Phenolic Profile and Antioxidant Activity of the Edible Tree Peony Flower and Underlying Mechanisms of Preventive Effect on H2O2-Induced Oxidative Damage in Caco-2 Cells




Abstract
1. Introduction
2. Materials and Methods
2.1. Tree Peony Flower Samples
2.2. Chemicals
2.3. Sample Preparation
2.4. Determination of Total Phenolic Content and Total Flavonoid Content
2.5. Determination of Antioxidant Capacity In Vitro
2.6. Identification and Quantification of Phenolic Compounds by HPLC- Quadrupole Time-of-Flight Mass Spectrometer (Q-TOF-MS2)
2.7. Cell Culture
2.8. Cell Viability Assay
2.9. Transepithelial Electrical Resistant (TEER) Measurement
2.10. Reactive Oxygen Species (ROS) Assay
2.11. Glutathione (GSH) Content Determination
2.12. RNA Extraction and Real-Time PCR
2.13. Immunofluorescent Staining
2.14. Statistical Analysis
3. Results
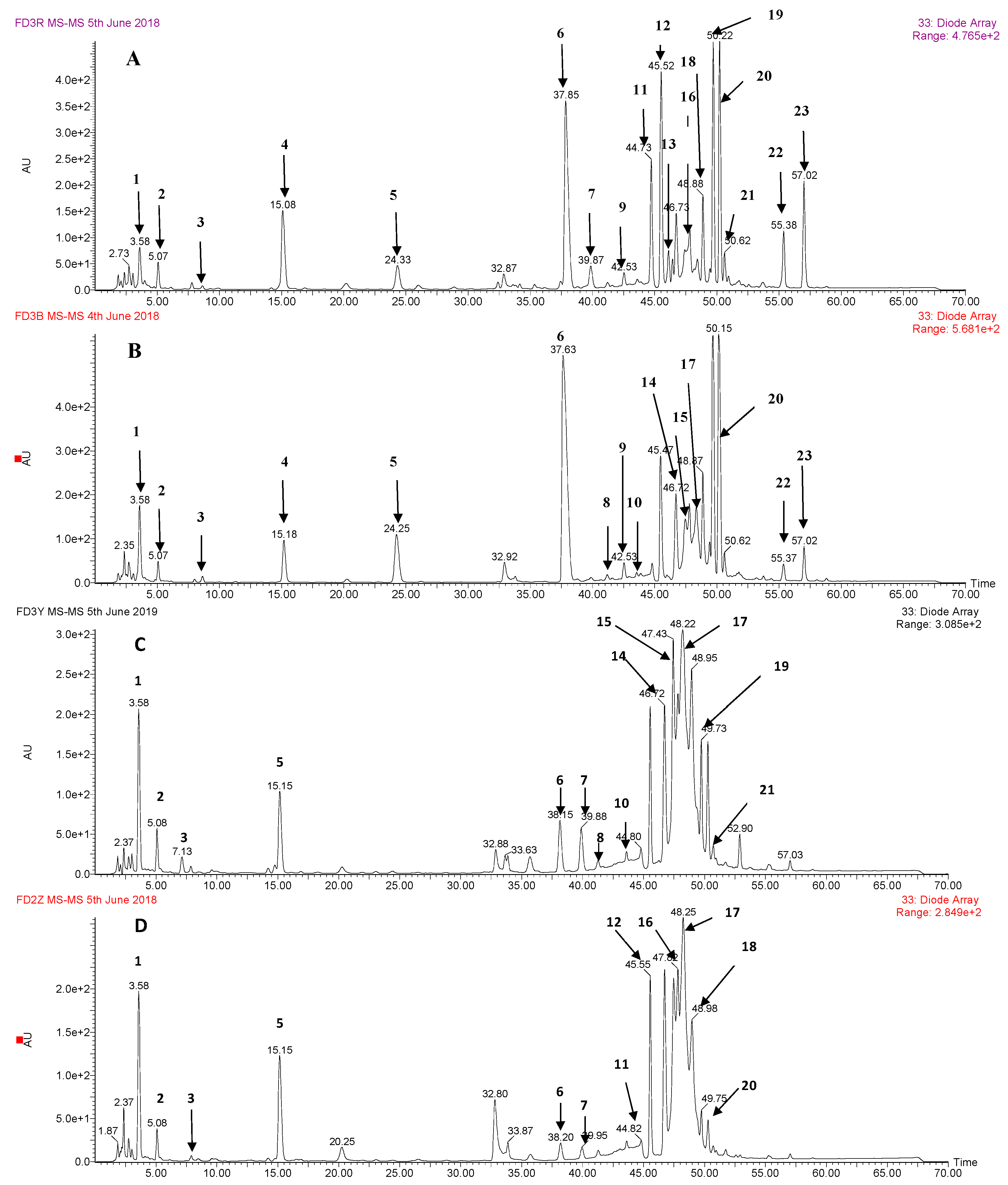
3.1. Identification of Phenolic Compounds by HPLC-DAD-ESI-MS2
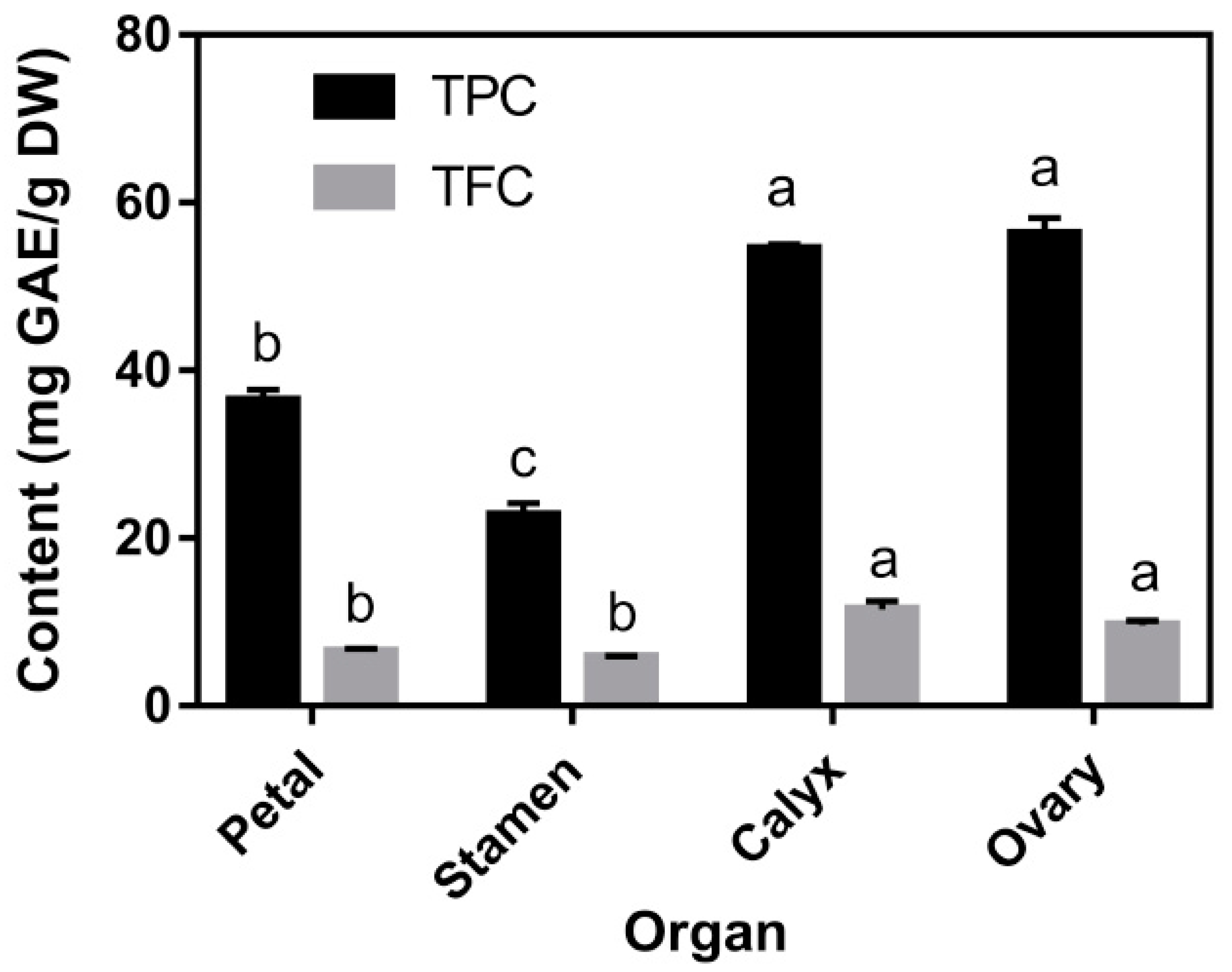
3.2. TPC, TFC, and Individual Phenolic Compounds Content
3.3. Antioxidant Properties In Vitro
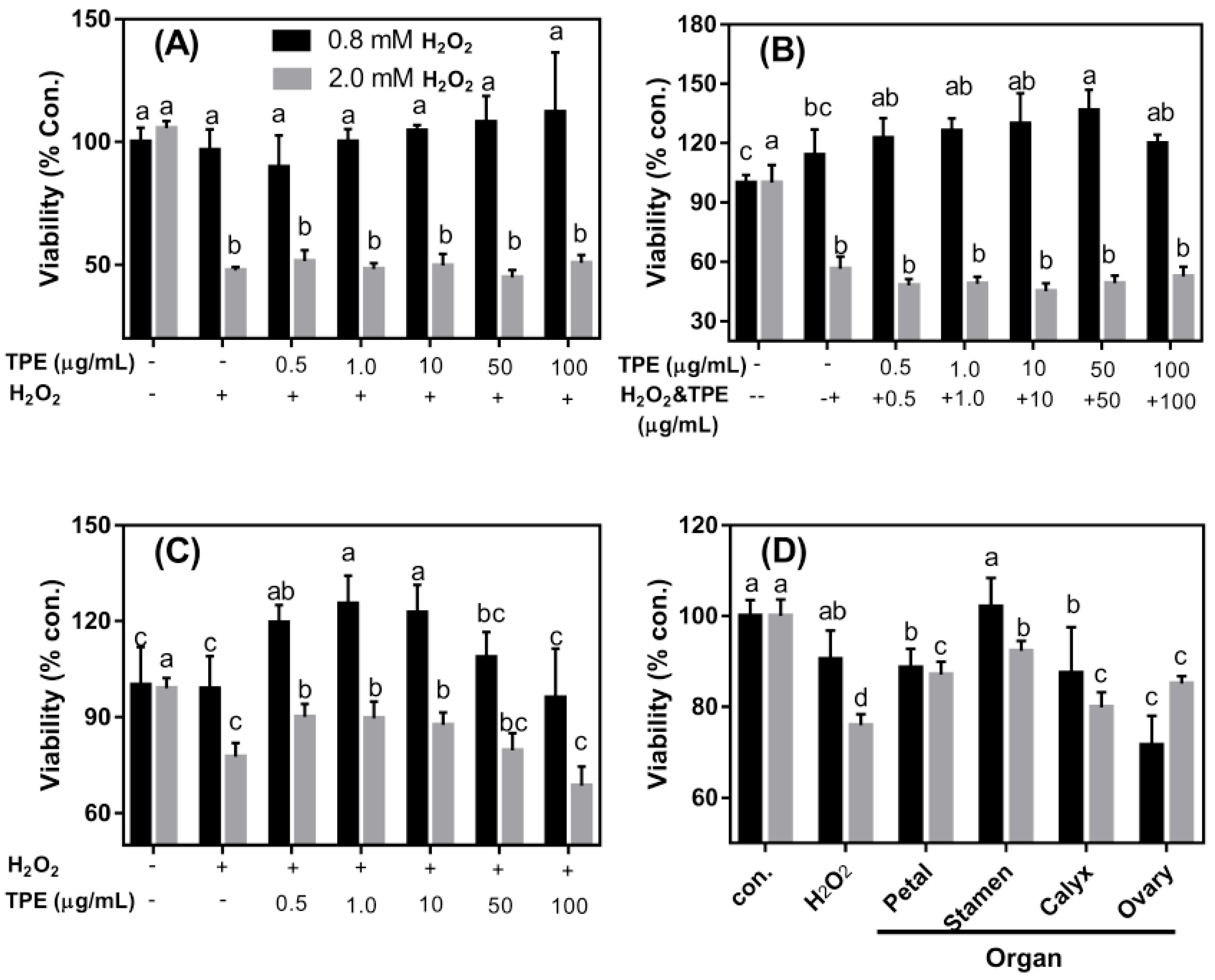
3.4. The Protective Effects of TPE against Oxidative Damage Induced by H2O2 on Caco-2 Cells
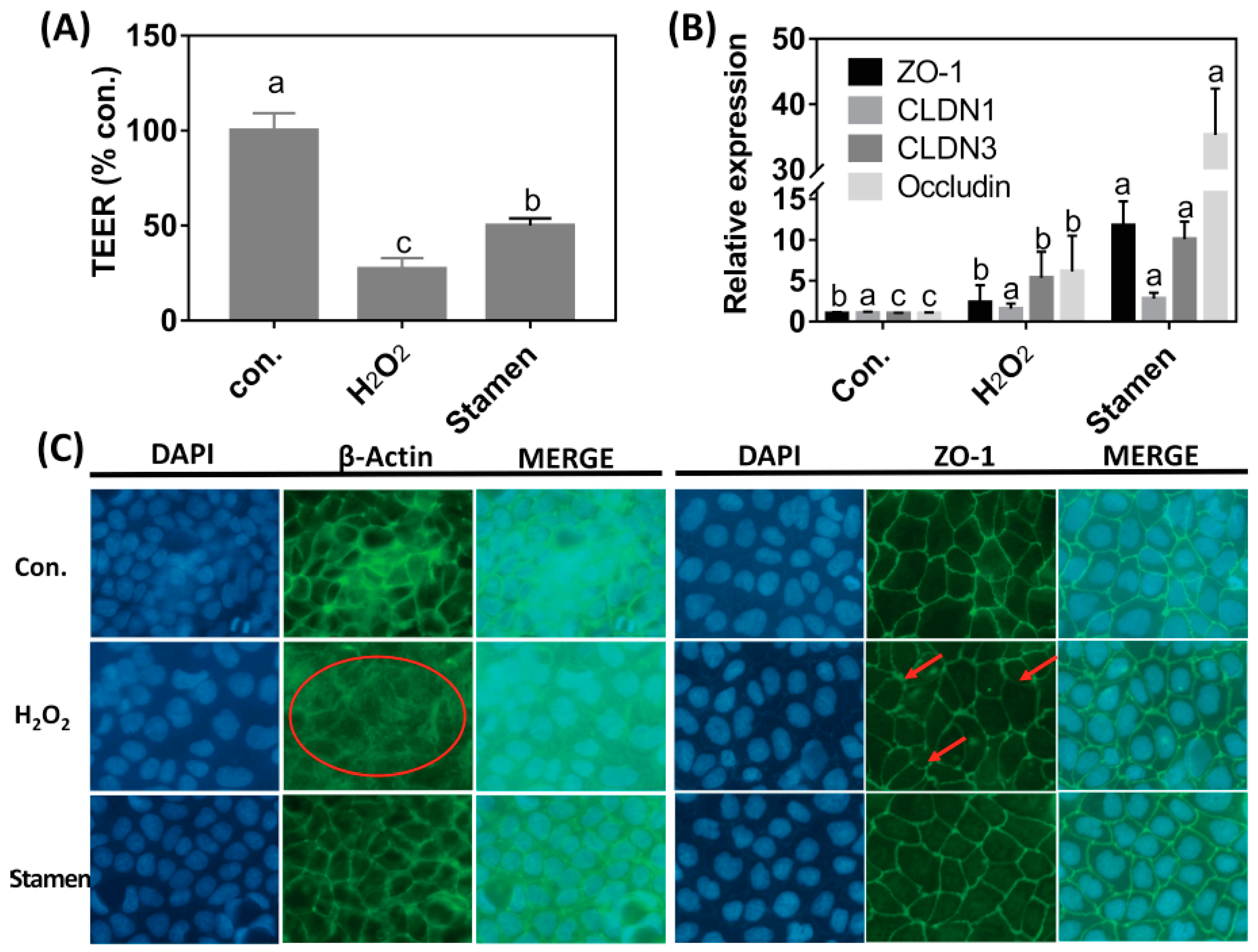
3.5. Effect of TPE from Stamen on Redox State of Caco-2 Cell under H2O2 Treatment
3.6. Effect of TPE from Stamen on Barrier Function of Caco-2 Cell under H2O2 Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.J.; Gan, R.Y.; Li, S.; Zhou, Y.; Li, A.N.; Xu, D.P.; Li, H.B. Antioxidant Phytochemicals for the Prevention and Treatment of Chronic Diseases. Molecules 2015, 20, 21138–21156. [Google Scholar] [CrossRef] [PubMed]
- González-Barrio, R.; Periago, M.J.; Luna-Recio, C.; Garcia-Alonso, F.J.; Navarro-González, I. Chemical composition of the edible flowers, pansy (Viola wittrockiana) and snapdragon (Antirrhinum majus) as new sources of bioactive compounds. Food Chem. 2018, 252, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Pires, T.C.S.P.; Dias, M.I.; Barros, L.; Calhelha, R.C.; Alves, M.J.; Oliveira, M.B.P.P.; Santos-Buelga, C.; Ferreira, I.C.F.R. Edible flowers as sources of phenolic compounds with bioactive potential. Food Res. Int. 2018, 105, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Kaisoon, O.; Siriamornpun, S.; Weerapreeyakul, N.; Meeso, N. Phenolic compounds and antioxidant activities of edible flowers from Thailand. J. Funct. Foods 2011, 3, 88–99. [Google Scholar] [CrossRef]
