
Quantification of Peptides in Food Hydrolysate from Vicia faba

Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
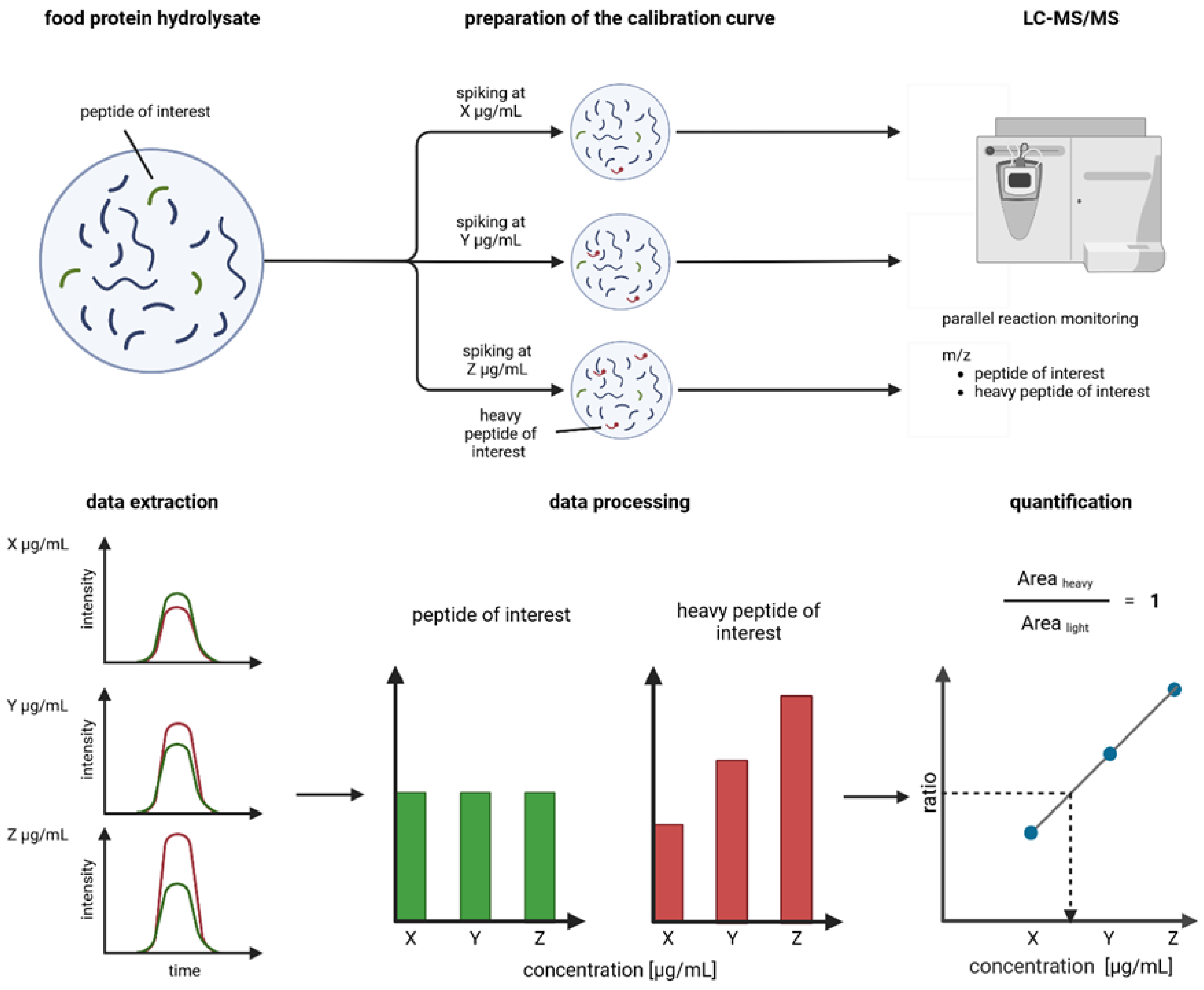
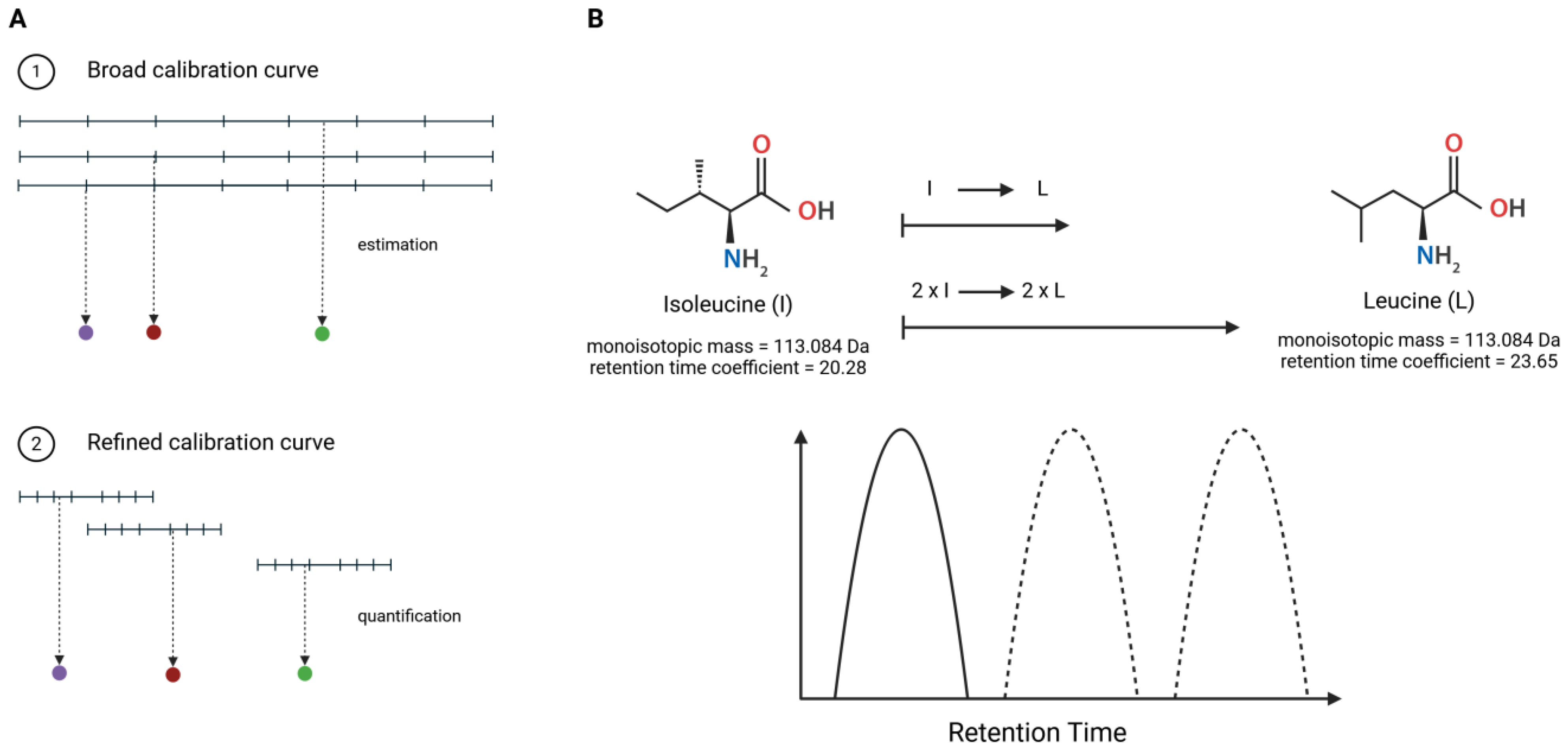
2.2. Preparation of Broad and Refined Internal Calibration Curves
2.3. Parallel Reaction Monitoring Mass Spectrometry Method and Inclusion List
2.4. Peak Integration
2.5. Peptide Sequence Validation
2.6. Linear Regression and Peptide Quantification
3. Results
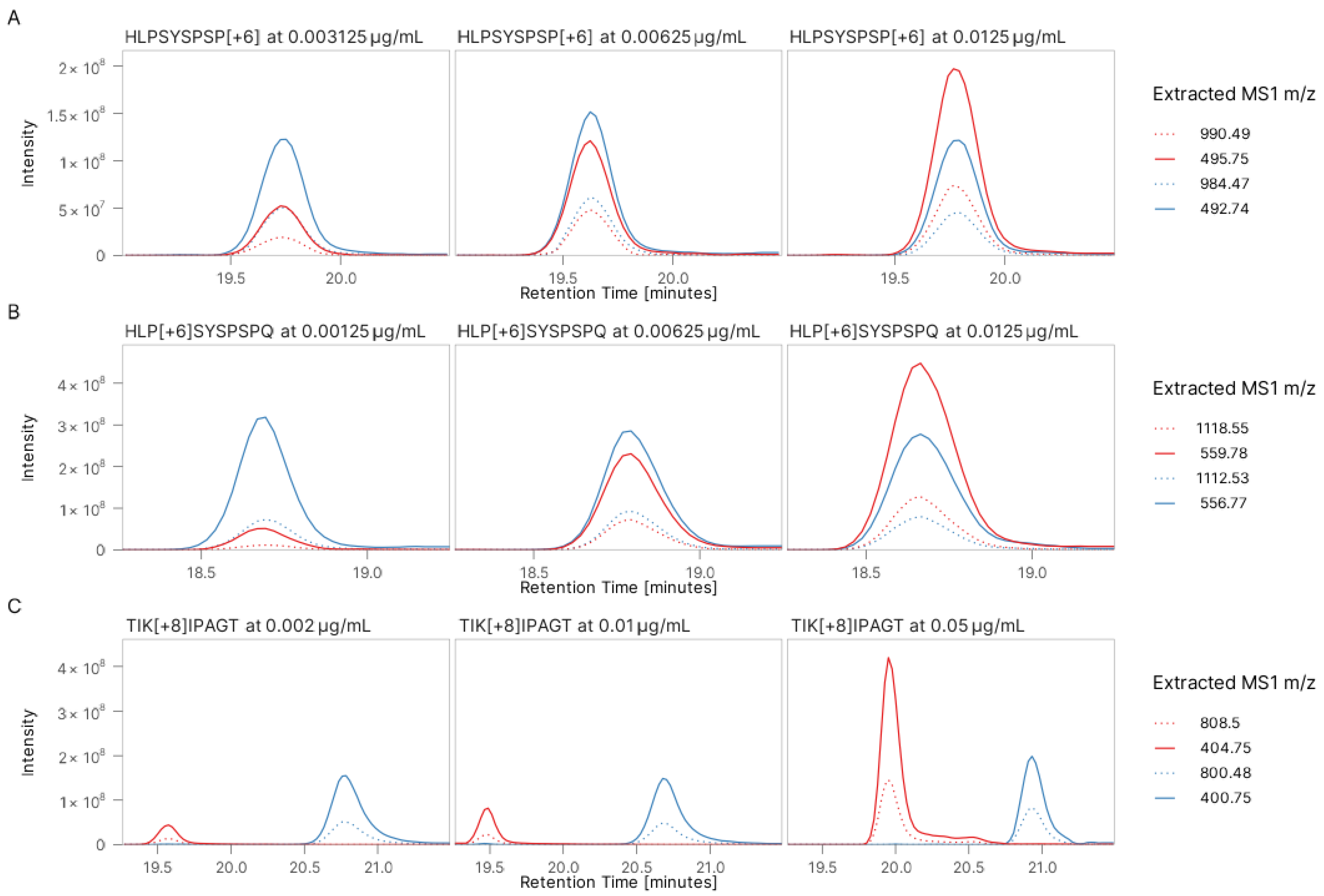
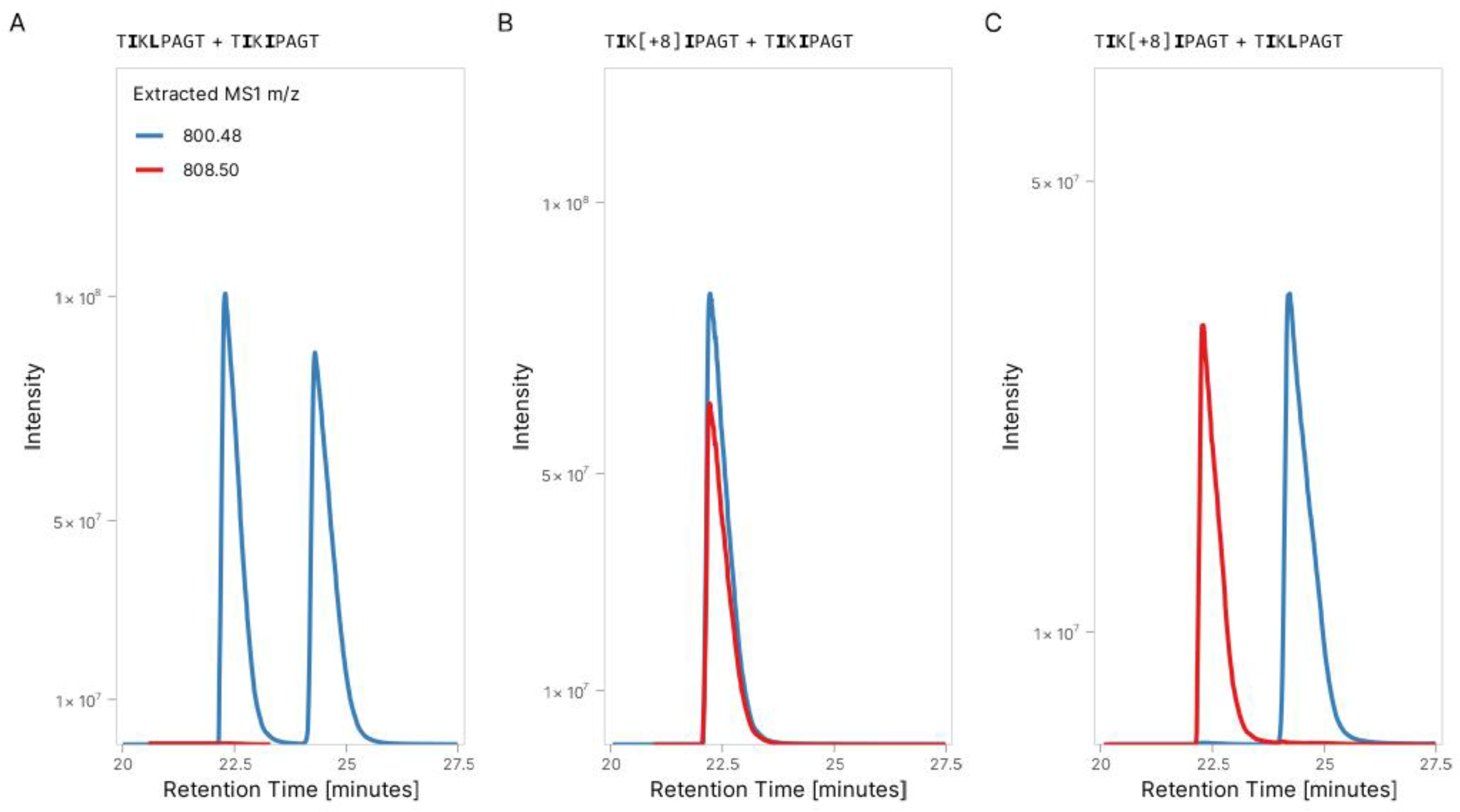
