Distribution of β-Glucan, Phenolic Acids, and Proteins as Functional Phytonutrients of Hull-Less Barley Grain

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Milling Procedure
2.3. Composition of Hull-Less Barley Fractions
2.4. β-Glucan Content
2.5. Extraction of Samples
2.6. Total Phenolic Content (TPC)
2.7. Antioxidant Activity (DPPH Radical Scavenging Activity)
2.8. Extraction of Albumins/Globulins and Hordeins
2.9. HPLC Analysis of Protein Fractions
2.10. Extraction of Phenolic Acids
2.11. HPLC Analysis of Phenolic Acids
2.12. Statistical Analysis
3. Results and Discussion
3.1. Hull-Less Barley Grain Characteristics
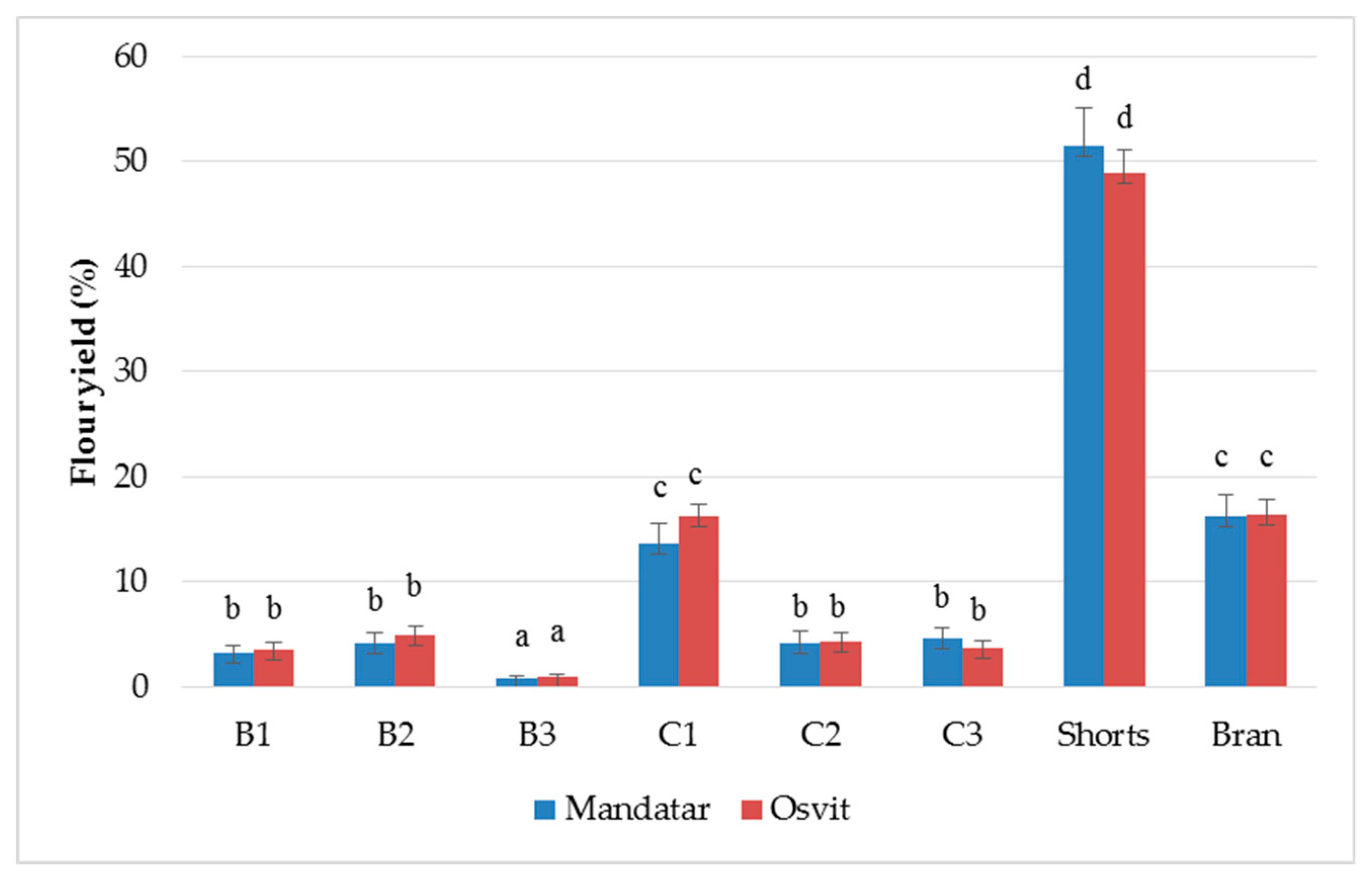
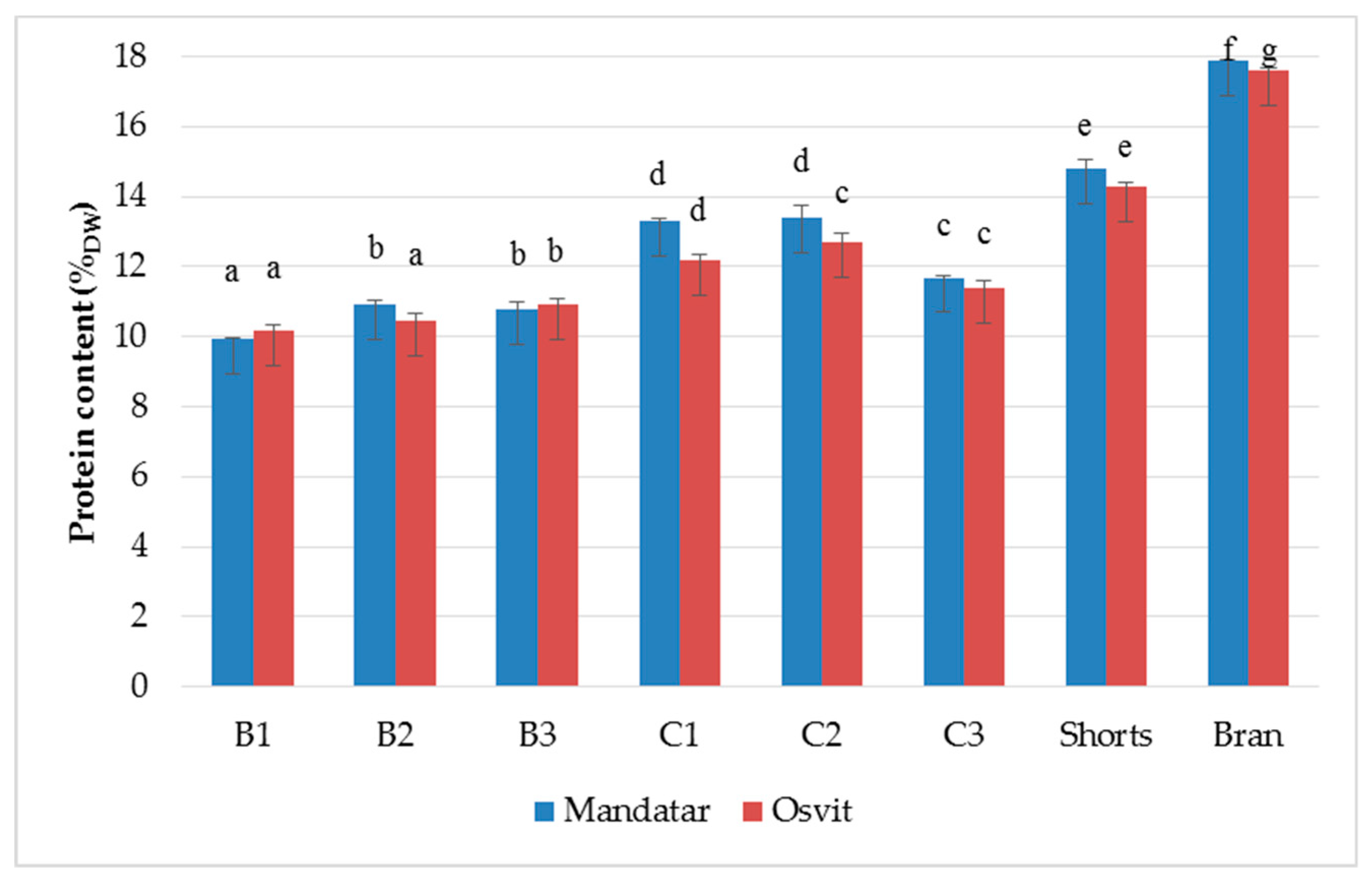
3.2. Yield and Chemical Characterisation of Milling Fractions
3.3. HPLC Albumins/Globulins and Hordeins
3.4. β-Glucan Content in Milling Fractions
3.5. Total Phenolic Content and Antioxidant Activity of Milling Fractions
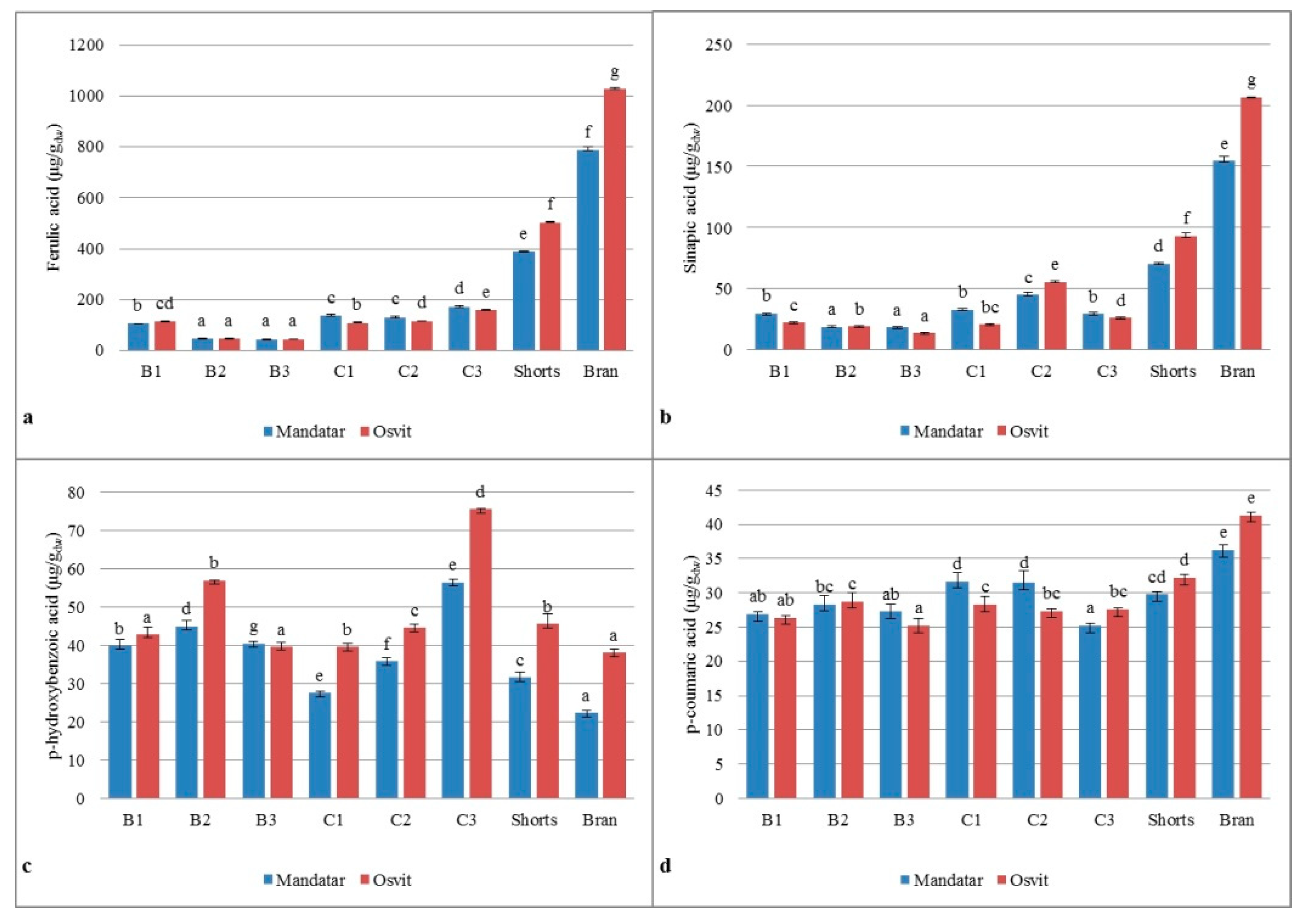
3.6. Phenolic Acids in Milling Fractions
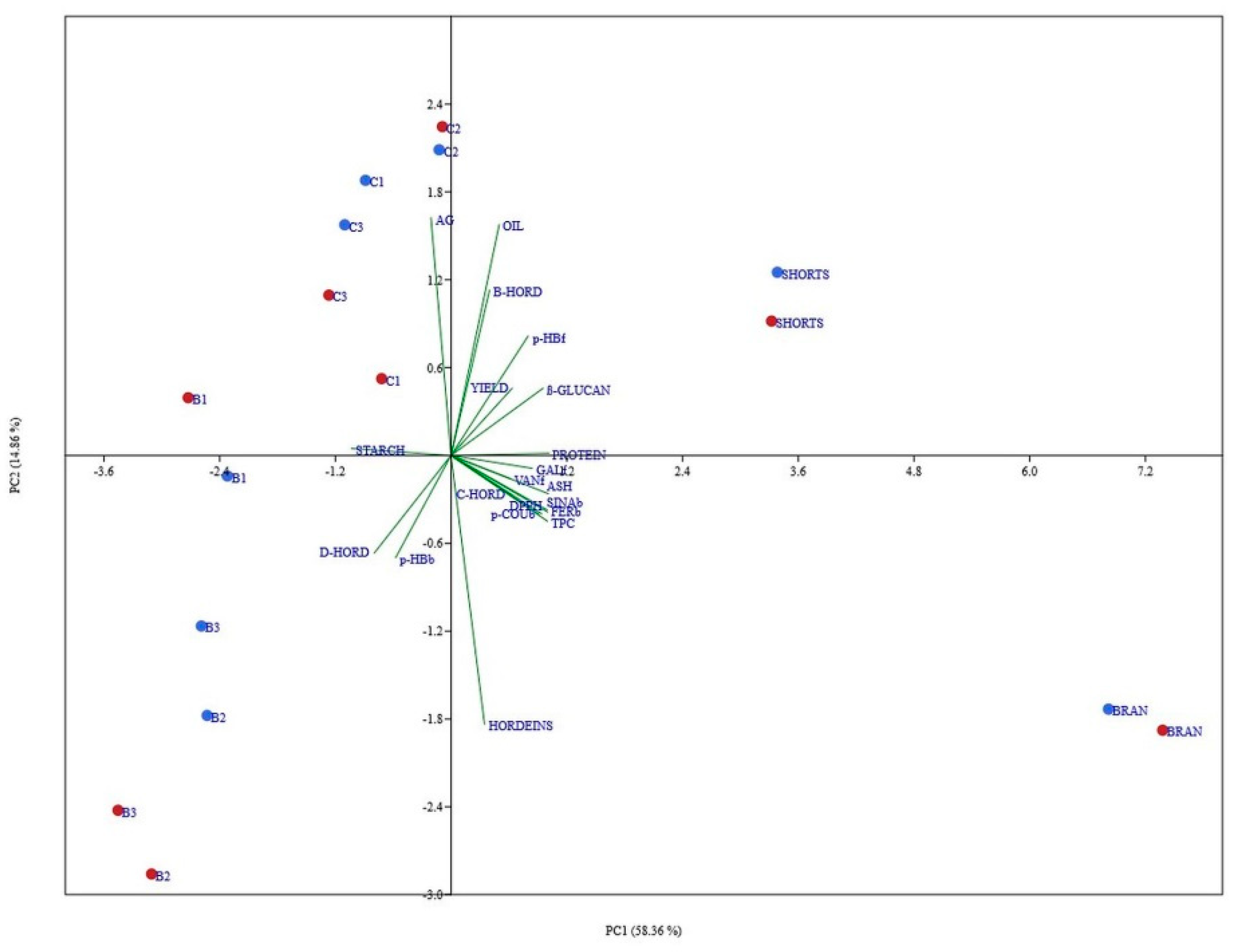
3.7. Principal Component Analysis of All Data
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martínez, M.; Motilva, M.-J.; López de las Hazas, M.-C.; Romero, M.-P.; Vaculova, K.; Ludwig, I.A. Phytochemical composition and β-glucan content of barley genotypes from two different geographic origins for human health food production. Food Chem. 2018, 245, 61–70. [Google Scholar] [CrossRef]
- Baik, B.-K.; Ullrich, S.E. Barley for food: Characteristics, improvement, and renewed interest. J. Cereal Sci. 2008, 48, 233–242. [Google Scholar] [CrossRef]
- Komeili, H.R.; Sheikholeslami, Z. Replacement effect of wheat flour with barley flour and hull-less barley flour on the bread porosity and color. Adv. Agric. Biol. 2014, 2, 39–43. [Google Scholar]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef]
- Holtekjølen, A.K.; Uhlen, A.K.; Bråthen, E.; Sahlstrøm, S.; Knutsen, S.H. Contents of starch and non-starch polysaccharides in barley varieties of different origin. Food Chem. 2006, 94, 348–358. [Google Scholar] [CrossRef]
- Moza, J.; Gujral, H.S. Influence of non-starchy polysaccharides on barley milling behavior and evaluating bioactive composition of milled fractions. Food Chem. 2017, 218, 137–143. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Lagassé, S.L.; Hatcher, D.W.; Dexter, J.E.; Rossnagel, B.G. The enrichment of Asian noodles with fibre-rich fractions derived from roller milling of hull-less barley. J. Sci. Food Agric. 2005, 85, 2094–2104. [Google Scholar] [CrossRef]