- Zhao, D.; Tang, W.; Hao, Z.; Tao, J. Identification of flavonoids and expression of flavonoid biosynthetic genes in two coloured tree peony flowers. Biochem. Biophys. Res. Commun. 2015, 459, 450–456. [Google Scholar] [CrossRef]
- Fan, J.; Zhu, W.; Kang, H.; Ma, H.; Tao, G. Flavonoid constituents and antioxidant capacity in flowers of different Zhongyuan tree penoy cultivars. J. Funct. Foods 2012, 4, 147–157. [Google Scholar] [CrossRef]
- Wang, L.-S.; Shiraishi, A.; Hashimoto, F.; Aoki, N.; Shimizu, K.; Sakata, Y. Analysis of Petal Anthocyanins to Investigate Flower Coloration of Zhongyuan (Chinese) and Daikon Island (Japanese) Tree Peony Cultivars. J. Plant Res. 2001, 114, 33–43. [Google Scholar] [CrossRef]
- Li, C.; Du, H.; Wang, L.; Shu, Q.; Zheng, Y.; Xu, Y.; Zhang, J.; Zhang, J.; Yang, R.; Ge, Y. Flavonoid Composition and Antioxidant Activity of Tree Peony (Paeonia Section Moutan) Yellow Flowers. J. Agric. Food Chem. 2009, 57, 8496–8503. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, C.; Sun, Q.; Li, F.; Liu, J.; Zheng, C. Isolation and purification of four flavonoid constituents from the flowers of Paeonia suffruticosa by high-speed counter-current chromatography. J. Chromatogr. A 2005, 1075, 127–131. [Google Scholar] [CrossRef]
- Ma, L.; Jiao, K.; Luo, L.; Xiang, J.; Fan, J.; Zhang, X.; Yi, J.; Zhu, W. Characterization and macrophage immunomodulatory activity of two polysaccharides from the flowers of Paeonia suffruticosa Andr. Int. J. Biol. Macromol. 2019, 124, 955–962. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Shi, Q.-Q.; Ji, D.; Niu, L.-X.; Zhang, Y.-L. Determination of the phenolic content, profile, and antioxidant activity of seeds from nine tree peony (Paeonia section Moutan DC.) species native to China. Food Res. Int. 2017, 97, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, X.; Wu, K.; Wang, M.; Liu, P.; Wang, X.; Deng, R. Antioxidant Activities and Chemical Constituents of Flavonoids from the Flower