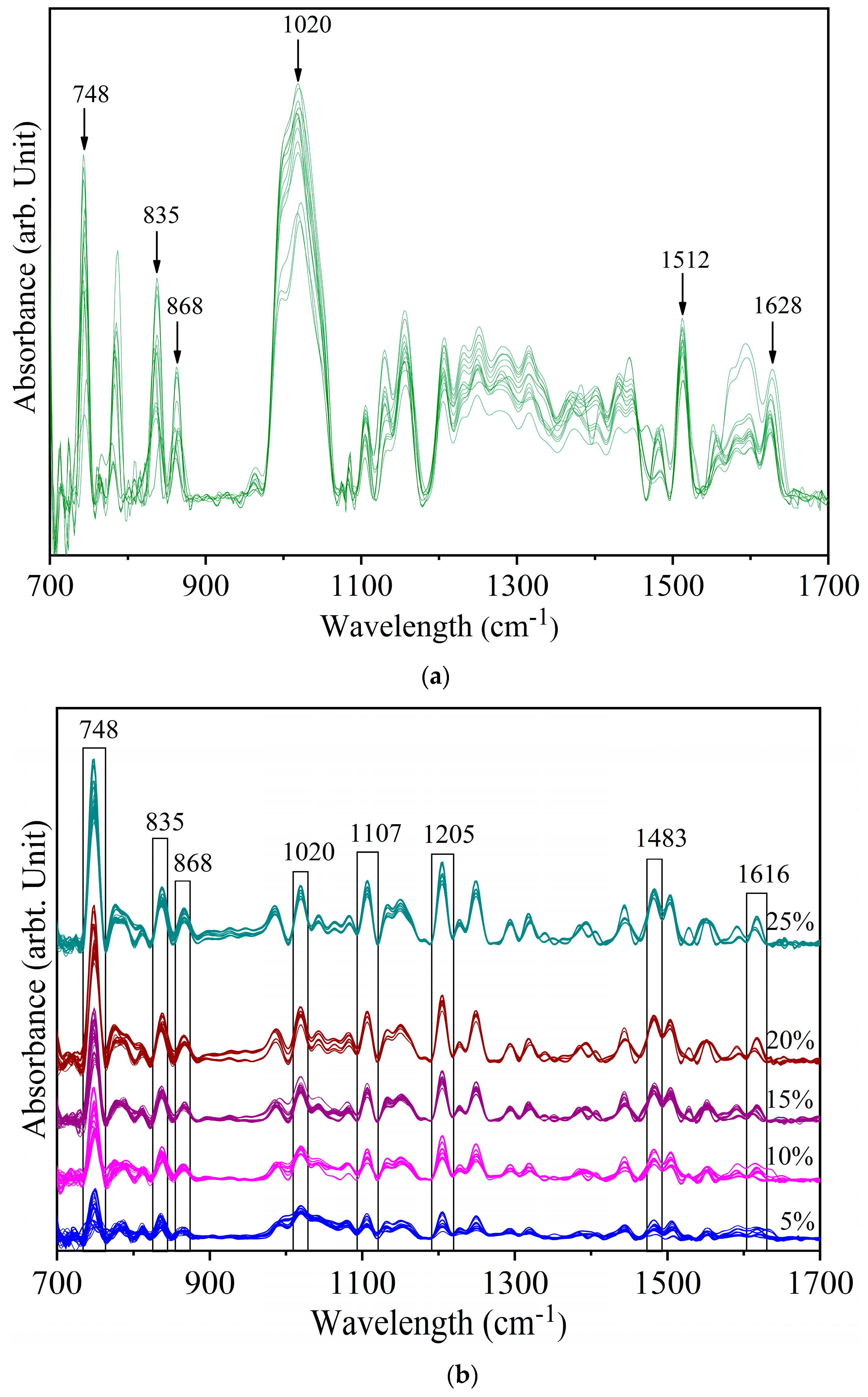
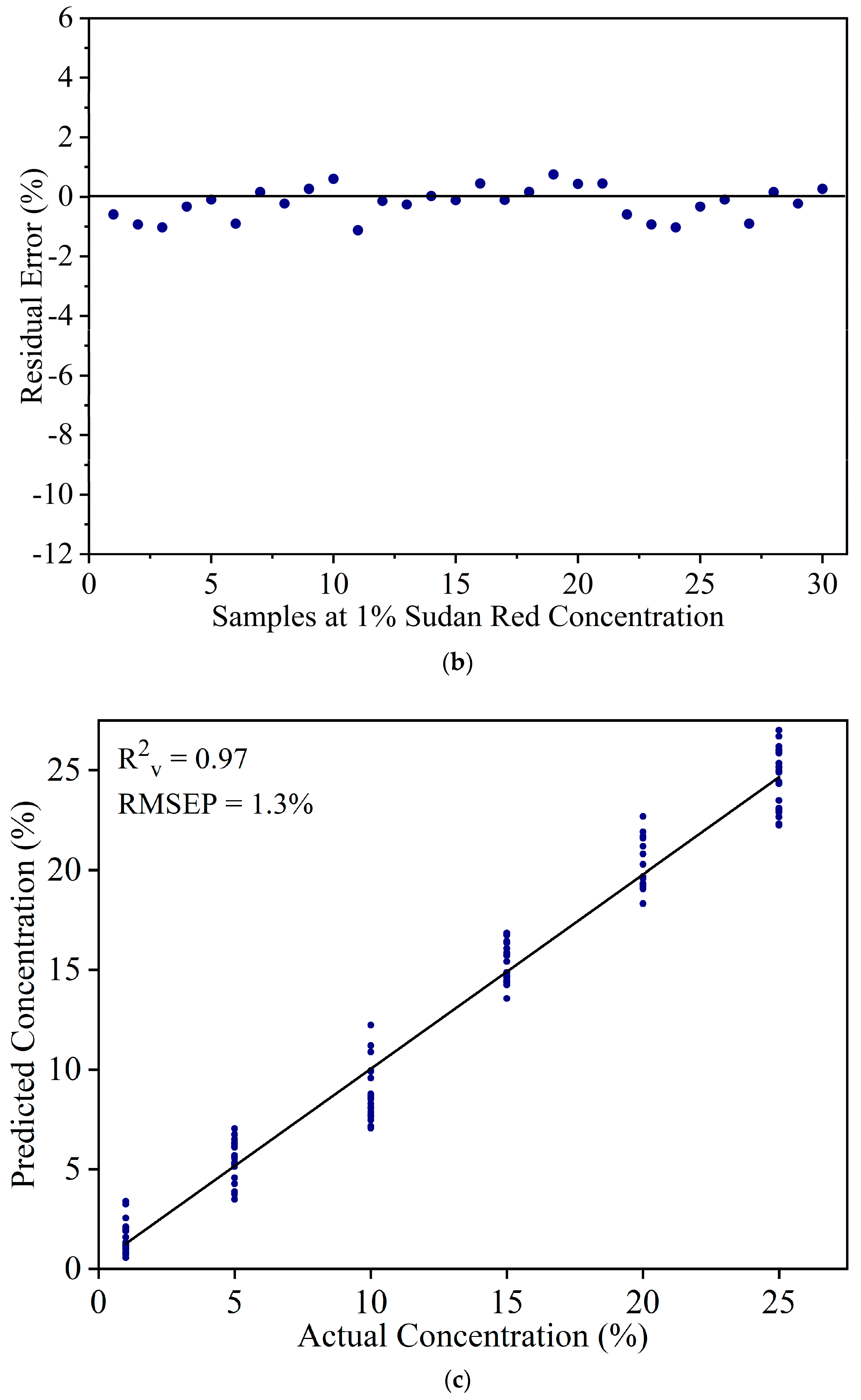
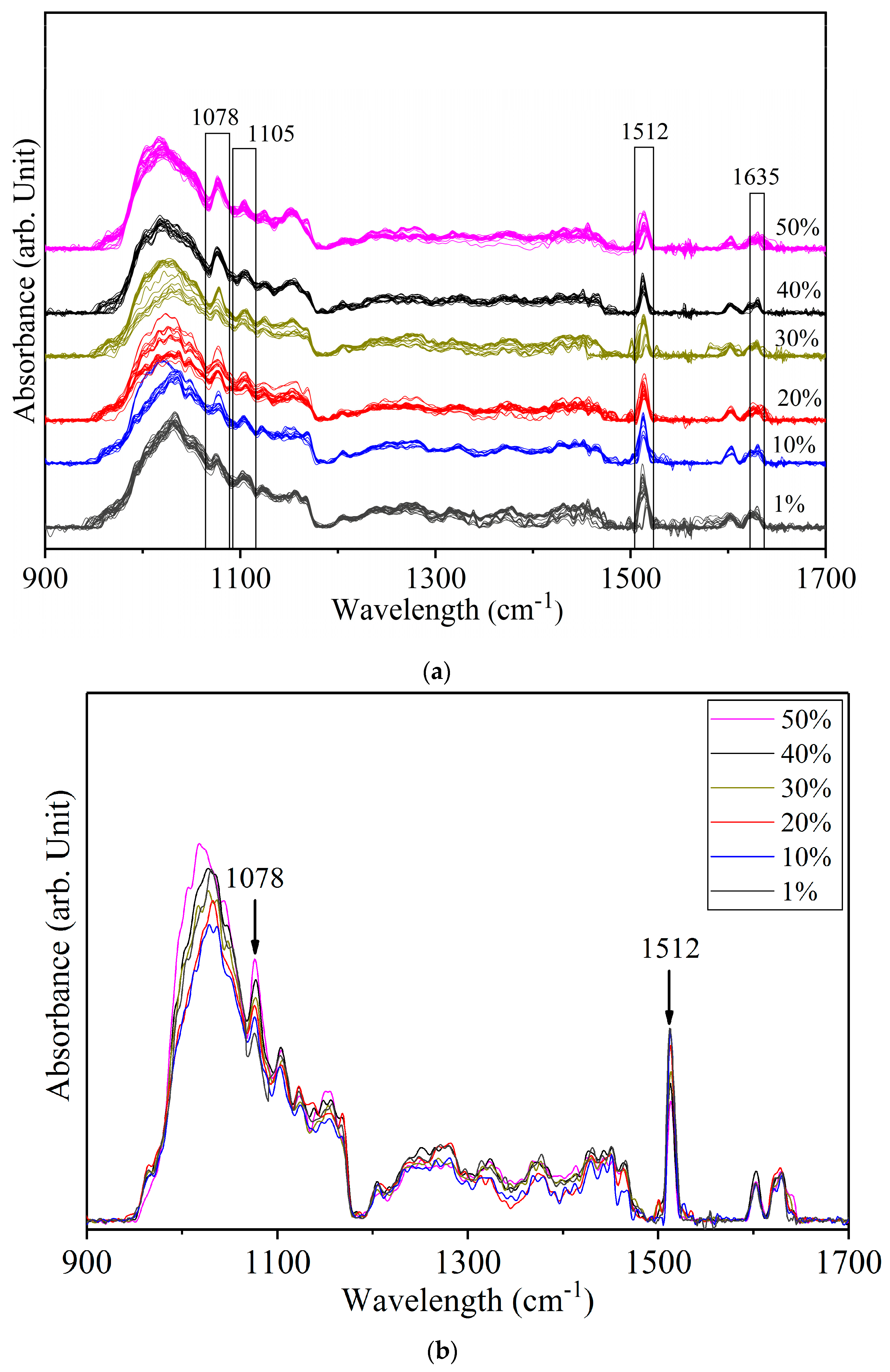
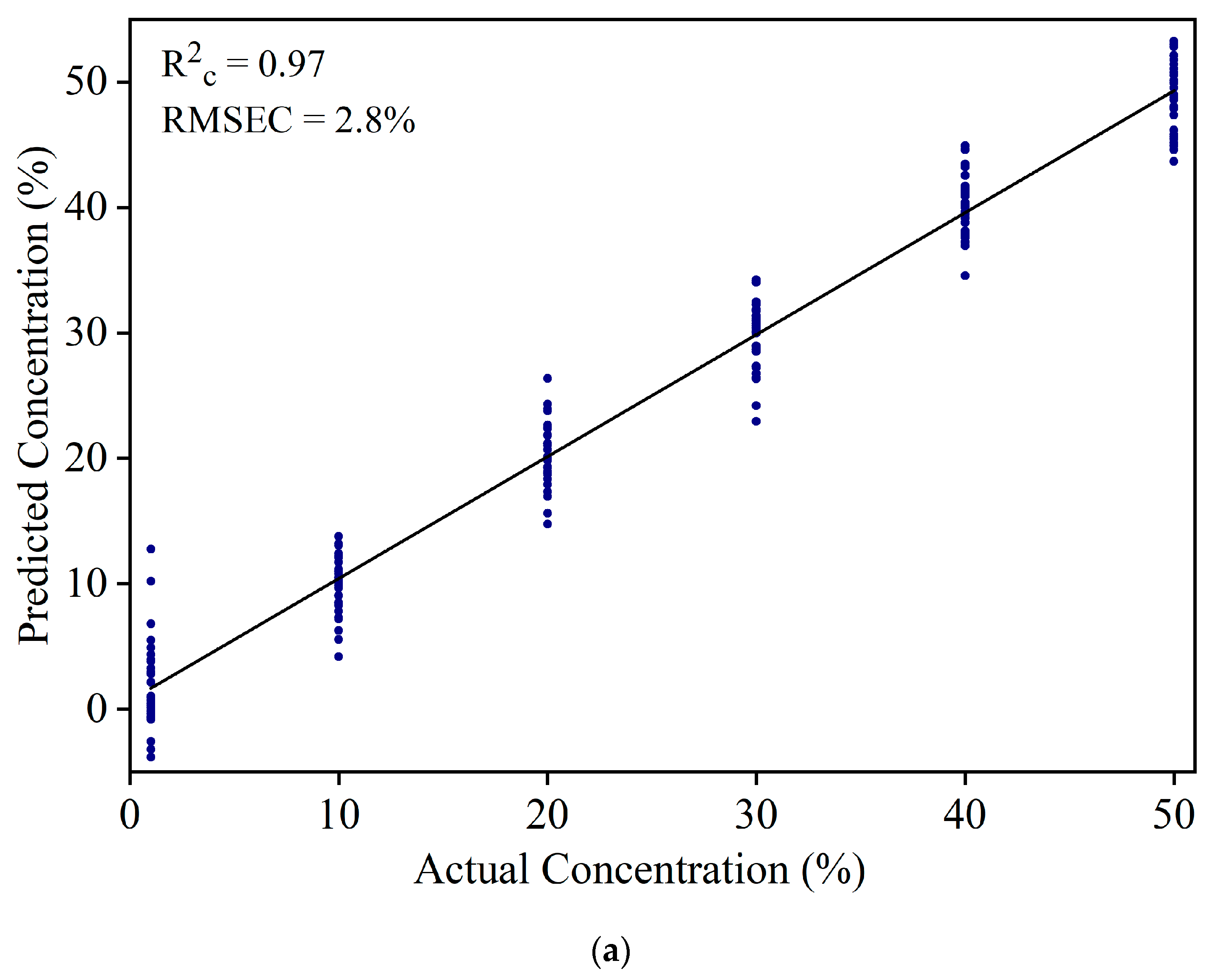
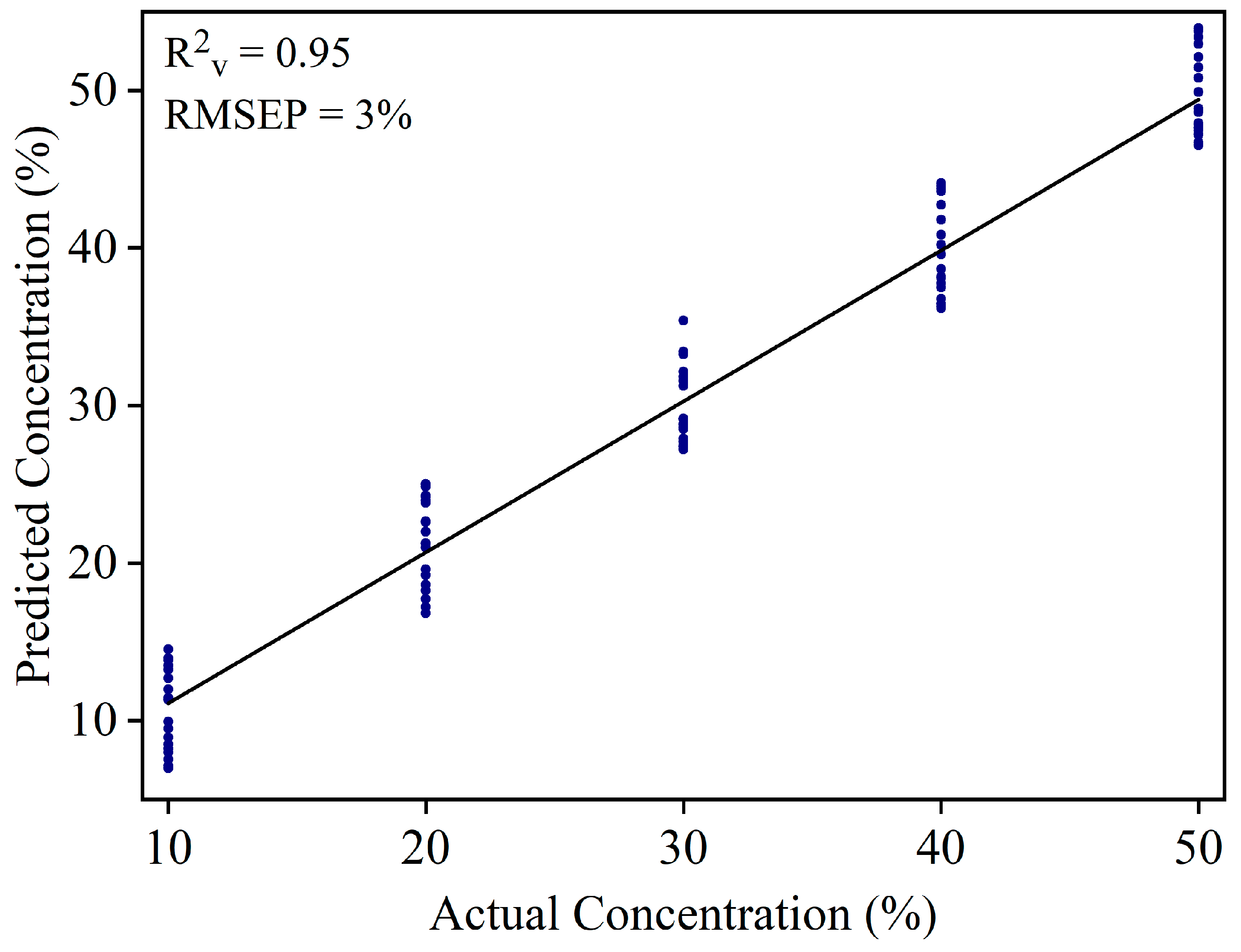
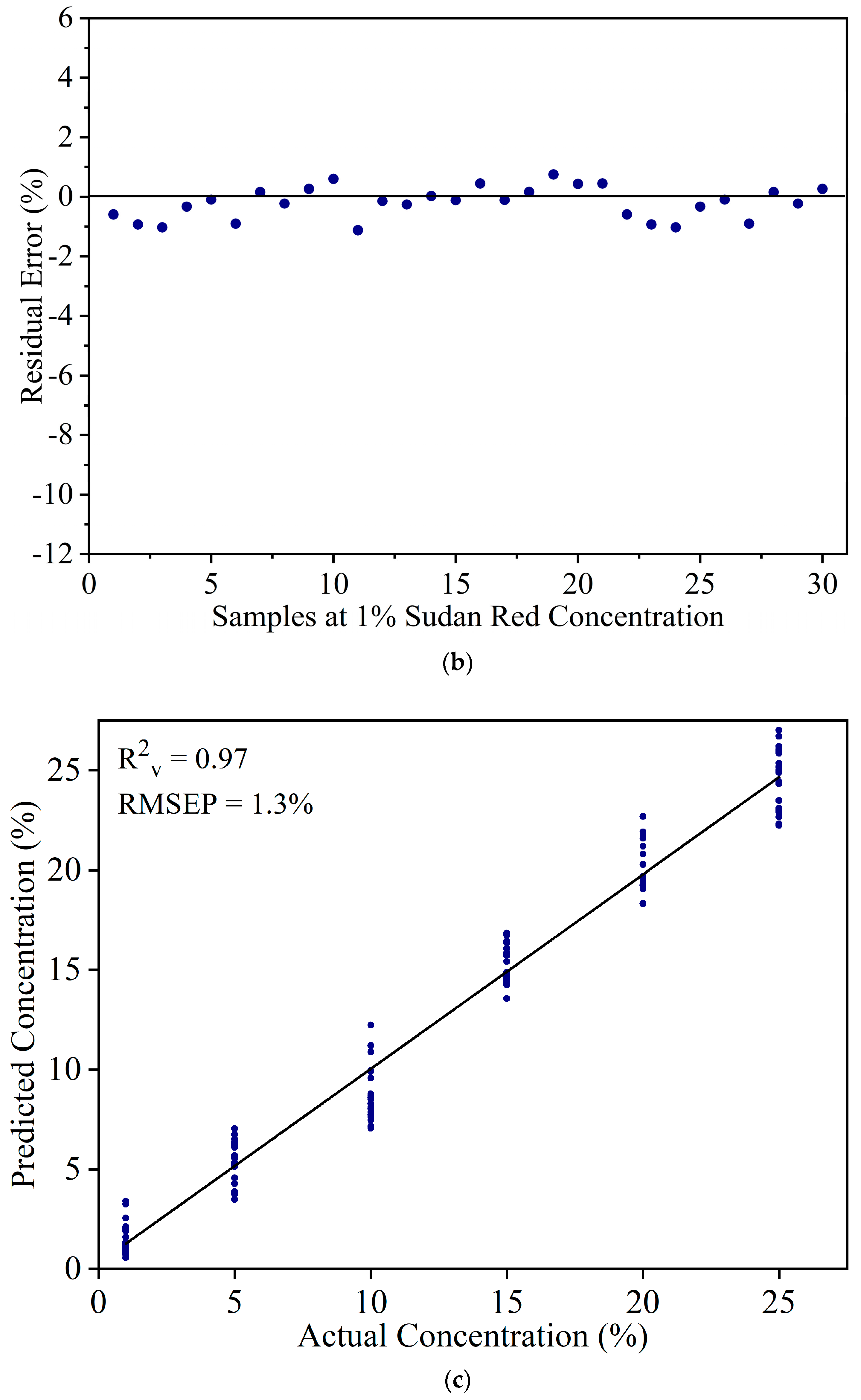
3.2. Detection of Sudan Red Contamination in Yellow Turmeric
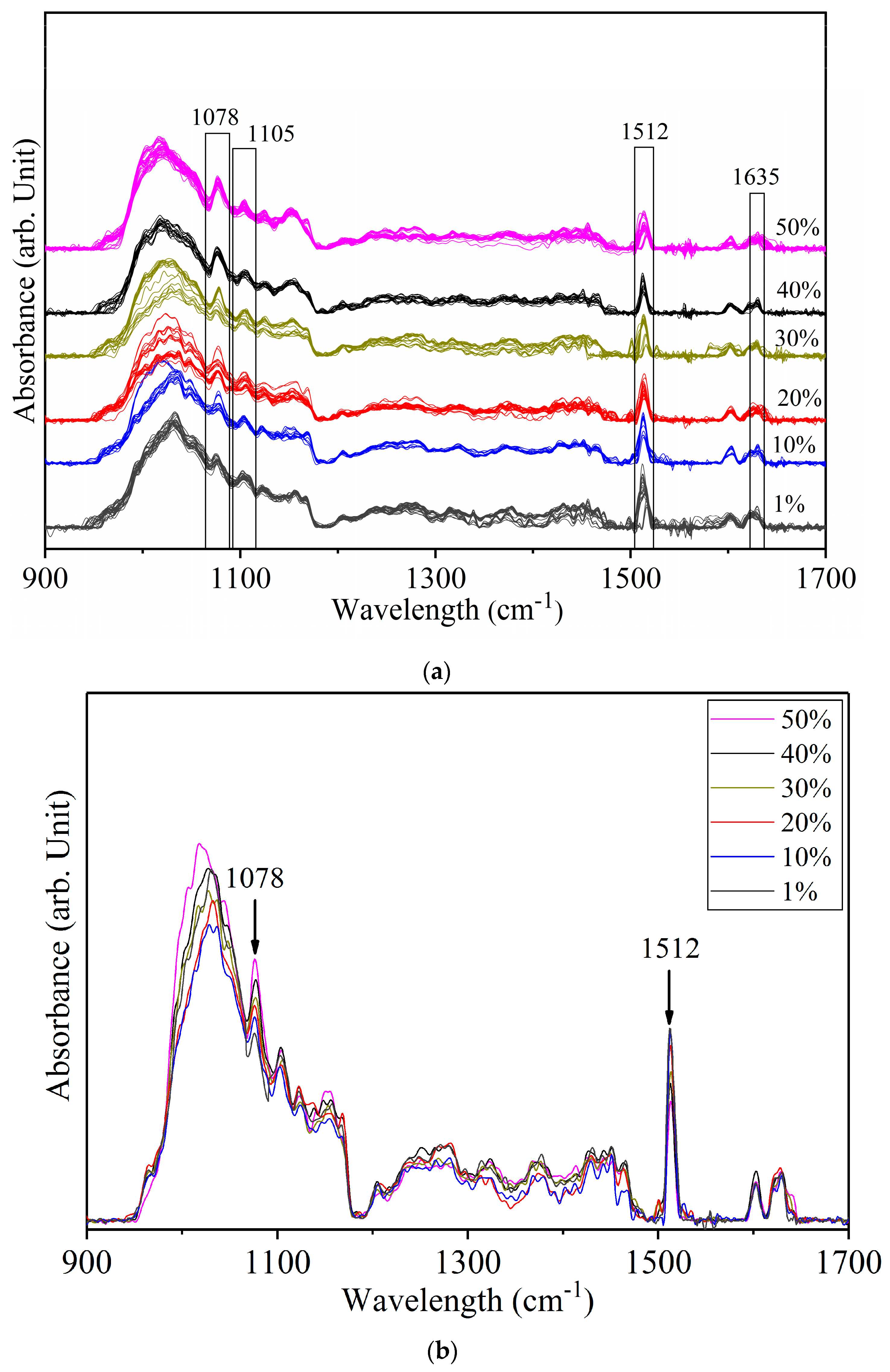
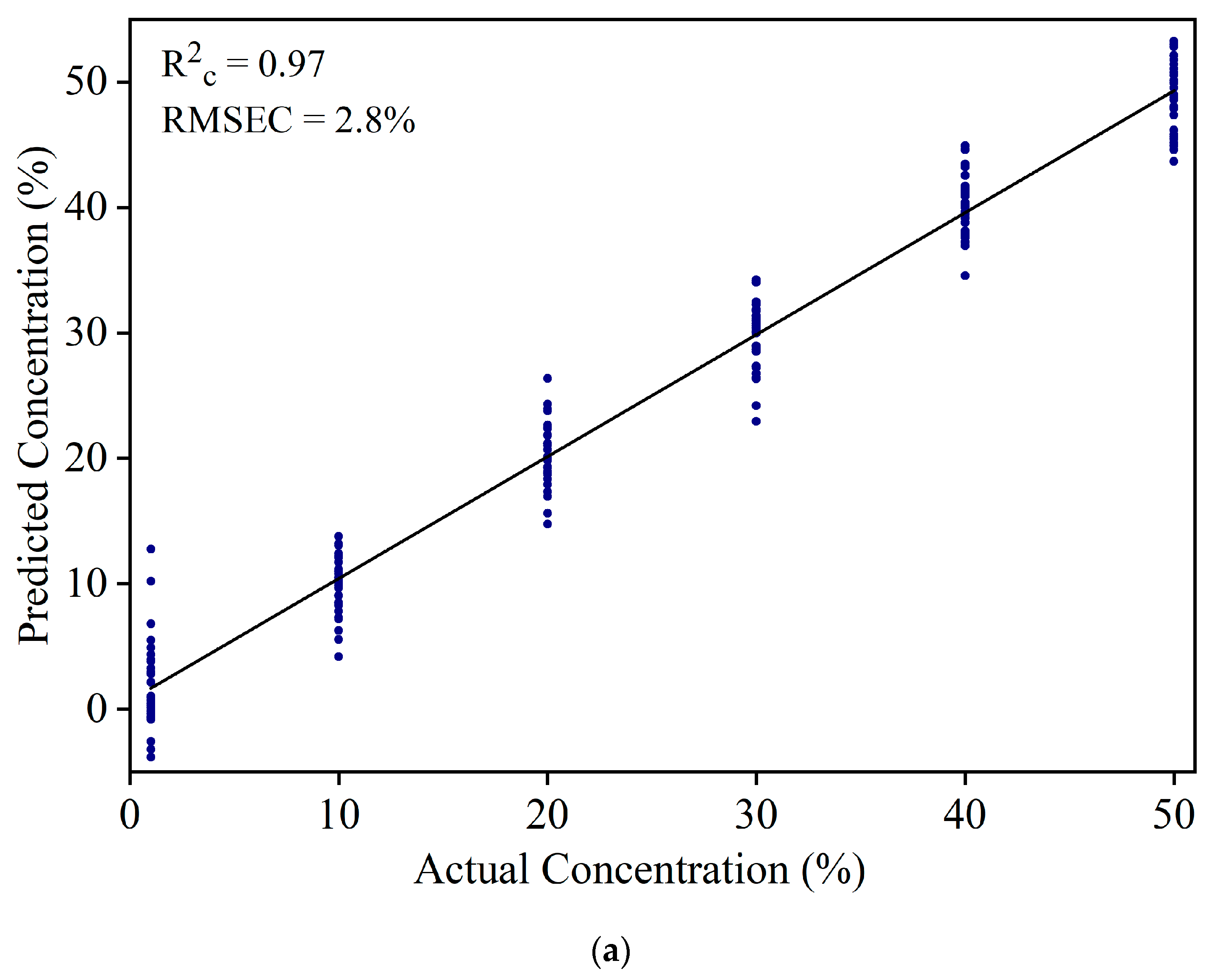
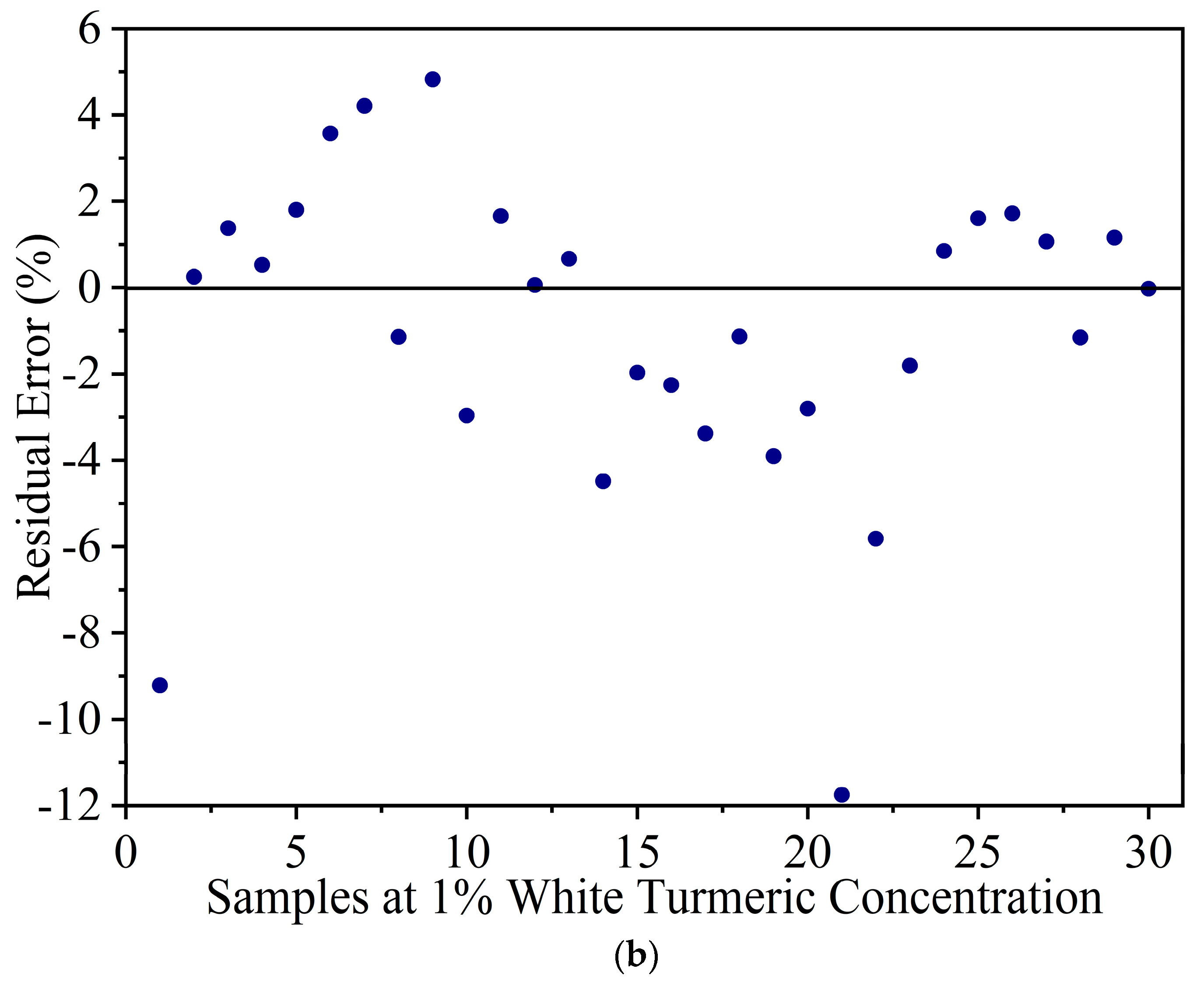
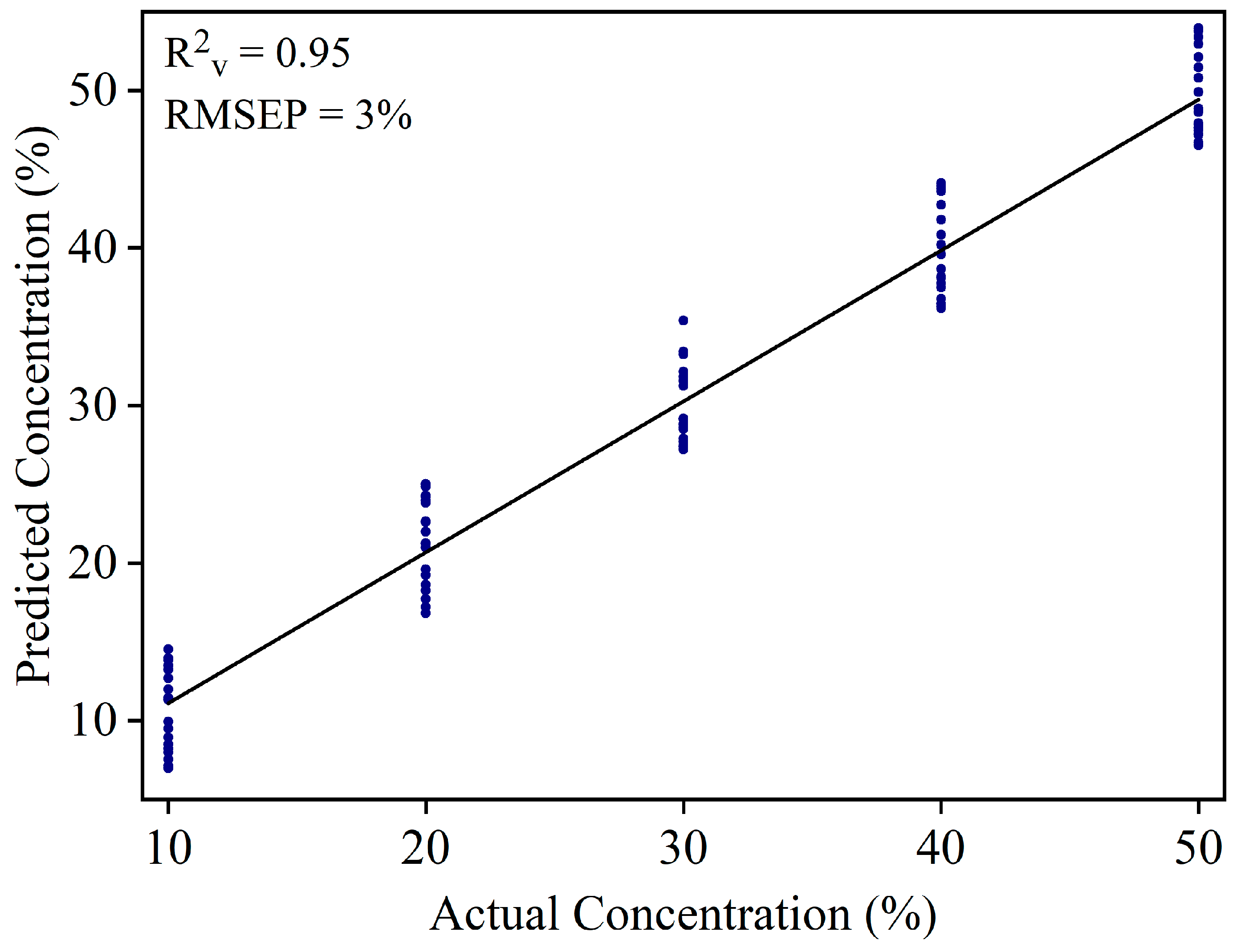
3.3. Detection of White Turmeric Adulteration in Yellow Turmeric
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Tayyem, R.F.; Heath, D.D.; Al-Delaimy, W.K.; Rock, C.L. Curcumin Content of