3.1. Validation of Peptide Sequence Using Synthetic Heavy Peptides
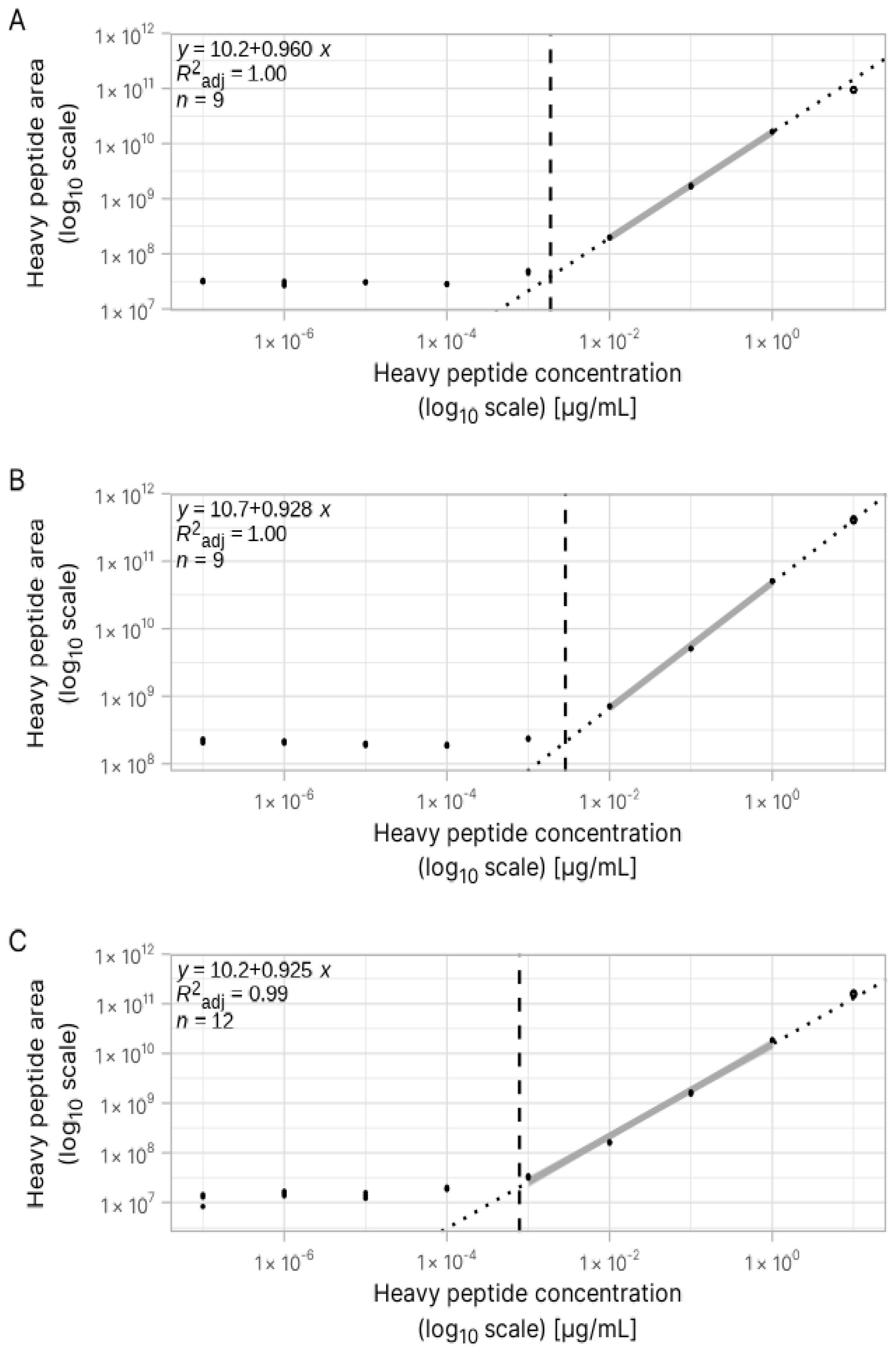
3.2. Estimation of Concentration for Multiple Peptides with Broad Internal Calibration Curves
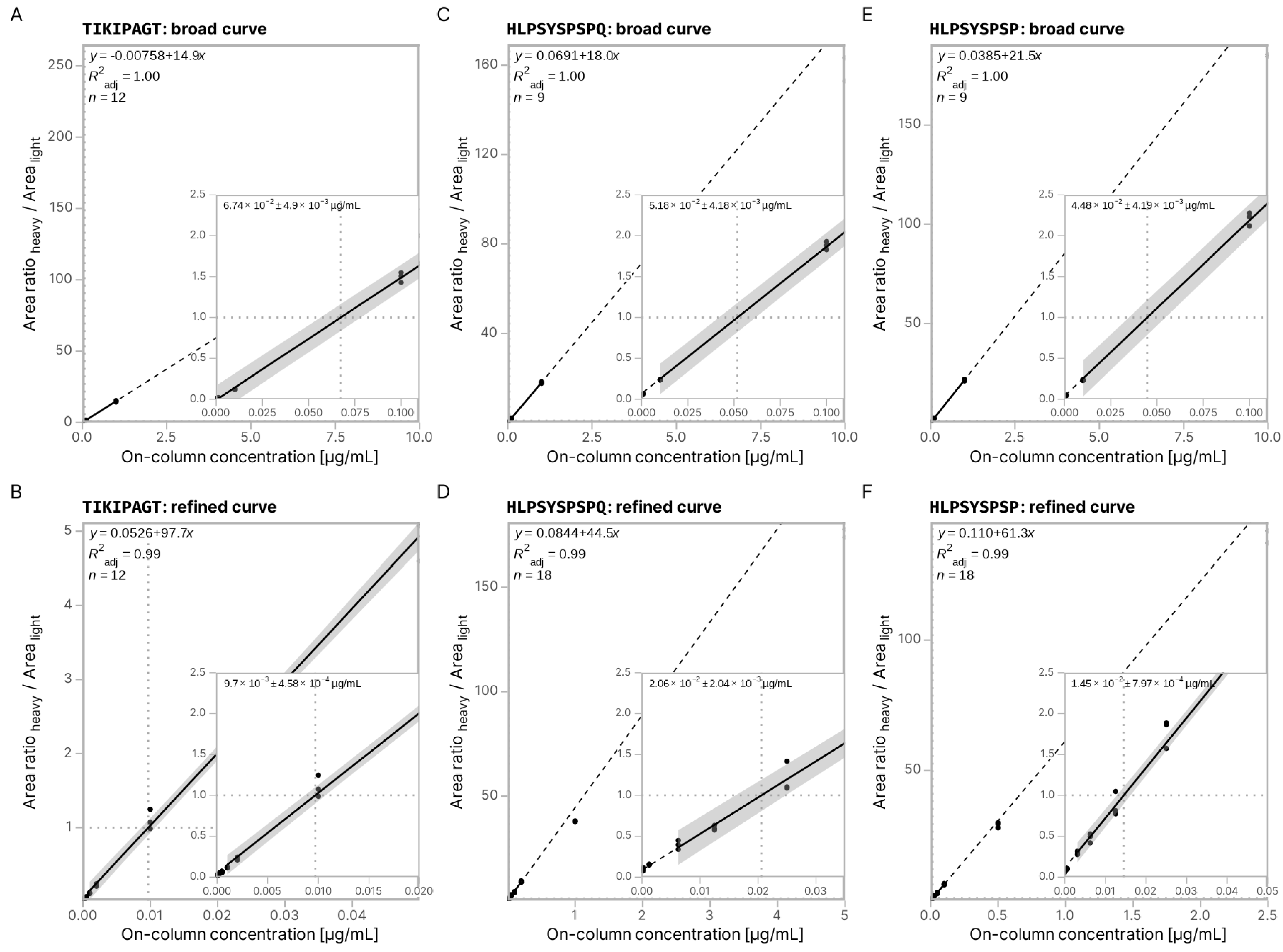
3.3. Absolute Quantification Using Peptide-Specific, Refined Calibration Curves
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PRM | Parallel reaction monitoring |
| MRM | Multiple reaction monitoring |
| SRM | Selected reaction monitoring |
| XIC | Extracted ion chromatogram |