of Paeonia ostii. Molecules 2016, 22, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, P.; Gao, J.; Wang, X.; An, J.; Xu, S.; Deng, R.X. Profiling and simultaneous quantitative determination of oligostilbenes in Paeonia ostii seed shell from different geographical areas in China and their comparative evaluation. Phytochem. Anal. 2019, 30, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, Y.; Gao, J.-Y.; Du, M.-Z.; Zhang, K.; Zhang, J.-L.; Xue, N.-C.; Yan, M.; Qu, C.-X.; Deng, R.-X. HPLC-DAD analysis of 15 monoterpene glycosides in oil peony seed cakes sourced from different cultivation areas in China. Ind. Crops Prod. 2018, 118, 259–270. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, Y.; Xu, Y.-F.; Zhu, X.-Y.; Xu, X.-F.; Chang, S.; Deng, R.-X. Three new monoterpene glycosides from oil peony seed cake. Ind. Crops Prod. 2018, 111, 371–378. [Google Scholar] [CrossRef]
- Liu, P.; Xu, Y.-F.; Gao, X.-D.; Zhu, X.-Y.; Du, M.-Z.; Wang, Y.-X.; Deng, R.-X.; Gao, J.-Y. Optimization of ultrasonic-assisted extraction of oil from the seed kernels and isolation of monoterpene glycosides from the oil residue of Paeonia lactiflora Pall. Ind. Crops Prod. 2017, 107, 260–270. [Google Scholar] [CrossRef]
- Zhang, X.X.; Zhang, G.; Jin, M.; Niu, L.X.; Zhang, Y.L. Variation in Phenolic Content, Profile, and Antioxidant Activity of Seeds among Different Paeonia ostii Cultivated Populations in China. Chem. Biodivers. 2018, 15, e1800093. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Gao, J.; Lu, Z.; Yin, W.; Deng, R. Resveratrol Trimers from Seed Cake of Paeonia rockii. Molecules 2014, 19, 19549–19556. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, L.-N.; Wang, X.-S.; Gao, J.-Y.; Yi, J.-P.; Deng, R.-X. Characterization of Paeonia ostii seed and oil sourced from different cultivation areas in China. Ind. Crops Prod. 2019, 133, 63–71. [Google Scholar] [CrossRef]
- Yang, C.; Fischer, M.; Kirby, C.; Liu, R.; Zhu, H.; Zhang, H.; Chen, Y.; Sun, Y.; Zhang, L.; Tsao, R. Bioaccessibility, cellular uptake and transport of luteins and assessment of their antioxidant activities. Food Chem. 2018, 249, 66–76. [Google Scholar] [CrossRef]