Turmeric and Curry Powders. Nutr. Cancer 2006, 55, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Chao, K.; Schmidt, W.; Qin, J.; Kim, M.; Chan, D. Evaluation of turmeric powder adulterated with metanil yellow using FT-Raman and FT-IR spectroscopy. Foods 2016, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Joe, B.; Vijakumar, M.; Lokesh, B.R. Biological Properties of Curcumin-Cellular and Molecular Mechanisms of Action. Crit. Rev. Food Sci. Nutr. 2004, 44, 97–111. [Google Scholar] [CrossRef]
- Duvoix, A.; Blasius, R.; Delhalle, S.; Schnekenburger, M.; Morceau, F.; Henry, E.; Dicato, M.; Diederich, M. Chemopreventive and therapeutic effects of curcumin. Cancer Lett. 2005, 223, 181–190. [Google Scholar] [CrossRef]
- Ruby, A.J.; Kuttan, G.; Babu, K.D.; Rajasekharan, K.N.; Kuttan, R. Anti-tumour and antioxidant activity of natural curcuminoids. Cancer Lett. 1995, 94, 79–83. [Google Scholar] [CrossRef]
- Sidhu, G.S.; Singh, A.K.; Thaloor, D.; Banaudha, K.K.; Patnaik, G.K.; Srimal, R.C.; Maheshwari, R.K. Enhancement of wound healing by curcumin in animals. Wound Repair Regen. 1998, 6, 167–177. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Yamada, M. Review: Curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 2010, 16, 285–297. [Google Scholar] [CrossRef]