- Zhang, G.; Xue, W.; Dai, J.; Xu, Q.; Wang, Y.; Yuan, H.; Yang, K.; Qi, Y.; Zeng, X.; Nyima, T. Quantitative proteomics analysis reveals proteins and pathways associated with anthocyanin accumulation in barley. Food Chem. 2019, 298, 124973. [Google Scholar] [CrossRef]
- Abdel-Aal, E.S.M.; Choo, T.-M. Differences in compositional properties of a hulless barley cultivar grown in 23 environments in eastern Canada. Can. J. Plant Sci. 2014, 94, 807–815. [Google Scholar] [CrossRef]
- Brennan, C.S.; Smith, D.B.; Harris, N.; Shewry, P.R. The production and characterisation of Hor3 null lines of barley provides new information on the relationship of D hordein to malting performance. J. Cereal Sci. 1998, 28, 291–299. [Google Scholar] [CrossRef]
- Šimić, G.; Sudar, R.; Lalić, A.; Jurković, Z.; Horvat, D.; Babić, D. Relationship between hordein proteins and malt quality in barley cultivars grown in Croatia. Cereal Res. Commun. 2007, 35, 1487–1496. [Google Scholar] [CrossRef]
- Yang, J. Investigation of Barley Proteins Interfacial Properties and Their Applications as Nanoscaled Materials. Ph.D. Thesis, University of Alberta, Department of Agricultural, Food and Nutritional Science, Edmonton, AB, Canada, 2016. [Google Scholar]
- Idehen, E.; Tang, Y.; Sang, S. Bioactive phytochemicals in barley. J. Food Drug Anal. 2017, 25, 148–161. [Google Scholar] [CrossRef]
- Zhao, Z.; Moghadasian, M.H. Chemistry, natural sources, dietary intake and pharmacokinetic properties of ferulic acid: A review. Food Chem. 2008, 109, 691–702. [Google Scholar] [CrossRef]
- Madhujith, T.; Shahidi, F. Antioxidative and antiproliferative properties of selected barley (Hordeum vulgarae L.) cultivars and their potential for inhibition of low-density lipoprotein (LDL) cholesterol oxidation. J. Agric. Food Chem. 2007, 55, 5018–5024. [Google Scholar] [CrossRef]
- Reis Giada, M.D.L. Food Phenolic Compounds: Main Classes, Sources and Their Antioxidant Power. In Oxidative Stress and Chronic Degenerative Diseases—A Role for Antioxidants, 1st ed.; Morales-González, J.A., Ed.; IntechOpen: London, UK, 2013; pp. 87–112. [Google Scholar]