- Bao, Y.; Qu, Y.; Li, J.; Li, Y.; Ren, X.; Maffucci, K.G.; Li, R.; Wang, Z.; Zeng, R. In Vitro and In Vivo Antioxidant Activities of the Flowers and Leaves from Paeonia rockii and Identification of Their Antioxidant Constituents by UHPLC-ESI-HRMS(n) via Pre-Column DPPH Reaction. Molecules 2018, 23, 392. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Apea-Bah, F.B.; Ndolo, V.U.; Katundu, M.C.; Beta, T. Profile of phenolic compounds and antioxidant activity of finger millet varieties. Food Chem. 2019, 275, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Biney, K.; Beta, T. Phenolic profile and carbohydrate digestibility of durum spaghetti enriched with buckwheat flour and bran. LWT Food Sci. Technol. 2014, 57, 569–579. [Google Scholar] [CrossRef]
- Xiang, J.; Zhang, M.; Apea-Bah, F.B.; Beta, T. Hydroxycinnamic acid amide (HCAA) derivatives, flavonoid C-glycosides, phenolic acids and antioxidant properties of foxtail millet. Food Chem. 2019, 295, 214–223. [Google Scholar] [CrossRef]
- Qiu, Y.; Liu, Q.; Beta, T. Antioxidant properties of commercial wild rice and analysis of soluble and insoluble phenolic acids. Food Chem. 2010, 121, 140–147. [Google Scholar] [CrossRef]
- Xiang, J.; Li, W.; Ndolo, V.U.; Beta, T. A comparative study of the phenolic compounds and in vitro antioxidant capacity of finger millets from different growing regions in Malawi. J. Cereal Sci. 2019, 87, 143–149. [Google Scholar] [CrossRef]
- Yang, R.; Hui, Q.; Jiang, Q.; Liu, S.; Zhang, H.; Wu, J.; Lin, F.; O, K.; Yang, C. Effect of Manitoba-Grown Red-Osier Dogwood Extracts on Recovering Caco-2 Cells from H2O2-Induced Oxidative Damage. Antioxidants 2019, 8, 250. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Prior, R.L. The Chemistry behind Antioxidant Capacity Assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef]
- Serra, G.; Incani, A.; Serreli, G.; Porru, L.; Melis, M.P.; Tuberoso, C.I.G.; Rossin, D.; Biasi, F.; Deiana, M. Olive oil polyphenols reduce oxysterols -induced redox imbalance and pro-inflammatory response in intestinal cells. Redox Biol. 2018, 17, 348–354. [Google Scholar] [CrossRef]