- Jayaprakasha, G.K.; Rao, L.J.M.; Sakariah, K.K. Improved HPLC Method for the Determination of Curcumin, Demethoxycurcumin, and Bisdemethoxycurcumin. J. Agric. Food Chem. 2002, 50, 3668–3672. [Google Scholar] [CrossRef]
- Velagudhan, K.C.; Muralidharan, V.K.; Amalraj, V.A.; Gautam, P.L.; Mandal, S.; Kumar, D. Curcuma genetic resources. National Bureau of Plant Genetic Resources; ICAR, Regional Station: Thrissur, India, 1999. [Google Scholar]
- Milobedzka, J.; Kostanecki, S.V.; Lampe, V. Zur Kenntnis des Curcumins. Ber. Dtsch. Chem. Ges. 1910, 43, 2163–2170. [Google Scholar] [CrossRef]
- Heath, D.D.; Khwaja, F.; Rock, C.L. Curcumin content of turmeric and curry powders. FASEB 2004, 18, A125. [Google Scholar]
- Hossain, M.A.; Ishimine, Y. Growth, yield and quality of turmeric (Curcuma longa L.) cultivated on dark-red soil, gray soil and red soil in Okinawa, Japan. Plant Prod. Sci. 2005, 8, 482–486. [Google Scholar] [CrossRef]
- Siviero, A.; Gallo, E.; Maggini, V.; Gori, L.; Mugelli, A.; Firenzuoli, F.; Vannacci, A. Curcumin, a golden spice with a low bioavailability. J. Herb. Med. 2015, 5, 57–70. [Google Scholar] [CrossRef]
- Akamine, H.; Hossain, M.A.; Ishimine, Y.; Yogi, K.; Hokama, K.; Iraha, Y.; Aniya, Y. Effects of application of N, P and K alone or in combination on growth, yield and curcumin content of turmeric (Curcuma longa L.). Plant Prod. Sci. 2007, 10, 151–154. [Google Scholar] [CrossRef]
- Sasikumar, B. Genetics resources of Curcuma: Diversity, characterization and utilization. Plant Genet. Resour. 2005, 3, 230–251. [Google Scholar] [CrossRef]
- Blumenthal, M.; Lindstrom, A.; Lynch, M.E.; Rea, P. Herb sales continue growth-up 3.3% in 2010. HerbalGram 2011, 90, 64–67. [Google Scholar]
- Curcumin Market Size, Share and Trends Analysis Report by Application (Pharmaceutical, Food, Cosmetics), by Region (Asia Pacific, North America, CSA, Europe, MEA) and Segment Forecasts 2018–2028. Available online: https://www.grandviewresearch.com/industry-analysis/turmeric-extract-curcumin-market (accessed on 28 March 2019).