- Gamel, T.H.; Abdel-Aal, E.-S. Phenolic acids and antioxidant properties of barley wholegrain and pearling fractions. Agric. Food Sci. 2012, 21, 118–131. [Google Scholar] [CrossRef]
- Holtekjølen, A.K.; Kinitz, C.; Knutsen, S.H. Flavanol and bound phenolic acid contents in different barley varieties. J. Agric. Food Chem. 2006, 54, 2253–2260. [Google Scholar] [CrossRef]
- Abdel-Aal, E.S.M.; Choo, T.-M.; Dhillon, S.; Rabalski, I. Free and bound phenolic acids and total phenolics in black, blue, and yellow barley and their contribution to free radical scavenging capacity. Cereal Chem. 2012, 89, 198–204. [Google Scholar] [CrossRef]
- Andersson, A.A.; Lampi, A.M.; Nyström, L.; Piironen, V.; Li, L.; Ward, J.L.; Gebruers, K.; Courtin, C.M.; Delcour, J.A.; Boros, D.; et al. Phytochemical and dietary fiber components in barley varieties in the Healthgrain diversity screen. J. Agric. Food Chem. 2008, 56, 9767–9776. [Google Scholar] [CrossRef]
- Ward, J.L.; Poutanen, K.; Gebruers, K.; Piironen, V.; Lampi, A.M.; Nyström, L.; Andersson, A.A.M.; Åman, P.; Boros, D.; Rakszegi, M.; et al. The Healthgrain cereal diversity screen: Concept, results, and prospects. J. Agric. Food Chem. 2008, 56, 9699–9709. [Google Scholar] [CrossRef]
- McKevith, B. Nutritional aspects of cereals. Nutr. Bull. 2004, 29, 111–142. [Google Scholar] [CrossRef]
- Bhatty, R.S. Milling yield and flour quality of hull-less barley. Cereal Food World 1987, 32, 268–272. [Google Scholar]
- Klamczynski, A.P.; Czuchajowska, Z. Quality of flours from waxy and nonwaxy barley for production of baked products. Cereal Chem. 1999, 76, 530–535. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Dexter, J.E.; Desjardins, R.G.; Rossnagel, B.G.; Lagasse, S.L.; Hatcher, D.W. Roller milling of Canadian hull-less barley: Optimization of roller milling conditions and composition of mill streams. Cereal Chem. 2003, 80, 637–644. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; McMillan, T.L.; Kletke, J.B.; Dexter, J.E. Effects of pearling, grinding conditions, and roller mill flow on the yield and composition of milled products from hull-less barley. Cereal Chem. 2011, 88, 375–384. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; McMillan, T.; Bazin, S.; Kletke, J.; Dushnicky, L.; Dexter, J.; Chepurna, A.; Rossnagel, B. Milling of Canadian oats and barley for functional food ingredients: Oat bran and barley fibre-rich fractions. Can. J. Plant Sci. 2014, 94, 573–586. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Symons, S.J.; Dexter, J.E. Fractionation of wheat and barley. In Whole Grain Foods in Health and Disease; Marquart, L., Slavin, J.L., Fulcher, R.G., Eds.; AACC International: St. Paul, MN, USA, 2004; pp. 47–82. [Google Scholar]
- Andersson, A.M.; Courtin, C.M.; Delcour, J.A.; Fredrikson, H.; Schofield, J.D.; Trogh, I.; Tsiami, A.A.; Åman, P. Milling performance of North European hull-less barleys and characterization of resultant millstreams. Cereal Chem. 2003, 80, 667–673. [Google Scholar] [CrossRef]
- Wiege, I.; Sluková, M.; Vaculová, K.; Panciková, B.; Wiege, B. Characterization of milling fractions from new sources of barley for use in food industry. Starch-Stärke 2016, 68, 321–328. [Google Scholar] [CrossRef]
- HRN ISO 5984. Animal Feeding Stuffs—Determination of Crude Ash; International Organization for Standardization: Geneva, Switzerland, 2004. [Google Scholar]
- HRN EN ISO 20483. Cereals and Pulses—Determination of the Nitrogen Content and Calculation of the Crude Protein Content—Kjeldahl Method; International Organization for Standardization: Geneva, Switzerland, 2014. [Google Scholar]
- HRN EN ISO 10520. Native Starch—Determination of Starch Content—Ewers Polarimetric Method from SAI Global; International Organization for Standardization: Geneva, Switzerland, 2002. [Google Scholar]
- Mc Cleary, B.V.; Glennie-Holmes, M. Enzymatic quantification of (1→3)(1→4)-β-D-glucan in barley and malt. J. Inst. Brew. 1985, 91, 285–295. [Google Scholar] [CrossRef]
- Singleton, V.L.; Rossi, J.A. Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagent. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT-Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Celus, I.; Brijs, K.; Delcour, J.A. The effects of malting and mashing on barley protein extractability. J. Cereal Sci. 2006, 44, 203–211. [Google Scholar] [CrossRef]
- Zavala‑Lopez, M.; Silverio Garcia, L. An improved microscale method for extraction of phenolic acids from maize. Plant Methods 2017, 13, 81. [Google Scholar] [CrossRef]
- Mattila, P.; Pihlava, J.M.; Hellström, J. Contents of phenolic acids, alkyl- and alkenylresorcinols, and avenanthramides in commercial grain products. J. Agric. Food Chem. 2005, 53, 8290–8295. [Google Scholar] [CrossRef]