- Wu, H.; Luo, T.; Li, Y.M.; Gao, Z.P.; Zhang, K.Q.; Song, J.Y.; Xiao, J.S.; Cao, Y.P. Granny Smith apple procyanidin extract upregulates tight junction protein expression and modulates oxidative stress and inflammation in lipopolysaccharide-induced Caco-2 cells. Food Funct. 2018, 9, 3321–3329. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Germoush, M.O.; Al-Anazi, K.M.; Mahmoud, A.H.; Farah, M.A.; Allam, A.A. Commiphora molmol protects against methotrexate-induced nephrotoxicity by up-regulating Nrf2/ARE/HO-1 signaling. Biomed. Pharmacother. 2018, 106, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Suzuki, T.; Taylor, W.L.; Bhargava, A.; Rao, R.K. Contrasting effects of ERK on tight junction integrity in differentiated and under-differentiated Caco-2 cell monolayers. Biochem. J. 2011, 433, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Gong, E.S.; Gao, N.; Li, T.; Chen, H.; Wang, Y.; Si, X.; Tian, J.; Shu, C.; Luo, S.; Zhang, J.; et al. Effect of In Vitro Digestion on Phytochemical Profiles and Cellular Antioxidant Activity of Whole Grains. J. Agric. Food Chem. 2019, 67, 7016–7024. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, H.; Cheng, L.; Wang, L.; Qian, H.; Qi, X. In vitro and in vivo antioxidant activity of polyphenols extracted from black highland barley. Food Chem. 2016, 194, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, J.; Zhang, X.; Wang, L.; Zhang, W. Pomegranate peel polyphenols inhibits inflammation in LPS-induced RAW264.7 macrophages via the suppression of MAPKs activation. J. Funct. Foods 2018, 43, 62–69. [Google Scholar] [CrossRef]
- Omonijo, F.A.; Liu, S.; Hui, Q.; Zhang, H.; Lahaye, L.; Bodin, J.-C.; Gong, J.; Nyachoti, M.; Yang, C. Thymol improves barrier function and attenuates inflammatory responses in porcine intestinal epithelial cells during lipopolysaccharide (LPS)-induced inflammation. J. Agric. Food Chem. 2019, 67, 615–624. [Google Scholar] [CrossRef]
- Wijeratne, S.S.K.; Cuppett, S.L.; Schlegel, V. Hydrogen peroxide induced oxidative stress damage and antioxidant enzyme response in Caco-2 human colon cells. J. Agric. Food Chem. 2005, 53, 8768–8774. [Google Scholar] [CrossRef]
- Vreeburg, R.A.; van Wezel, E.E.; Ocana-Calahorro, F.; Mes, J.J. Apple extract induces increased epithelial resistance and claudin 4 expression in Caco-2 cells. J. Sci. Food Agric. 2012, 92, 439–444. [Google Scholar] [CrossRef]