- Sasikumar, B.; Syamkumar, S.; Remya, R.; Zachariah, T.J. PCR based detection of adulteration in the market samples of turmeric powder. Food Biotechnol. 2004, 18, 299–306. [Google Scholar] [CrossRef]
- Sen, A.R.; Gupta, P.S.; Dastidar, N.G. Detection of Curcuma zedoaria and Curcuma aromatic in Curcuma longa (Turmeric) by thin-layer chromatography. Analyst 1974, 99, 153–155. [Google Scholar] [CrossRef]
- Dhanya, K.; Syamkumar, S.; Siju, S.; Sasikumar, B. Sequence characterized amplified region markers: A reliable tool for adulterant detection in turmeric powder. Food Res. Int. 2011, 44, 2889–2895. [Google Scholar] [CrossRef]
- Di Anibal, C.V.; Odena, M.; Ruisanchez, I.; Callao, M.P. Determining the adulteration of spices with Sudan I-II-III-IV dyes by UV-visible spectroscopy and multivariate classification techniques. Talanta 2009, 79, 887–892. [Google Scholar] [CrossRef]
- Lohumi, S.; Joshi, R.; Kandpal, L.M.; Lee, H.; Kim, M.S.; Cho, H.; Mo, C.; Seo, Y.W.; Rahman, A.; Cho, B.K. Quantitative analysis of Sudan dye adulteration in paprika using FTIR spectroscopy. Food Addit. Contam. Part A 2017, 34, 678–686. [Google Scholar] [CrossRef]
- Tateo, F.; Bononi, M. Fast determination of Sudan I by HPLC/APCI-MS in hot chili, spices, and oven-baked foods. J. Agric. Food Chem. 2004, 52, 655–658. [Google Scholar] [CrossRef]
- Chen, L.; Hu, J.; Zhang, W.; Zhang, J.; Guo, P.; Sun, C. Simultaneous determination of nine banned azo dyes in foodstuffs and beverages by high-performance capillary electrophoresis. Food Anal. Methods 2015, 8, 1903–1910. [Google Scholar] [CrossRef]
- Zhao, S.; Yin, J.; Zhang, J.; Ding, X.; Wu, Y.; Shao, B. Determination of 23 dyes in chili powder and paste by high-performance liquid chromatography-electrospray ionization tandem mass spectrometry. Food Anal. Methods 2012, 5, 1018–1026. [Google Scholar] [CrossRef]
- Parvathy, V.A.; Swetha, V.P.; Sheeja, T.E.; Sasikumar, B. Detection of plant-based adulterants in turmeric powder using DNA barcoding. Pharm. Biol. 2015, 53, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Di Anibal, C.V.; Ruisanchez, I.; Callao, M.P. High-resolution 1H nuclear resonance spectrometer combined with chemometrics treatment to identify adulteration of culinary spices with Sudan dyes. Food Chem. 2011, 124, 1139–1145. [Google Scholar] [CrossRef]
- Di Anibal, C.; Rodriguez, M.S.; Albertengo, L. UV-visible spectroscopy and multivariate classification as a screening tool to identify adulteration of culinary spices with Sudan I and blends of Sudan I + IV dyes. Food Anal. Methods 2014, 7, 1090–1096. [Google Scholar] [CrossRef]
- Horn, B.; Esslinger, S.; Pfister, M.; Fauhl-Hassek, C.; Riedl, J. Non-targeted detection of paprika adulteration using mid-infrared spectroscopy and one-class classification-Is it the data preprocessing that makes the performance? Food Chem. 2018, 257, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, E.A.; Polissiou, M.G. Assessing saffron (Crocus sativus L.) adulteration with plant-derived adulterants by diffuse reflectance infrared Fourier transform spectroscopy coupled with chemometrics. Talanta 2017, 162, 558–566. [Google Scholar] [CrossRef]
- Barbosa-Canovas, G.V.; Ortega-Rivas, E.; Juliano, P.; Yan, H. Food Powders. Physical Properties, Properties and Functionality; Springer: New York, NY, USA, 2005; pp. 221–244. [Google Scholar]
- Vazhnova, T.; Lukyanov, D.B. Fourier self-deconvolution of the IR spectra as a tool for investigation of distinct function groups in porous materials: Bronsted acid sites in zeolites. Anal. Chem. 2013, 85, 11291–11296. [Google Scholar] [CrossRef]
- Kaupplnen, J.K.; Moffatt, D.J.; Mantsch, H.H.; Cameron, D.G. Fourier self-deconvolution: A method for resolving intrinsically overlapped bands. Appl. Spectrosc. 1981, 35, 271–276. [Google Scholar] [CrossRef]
- Tooke, P.B. Fourier self-deconvolution in IR spectroscopy. Trends Anal. Chem. 1988, 7, 130–136. [Google Scholar] [CrossRef]
- Kaupplnen, J.K.; Moffatt, D.J.; Mantsch, H.H.; Cameron, D.G. Fourier transforms in the computation of self-deconvoluted and first-order derivative spectra of overlapped band contours. Anal. Chem. 1981, 53, 1454–1457. [Google Scholar] [CrossRef]
- Song, S.; Asher, S.A.; Krimm, S.; Bandehar, J. Assignment of a New Conformation-Sensitive UV Resonance Raman Band in Peptides and Proteins. J. Am. Chem. Soc. 1988, 110, 8547–8548. [Google Scholar] [CrossRef]
- Edwards, H.G.M.; Johnson, A.F.; Lewis, I.R.; Wheelwright, S.J. Raman and FTIR spectroscopic studies of copolymers of methyl methacrylate with butadiene. Spectrochim. Acta 1993, 49, 457–464. [Google Scholar] [CrossRef]
- Synytsya, A.; Čopíková, C.J.; Matéjka, P.; Machovič, V. Fourier transform Raman and infrared spectroscopy of pectins. Carbohydr. Polym. 2003, 54, 97–106. [Google Scholar] [CrossRef]
- Schulz, H.; Baranska, M. Identification and quantification of valuable plant substances by IR and Raman spectroscopy. Vib. Spectrosc. 2007, 43, 13–25. [Google Scholar] [CrossRef]
- Sundaraganesan, N.; Anand, B.; Jian, F.F.; Zhao, P. FT-Raman and FT-IR spectra, ab initio and density functional studies of 3,4-dichlorobenyzl alcohol. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006, 65, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Currie, D.J.; Lough, C.E.; McClusky, F.K.; Holmes, H.L. Effect of functional group conformation on infrared spectra of some gen dysfunctional phenylethylene derivatives. Can. J. Chem. 1969, 47, 3147–3152. [Google Scholar] [CrossRef]