| DDA | Data-dependent acquisition |
| DIA | Data-independent acquisition |
| BCA | Bicinchoninic acid assay |
References
- Chalamaiah, M.; Yu, W.; Wu, J. Immunomodulatory and Anticancer Protein Hydrolysates (Peptides) from Food Proteins: A Review. Food Chem. 2018, 245, 205–222. [Google Scholar] [CrossRef]
- Chalamaiah, M.; Keskin Ulug, S.; Hong, H.; Wu, J. Regulatory Requirements of Bioactive Peptides (Protein Hydrolysates) from Food Proteins. J. Funct. Foods 2019, 58, 123–129. [Google Scholar] [CrossRef]
- Hayes, M.; Bleakley, S. Applications. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos, S., Ed.; Woodhead Publishing: Sawston, UK, 2018; pp. 603–622. ISBN 978-0-08-100736-5. [Google Scholar]
- He, R.; Girgih, A.T.; Rozoy, E.; Bazinet, L.; Ju, X.-R.; Aluko, R.E. Selective Separation and Concentration of Antihypertensive Peptides from Rapeseed Protein Hydrolysate by Electrodialysis with Ultrafiltration Membranes. Food Chem. 2016, 197, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, Q.; Xue, B.; Chen, L.; Wang, Y.; Ou, S.; Peng, X. Angiotensin-I-Converting