- Izydorczyk, M.S.; Dexter, J.E. Barley/Milling and Processing. In Encyclopedia of Grain Science; Wrigley, C., Ed.; Elsevier Academic Press: London, UK, 2004; pp. 57–68. [Google Scholar]
- Lahouar, L.; Ghrairi, F.; Arem, A.E.; Medimagh, S.; Felah, M.E.; Salem, H.B.; Achour, L. Biochemical composition and nutritional evaluation of barley rihane (Hordeum vulgare L.). Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 310–317. [Google Scholar] [CrossRef]
- Siebenhandl-Ehn, S.; Kinner, M.; Leopold, L.F.; Poppernitsch, M.B.; Prückler, M.; Wurbs, P.; Poisinger, S.; Kalas, E.; Berghofer, E.; Grausgruber, H. Hulless barley—A rediscovered source for functional foods phytochemical profile and soluble dietary fibre content in naked barley varieties and their antioxidant properties. In Phytochemicals—Bioactivities and Impact on Health; Rasooli, I., Ed.; InTech Europe: Rijeka, Croatia, 2011; pp. 269–294. [Google Scholar]
- Newman, R.K.; Newman, C.W. Barley for Food and Human Nutrition: Science, Technology, and Products, 1st ed.; Willey: Hoboken, NJ, USA, 2008. [Google Scholar]
- Shewry, P.R.; Halford, N.G. Cereal seed storage proteins: Structures, properties and role in grain utilization. J. Exp. Bot. 2002, 53, 947–958. [Google Scholar] [CrossRef]
- Edney, M.J.; Tkachuk, R.; Macgregor, A.W. Nutrient composition of the hull-less barley cultivar, Condor. J. Sci. Food Agric. 1992, 60, 451–456. [Google Scholar] [CrossRef]
- Sikdar, M.S.; Bowra, S.; Schmidt, D.; Dionisio, G.; Holm, P.B.; Vincze, E. Targeted modification of storage protein content resulting in improved amino acid composition of barley grain. Transgenic Res. 2016, 25, 19–31. [Google Scholar] [CrossRef]
- Šimić, G.; Lalić, A.; Kovačević, J.; Horvat, D. Effect of genotype and environment on spring barley hordeins. Cereal Res. Commun. 2008, 36 (Suppl. 1), 1491–1494. [Google Scholar]
- Wood, P.J. Oat and rye β-glucan: Properties and function. Cereal Chem. 2010, 87, 315–330. [Google Scholar] [CrossRef]
- Slavin, J.L.; Marquart, L.; Jacobs, D., Jr. Consumption of whole-grain foods and decreased risk of cancer: Proposed mechanisms. Cereal Foods World 2000, 45, 54–58. [Google Scholar]