- Smulevich, G.; Angeloni, L.; Giovannarde, S.; Marzocchi, M.P. Resonance Raman and polarized light infrared spectra of 1,4-dihydroxyanthraquinone. vibrational studies of the ground and excited electronic states. Chem. Phys. 1982, 65, 313–322. [Google Scholar] [CrossRef]
- Feng, J.; YuHong, Q.; Green, A.E.S. Analytical model of corn cob Pyroprobe-FTIR data. Biomass Bioenergy 2006, 30, 486–492. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.; Chung, H. Direct on-line Raman measurement of flying solid samples: Determination of polyethylene pellet density. Talanta 2011, 83, 879–884. [Google Scholar] [CrossRef]
- Amiri, A.; Zardini, H.Z.; Shanbedi, M.; Maghrebi, M.; Baniadam, M.; Tolueinia, B. Efficient method for functionalization of carbon nanotubes by lysine and improved antimicrobial activity and water-dispersion. Mater. Lett. 2012, 72, 153–156. [Google Scholar] [CrossRef]
- Çelekli, A.; Çelekli, F.; Çiçek, E.; Bozkurt, H. Predictive modeling of sorption and desorption of a reactive azo dye by pumpkin husk. Environ. Sci. Pollut. Res. 2014, 21, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Sarıkaya, E.K.; Dereli, Ö. Molecular structure and vibrational spectra of 7-Methoxy- 4-methylcoumarin by density functional method. J. Mol. Struct. 2013, 1052, 214–220. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristics Group Frequencies, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2001; pp. 22–23. [Google Scholar]
- Liang, Y.; Zheng, M.; Park, K.H.; Lee, H.S. Thickness-dependent crystal orientation in poly (trimethylene 2,6-naphthalate) films studied with GIWAXD and RA-FTIR methods. Polymer 2008, 49, 1961–1967. [Google Scholar] [CrossRef]
- Guan, X.-H.; Shang, C.; Chen, G.-H. ATR-FTIR investigation of the role of phenolic groups in the interaction of some NOM model compound with aluminum hydroxide. Chemosphere 2006, 65, 2074–2081. [Google Scholar] [CrossRef]
- Darvin, M.E.; Gersonde, I.; Ey, S.; Brand, N.N.; Albrecht, H.; Gonchukov, S.A.; Sterry, W.; Lademann, J. Noninvasive Detection of beta-Carotene and Lycopene in Human Skin using Raman spectroscopy. Laser Phys. 2004, 14, 231–233. [Google Scholar]
- Kunov-Kruse, A.J.; Kristensen, S.B.; Liu, C.; Berg, R.W. Experimental and ab initio DFT calculated Raman spectrum of Sudan, I., a red dye. J. Raman Spectrosc. 2011, 42, 1470–1478. [Google Scholar] [CrossRef]
- Dhakal, S.; Chao, K.; Schmidt, W.; Qin, J.; Kim, M.; Huang, Q. Detection of azo dyes in curry powder using a 1064-nm dispersive point-scan Raman system. Appl. Sci. 2018, 8, 564. [Google Scholar] [CrossRef]
- Mishra, K.K.; Dixit, S.; Purshottam, S.K.; Pandey, R.C.; Das, M.; Khanna, S.K. Exposure assessment to Sudan dyes through consumption of artificially coloured chilli powers in India. Int. J. Food Sci. Technol. 2007, 42, 1363–1366. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sudan Red G | Yellow Turmeric | White Turmeric | Special Assignments |
|---|---|---|---|
| 1635 | ν (C=O) stretching polycyclic quinones | ||
| 1628 | ν (C=C) stretching trans form | ||
| 1616 | ν (C–N) stretching in C=C–N=N | ||
| 1603 | 1605 | ν (C=C) stretching out of plane | |
| 1554 | δ (N-H) bending in Ar–N=N··H–O–Ar’ | ||
| 1512 | 1514 | δ (Ar–O + Ar–O–R) bending asym | |
| 1500 | ν (N=N) stretching cis | ||
| 1483 | ν (N=N) stretching trans | ||
| 1448 | ν (N=N) stretching + δ (H–C) | ||
| 1385 | δ (Ar–O + Ar–N–R) bending sym | ||
| 1381 | δ (OH) bending alcoholic | ||
| 1369 | δ (H–C) bending in O=C–CH2–C=O | ||
| 1317 | ν (Ar–O) stretching sym in Ar–N=N··H–O–Ar’ | ||
| 1294 | ν (C–O)− stretching on deprotonated Ar–O− | ||
| 1282 | ν (C–O) stretching phenolic | ||
| 1252 | 1252 | CH bending | |
| 1234 | CH bending | ||
| 1223 | δ (O–H) bending asym | ||
| 1205 | CH3 deformation | ||
| 1161 | ν (C–Nazo) δ(CH) | ||
| 1149 | 1155 | 1157 | δ (CH3) rocking, methoxy |
| 1107 | aromatic CH bending in-plane | ||
| 1078 | ρ (CH3) rocking | ||
| 1020 | 1028 | 1024 | ring breathing |
| 987 | δ (C–N=N) out of plane bending (II) | ||
| 868 | ring breathing and stretching C–O | ||
| 835 | γ(C–O)ring out of ring plane OH bending | ||
| 748 | γ(N–C–C–O)oop resonance form of Amide V |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhakal, S.; Schmidt, W.F.; Kim, M.; Tang, X.; Peng, Y.; Chao, K. Detection of Additives and Chemical Contaminants in Turmeric Powder Using FT-IR Spectroscopy. Foods 2019, 8, 143. https://doi.org/10.3390/foods8050143
Dhakal S, Schmidt WF, Kim M, Tang X, Peng Y, Chao K. Detection of Additives and Chemical Contaminants in Turmeric Powder Using FT-IR Spectroscopy. Foods. 2019; 8(5):143. https://doi.org/10.3390/foods8050143
Chicago/Turabian StyleDhakal, Sagar, Walter F. Schmidt, Moon Kim, Xiuying Tang, Yankun Peng, and Kuanglin Chao. 2019. "Detection of Additives and Chemical Contaminants in Turmeric Powder Using FT-IR Spectroscopy" Foods 8, no. 5: 143. https://doi.org/10.3390/foods8050143
APA StyleDhakal, S., Schmidt, W. F., Kim, M., Tang, X., Peng, Y., & Chao, K. (2019). Detection of Additives and Chemical Contaminants in Turmeric Powder Using FT-IR Spectroscopy. Foods, 8(5), 143. https://doi.org/10.3390/foods8050143