- Suzuki, T.; Hara, H. Quercetin enhances intestinal barrier function through the assembly of zonula [corrected] occludens-2, occludin, and claudin-1 and the expression of claudin-4 in Caco-2 cells. J. Nutr. 2009, 139, 965–974. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Retention Time | (M – H)− (m/z) | UV λmax (nm) | Formula | m/z of Main Fragments (Relative Intensity, %), MS/MS | Compound Identified |
|---|---|---|---|---|---|---|
| 1 | 3.57 | 331 | 278 | C13H16O10 | 271(18), 211(18), 169(45), 125(15) | galloyl hexose |
| 2 | 5.18 | 169 | 270 | C7H6O5 | 125 | gallic acid a |
| 3 | 8.70 | 299 | 260 | C13H16O8 | 239(40), 209(15), 179(75), 137(100), 119(27) | p-hydroxybenzoyl hexose |
| 4 | 15.13 | 183 | 270 | C8H8O5 | 168(25), 124(100) | methyl gallate a |
| 5 | 24.4 | 449 | 285/330 | C21H22O11 | 421(20), 287(100), 259(88), 243(8), 215(12), 179(10), 151(10), 125(23) | eriodictyol-O- glucoside |
| 6 | 37.73 | 609 | 265/345 | C27H30O16 | 447(60), 327(8), 285(70), 283(72), 255(10) | kaempferol-3,7-di-O-glucoside |
| 7 | 39.85 | 639 | 254/352 | C28H32O17 | 477(60), 519(5), 315(70), 313(66), 300(14) | isorhamnetin-3,7-di-O-glucoside |
| 8 | 41.30 | 335 | 278 | C15H12O9 | 183(100), 168(8), 124(40) | methyl digallate |
| 9 | 42.60 | 335 | 278 | C15H12O9 | 183(100), 168(8), 124(40) | methyl digallate |
| 10 | 43.65 | 335 | 278 | C15H12O9 | 183(100), 168(8), 124(40) | methyl digallate |
| 11 | 44.78 | 335 | 278 | C15H12O9 | 183(100), 168(8), 124(40) | methyl digallate |
| 12 | 45.50 | 939 | 274/359 | C41H32O26 | 787(6), 769(30), 617(12), 601(5), 465(6), 447(6), 431(6), 295(6), 277(6), 169(20) | penta-O-galloyl-glucose |
| 13 | 46.06 | 433 | 267/348 | C21H22O10 | 271(100), 177(5), 151(28), 119(13) | isosalipurposide |
| 14 | 46.68 | 1091 | 280 | C48H34O30 | 469(100), 393(6), 317(4), 295(5), 241(6), 169(65), 125(30) | hexa-O-galloyl-glucose |
| 15 | 47.43 | 1091 | 280 | C48H34O30 | 939(100), 787(8), 769(40), 617(10), 599(4), 447(6), 431(4), 169(5) | hexa-O-galloyl-glucose |
| 16 | 47.77 | 1091 | 280 | C48H34O30 | 939(100), 787(8), 769(40), 617(10), 599(4), 447(6), 431(4), 169(5) | hexa-O-galloyl-glucose |
| 17 | 48.43 | 1243 | 279 | C55H36O34 | 1091(48), 939(100), 787(4), 769(40), 617(2), 599(3), 447(3), 431(1), 169(1) | hepta-O-galloyl-glucose |