- Zheng, X.L.; Li, L.M.; Wang, Q. Distribution and molecular characterization of beta-glucans from hull-less barley bran, shorts and flour. Int. J. Mol. Sci. 2011, 12, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Arigò, A.; Česla, P.; Šilarová, P.; Calabrò, M.L.; Česlová, L. Development of extraction method for characterization of free and bonded polyphenols in barley (Hordeum vulgare L.) grown in Czech Republic using liquid chromatography-tandem mass spectrometry. Food Chem. 2018, 245, 829–837. [Google Scholar] [CrossRef]
- Yang, X.-J.; Bin Dang, B.; Fan, M.-T. Free and Bound Phenolic Compound Content and Antioxidant Activity of Different Cultivated Blue Highland Barley Varieties from the Qinghai-Tibet Plateau. Molecules 2018, 23, 879. [Google Scholar] [CrossRef] [PubMed]
- López-Pereaa, P.; Guzmán-Ortiz, F.A.; Román-Gutiérrez, A.D.; Castro-Rosasc, J.; Gómez-Aldapa, C.A.; Rodríguez-Marínb, M.L.; Falfán-Cortésb, R.N.; González-Olivaresc, L.G.; Torruco-Uco, J.G. Bioactive compounds and antioxidant activity of wheat Bran and barley husk in the extracts with different polarity. Int. J. Food Prop. 2019, 22, 646–658. [Google Scholar] [CrossRef]
- Ndolo, V.U.; Beta, T.; Fulcher, R.G. Ferulic acid fluorescence intensity profiles and concentration measured by HPLC in pigmented and non-pigmented cereals. Food Res. Int. 2013, 52, 109–118. [Google Scholar] [CrossRef]
- Vaher, M.; Matso, K.; Levandi, T.; Helmja, K.; Kaljurand, M. Phenolic Compounds and the Antioxidant Activity of the Bran, Flour and Whole Grain of Different Wheat Varieties. Procedia Chem. 2010, 2, 76–82. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | Ash (%) | Starch (%) | Oil (%) | |||
|---|---|---|---|---|---|---|
| Mandatar | Osvit | Mandatar | Osvit | Mandatar | Osvit | |
| B1 * | 1.06 b,** ± 0.03 | 1.33 c,** ± 0.01 | 77.75 d ± 0.86 | 78.23 e ± 0.27 | 1.80 b ± 0.06 | 1.41 a ± 0.07 |
| B2 | 0.85 a ± 0.02 | 0.92 a ± 0.02 | 80.78 e ±0,67 | 79.38 e ± 1.08 | 1.50 a ±0.09 | 2.96 cd ± 0.12 |
| B3 | 0.82 a ± 0.02 | 0.84 a ± 0.04 | 82.31 e ± 0.25 | 81.90 f ± 0.92 | 1.83 b ± 0.12 | 1.56 ab ± 0.14 |
| C1 | 1.18 c ± 0.03 | 1.31 c ± 0.05 | 75.66 cd ± 1.06 | 74.84 cd ± 0.44 | 2.45 c ± 0.15 | 2.31 bc ± 0.10 |
| C2 | 1.29 d ± 0.01 | 1.27 c ± 0.02 | 74.84 c ± 0.79 | 73.48 c ± 0.44 | 2.90 d ± 0.20 | 3.26 d ± 0.18 |
| C3 | 1.05 b ± 0.03 | 1.13 b ± 0.04 | 75.19 cd ± 0.37 | 75.94 d ± 0.56 | 2.70 d ± 0.15 | 2.63 cd ± 0.19 |
| Shorts | 1.71 e ± 0.04 | 2.09 d ± 0.05 | 55.74 b ± 0.55 | 59.10 b ± 0.28 | 2.75 d ± 0.12 | 2.41 c ± 0.23 |