| 18 | 48.85 | 447 | 266/365 | C21H20O11 | 285(65), 284(40), 257(20), 151(40) | luteolin-7-O-glucoside |
| 19 | 49.63 | 431 | 267/340 | C21H20O10 | 268(100), 269(40) | apigenin-7-O-glucoside |
| 20 | 50.22 | 577 | 267/340 | C27H30O14 | 269(100) | apigenin-7-O-neohesperidoside |
| 21 | 50.63 | 447 | 265/346 | C21H20O11 | 285(30), 284(52), 255(66), 227(52) | kaempferol-3-O-glucoside |
| Compound Identified | Petal | Stamen | Calyx | Ovary |
|---|---|---|---|---|
| galloyl hexose | 292.07 ± 15.75 b | 101.23 ± 7.22 c | 323.36 ± 2.18 a | 315.20 ± 2.70 a |
| gallic acid a | 48.34 ± 5.89 b | 53.11 ± 1.11 ab | 59.89 ± 4.11 a | 38.96 ± 1.97 c |
| p-hydroxybenzoyl hexose | 39.98 ± 1.51 a | 17.52 ± 0.96 b | 4.59 ± 0.17 c | 3.42 ± 0.11 c |
| methyl gallate a | 250.80 ± 12.37 c | 423.47 ± 18.54 a | 240.54 ± 3.39 c | 326.00 ± 3.55 b |
| methyl digallate | 21.67 ± 1.26 a | 14.41 ± 0.34 b | 22.83 ± 3.38 a | 14.34 ± 0.30 b |
| methyl digallate | 20.01 ± 1.33 a | 12.60 ± 0.33 b | n.d. | n.d. |
| methyl digallate | 12.37 ± 0.34 b | 19.18 ± 0.94 a | 19.02 ± 1.67 a | 12.80 ± 0.12 b |
| methyl digallate | 53.95 ± 8.89 b | 222.40 ± 8.14 a | 53.57 ± 6.13 b | 47.89 ± 8.66 b |
| penta-O-galloyl-glucose | 413.82 ± 9.83 b | 513.10 ± 31.68 a | 246.10 ± 1.38 c | 265.78 ± 1.03 c |
| hexa-O-galloyl-glucose | 328.36 ± 18.54 a | 198.72 ± 2.97 b | 308.29 ± 1.16 a | 329.73 ± 1.86 a |
| hexa-O-galloyl-glucose | 52.99 ± 2.46 c | 6.88 ± 0.11 d | 141.71 ± 3.64 a | 91.86 ± 2.42 b |
| hexa-O-galloyl-glucose | 91.80 ± 3.33 a | 62.28 ± 1.12 b | 19.59 ± 0.41 c | 47.39 ± 1.21 b |
| hepta-O-galloyl-glucose | 246.10 ± 17.91 b | 52.37 ± 3.88 d | 207.68 ± 9.27 c | 281.72 ± 4.87 a |
| Total identified phenolic acids content | 1872.26 ± 103.03 a | 1697.26 ± 31.40 b | 1647.18 ± 12.53 b | 1775.09 ± 11.36 ab |
| eriodictyol-O- glucoside | 45.73 ± 2.96 a | 16.35 ± 0.38 b | 1.34 ± 0.02 c | 0.99 ± 0.01 c |
| kaempferol- 3,7-di-O-glucoside | 1124.33 ± 41.99 a | 601.44 ± 13.24 b | 91.47 ± 2.93 c | 27.75 ± 0.61 d |
| isorhamnetin -3,7-di-O-glucoside | 11.58 ± 0.70 d | 56.35 ± 1.22 b | 74.82 ± 0.19 a | 18.03 ± 0.16 c |
| isosalipurposide | 3.49 ± 0.24 b | 33.22 ± 0.25 a | n.d. | n.d. |
| luteolin-7-O-glucoside | 155.43 ± 9.84 a | 116.35 ± 2.11 b | 10.23 ± 0.29 c | 5.51 ± 0.10 c |
| apigenin-7-O-glucoside | 475.08 ± 45.41 a | 414.69 ± 14.82 a | 115.13 ± 3.26 b | 31.41 ± 1.05 c |
| apigenin-7-O- neohesperidoside | 518.95 ± 5.37 a | 372.05 ± 12.05 b | 124.07 ± 2.99 c | 31.90 ± 0.34 d |
| kaempferol-3-O-glucoside | 27.27 ± 2.17 a | 27.63 ± 1.55 a | 21.18 ± 1.75 b | 10.64 ± 0.32 c |
| Total identified flavonoids content | 2361.87 ± 17.87 a | 1638.07 ± 42.54 b | 438.64 ± 11.34 c | 126.23 ± 7.18 d |
| Total identified phenolics content | 4234.12 ± 120.90 a | 3335.33 ± 45.20 b | 2085.83 ± 14.79 c | 1901.32 ± 11.67 d |
| Samples | DPPH (μmol trolox/g DW) | ABTS•+ (μmol trolox/g DW) | ORAC (μmol trolox/g DW) |
|---|---|---|---|
| Petal | 325.69 ± 9.99 b | 345.08 ± 14.18 c | 1061.03 ± 55.06 a |
| Stamen | 207.80 ± 7.73 c | 234.58 ± 7.67 d | 704.06 ± 91.57 c |
| Calyx | 433.57 ± 4.60 a | 582.33 ± 10.36 b | 828.84 ± 20.97 b |
| Ovary | 444.58 ± 13.84 a | 610.21 ± 24.33 a | 555.11 ± 43.23 d |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, J.; Yang, C.; Beta, T.; Liu, S.; Yang, R. Phenolic Profile and Antioxidant Activity of the Edible Tree Peony Flower and Underlying Mechanisms of Preventive Effect on H2O2-Induced Oxidative Damage in Caco-2 Cells. Foods 2019, 8, 471. https://doi.org/10.3390/foods8100471
Xiang J, Yang C, Beta T, Liu S, Yang R. Phenolic Profile and Antioxidant Activity of the Edible Tree Peony Flower and Underlying Mechanisms of Preventive Effect on H2O2-Induced Oxidative Damage in Caco-2 Cells. Foods. 2019; 8(10):471. https://doi.org/10.3390/foods8100471
Chicago/Turabian StyleXiang, Jinle, Chengbo Yang, Trust Beta, Shangxi Liu, and Runqiang Yang. 2019. "Phenolic Profile and Antioxidant Activity of the Edible Tree Peony Flower and Underlying Mechanisms of Preventive Effect on H2O2-Induced Oxidative Damage in Caco-2 Cells" Foods 8, no. 10: 471. https://doi.org/10.3390/foods8100471
APA StyleXiang, J., Yang, C., Beta, T., Liu, S., & Yang, R. (2019). Phenolic Profile and Antioxidant Activity of the Edible Tree Peony Flower and Underlying Mechanisms of Preventive Effect on H2O2-Induced Oxidative Damage in Caco-2 Cells. Foods, 8(10), 471. https://doi.org/10.3390/foods8100471