| Bran | 2.88 f ± 0.06 | 3.10 e ± 0.06 | 48.69 a ± 0.38 | 49.10 a ± 0.32 | 2.27 c ± 0.07 | 2.39 c ± 0.13 |
| Fraction | AG (g/100 gdw) | HORD (g/100 gdw) | ||
|---|---|---|---|---|
| Mandatar | Osvit | Mandatar | Osvit | |
| B1 * | 0.92 ab,** ± 0.07 | 1.27 f ± 0.09 | 7.39 c ± 0.18 | 8.07 c ± 0.16 |
| B2 | 0.95 ab ± 0.09 | 0.79 a ± 0.15 | 9.68 f ± 0.16 | 9.42 e ± 0.14 |
| B3 | 0.96 b ± 0.23 | 0.87 b ± 0.16 | 9.09 e ± 0.09 | 9.29 e ± 0.12 |
| C1 | 1.41 e ± 0.08 | 1.01 d ± 0.06 | 7.14 bc ± 0.08 | 7.23 b ± 0.18 |
| C2 | 1.22 d ± 0.19 | 1.08 e ± 0.18 | 6.96 ab ± 0.03 | 6.78 a ± 0.05 |
| C3 | 1.10 c ± 0.14 | 1.05 de ± 0.11 | 6.69 a ± 0.06 | 6.86 a ± 0.03 |
| Shorts | 1.14 c ± 0.26 | 0.95 c ± 0.19 | 8.19 d ± 0.20 | 8.39 d ± 0.16 |
| Bran | 0.90 a ± 0.20 | 0.78 a ± 0.12 | 9.76 f ± 0.19 | 10.49 f ± 0.22 |
| Fraction | D-HORD (%) * | C-HORD (%) | B-HORD (%) | |||
|---|---|---|---|---|---|---|
| Mandatar | Osvit | Mandatar | Osvit | Mandatar | Osvit | |
| B1 ** | 12.72 c,*** ± 0.02 | 12.26 c ± 0.04 | 28.43 a ± 0.14 | 28.33 a ± 0.10 | 58.86 a ± 0.12 | 59.42 abc ± 0.20 |
| B2 | 12.59 bc ± 0.11 | 12.65 c ± 0.09 | 29.05 ab ± 0.25 | 29.06 ab ± 0.24 | 58.37 a ± 0.26 | 58.29 a ± 0.14 |
| B3 | 12.28 b ± 0.12 | 12.31 c ± 0.19 | 29.36 ab ± 0.33 | 29.16 ab ± 0.19 | 58.36 a ± 0.15 | 58.53 ab ± 0.34 |
| C1 | 12.48 bc ± 0.06 | 11.46 b ± 0.08 | 28.96 ab ± 0.15 | 29.57 b ± 0.41 | 58.57 a ± 0.26 | 58.97 abc ± 0.21 |
| C2 | 11.48 a ± 0.07 | 11.37 b ± 0.03 | 29.31 ab ± 0.28 | 29.13 ab ± 0.22 | 59.20 a ± 0.32 | 59.50 abc ± 0.09 |
| C3 | 11.34 a ± 0.01 | 11.44 b ± 0.04 | 29.60 ab ± 0.45 | 29.59 b ± 0.35 | 59.06 a ± 0.41 | 58.97 abc ± 0.19 |
| Shorts | 11.34 a ± 0.10 | 11.49 b ± 0.06 | 29.62 ab ± 0.19 | 28.73 ab ± 0.10 | 59.03 a ± 0.19 | 60.07 c ± 0.45 |
| Bran | 11.32 a ± 0.04 | 10.60 a ± 0.01 | 30.24 b ± 0.39 | 29.71 b ± 0.26 | 58.44 a ± 0.18 | 59.70 bc ± 0.49 |
| Fraction | Total Phenolic Content (mg GAE/gdw) | DPPH Activity (%) | ||
|---|---|---|---|---|
| Mandatar | Osvit | Mandatar | Osvit | |
| B1 * | 1.04 b,** ± 0.01 | 1.05 bc ± 0.01 | 31.52 bc ± 0.06 | 31.27 b ± 0.22 |
| B2 | 1.15 c ± 0.03 | 0.93 a ± 0.01 | 30.77 b ± 0.09 | 37.34 c ± 0.31 |
| B3 | 0.91 a ± 0.01 | 1.02 b ± 0.02 | 34.70 d ± 0.18 | 36.19 c ± 0.38 |
| C1 | 1.02 b ± 0.02 | 1.04 b ± 0.01 | 32.71 c ± 0.21 | 28.81 a ± 0.25 |
| C2 | 1.18 d ± 0.05 | 1.05 bc ± 0.02 | 35.64 d ± 0.34 | 32.58 b ± 0.19 |
| C3 | 1.02 b ± 0.01 | 1.09 c ± 0.03 | 29.37 a ± 0. 28 | 32.09 b ± 0.24 |
| Shorts | 1.59 e ± 0.04 | 1.57 d ± 0.04 | 70.43 e ± 0.75 | 65.38 d ± 0.69 |
| Bran | 2.21 f ± 0.03 | 2.17 e ± 0.05 | 72.06 f ± 0.62 | 67.91 e ± 0.55 |
| Fraction | P-Hydroxybenzoic Acid | Gallic Acid | Vanilic Acid | |||
|---|---|---|---|---|---|---|
| Mandatar | Osvit | Mandatar | Osvit | Mandatar | Osvit | |
| B1 * | 10.24 b,** ± 0.21 | 4.23 b ± 0.23 | 4.62 a ± 0.15 | nd *** | 7.38 c ± 0.15 | 3.2 a ± 0.09 |
| B2 | 8.68 b ± 0.18 | 3.43 b ± 0.16 | nd | 3.5 a ± 0.12 | 3.57 a ± 0.02 | nd |
| B3 | 8.78 b ± 0.12 | nd | nd | 6.1 c ± 0.09 | 5.29 b ± 0.18 | nd |
| C1 | 13.79 c ± 0.18 | 4.39 b ± 0.17 | 5.72 b ± 0.16 | 4.0 ab ± 0.11 | 3.30 a ± 0.15 | 22.5 c ± 0.32 |
| C2 | 13.83 c ± 0.24 | 12.55 d ± 0.41 | 4.50 a ± 0.19 | 27.3 d ± 0.25 | 3.39 a ± 0.20 | nd |
| C3 | 13.45 c ± 0.09 | 7.89 c ± 0.15 | 5.35 ab ± 0.12 | 4.0 ab ± 0.17 | 7.69 c ± 0.08 | 3.5 a ± 0.14 |
| Shorts | 15.71 d ± 0.19 | 16.09 f ± 0.33 | 18.47 c ± 0.28 | 4.5 b ± 0.16 | 7.12 c ± 0.11 | 10.3 b ± 0.13 |
| Bran | 17.44 e ± 0.26 | 15.35 e ± 0.21 | 28.07 d ± 0.35 | 32.9 e ± 0.32 | 24.7 d ± 0.41 | 10.2 b ± 0.06 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimić, G.; Horvat, D.; Lalić, A.; Koceva Komlenić, D.; Abičić, I.; Zdunić, Z. Distribution of β-Glucan, Phenolic Acids, and Proteins as Functional Phytonutrients of Hull-Less Barley Grain. Foods 2019, 8, 680. https://doi.org/10.3390/foods8120680
Šimić G, Horvat D, Lalić A, Koceva Komlenić D, Abičić I, Zdunić Z. Distribution of β-Glucan, Phenolic Acids, and Proteins as Functional Phytonutrients of Hull-Less Barley Grain. Foods. 2019; 8(12):680. https://doi.org/10.3390/foods8120680
Chicago/Turabian StyleŠimić, Gordana, Daniela Horvat, Alojzije Lalić, Daliborka Koceva Komlenić, Ivan Abičić, and Zvonimir Zdunić. 2019. "Distribution of β-Glucan, Phenolic Acids, and Proteins as Functional Phytonutrients of Hull-Less Barley Grain" Foods 8, no. 12: 680. https://doi.org/10.3390/foods8120680
APA StyleŠimić, G., Horvat, D., Lalić, A., Koceva Komlenić, D., Abičić, I., & Zdunić, Z. (2019). Distribution of β-Glucan, Phenolic Acids, and Proteins as Functional Phytonutrients of Hull-Less Barley Grain. Foods, 8(12), 680. https://doi.org/10.3390/foods8120680