Enzyme Inhibitory Activities and In Vivo Antihypertensive Effects of Sardine Protein Hydrolysate. J. Food Sci. 2016, 81, H2831–H2840. [Google Scholar] [CrossRef]
- Zhuang, H.; Tang, N.; Yuan, Y. Purification and Identification of Antioxidant Peptides from Corn Gluten Meal. J. Funct. Foods 2013, 5, 1810–1821. [Google Scholar] [CrossRef]
- Ndiaye, F.; Vuong, T.; Duarte, J.; Aluko, R.E.; Matar, C. Anti-Oxidant, Anti-Inflammatory and Immunomodulating Properties of an Enzymatic Protein Hydrolysate from Yellow Field Pea Seeds. Eur. J. Nutr. 2012, 51, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Kim, H.H.; Kim, J.Y.; Kang, Y.I.; Woo, H.J.; Lee, H.J. Anticancer Activity of Hydrophobic Peptides from Soy Proteins. BioFactors 2000, 12, 151–155. [Google Scholar] [CrossRef]
- Li, J.-T.; Zhang, J.-L.; He, H.; Ma, Z.-L.; Nie, Z.-K.; Wang, Z.-Z.; Xu, X.-G. Apoptosis in Human Hepatoma HepG2 Cells Induced by Corn Peptides and Its Anti-Tumor Efficacy in H22 Tumor Bearing Mice. Food Chem. Toxicol. 2013, 51, 297–305. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, M.; Sun, C.; Brennan, M.; Li, H.; Wang, G.; Lai, F.; Wu, H. Enzymatic Preparation of Immunomodulatory Hydrolysates from Defatted Wheat Germ (Triticum Vulgare) Globulin. Int. J. Food Sci. Technol. 2016, 51, 2556–2566. [Google Scholar] [CrossRef]
- Katayama, S.; Corpuz, H.M.; Nakamura, S. Potential of Plant-Derived Peptides for the Improvement of Memory and Cognitive Function. Peptides 2021, 142, 170571. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, M.; Fan, H.; Wu, J. Emerging Proteins as Precursors of Bioactive Peptides/Hydrolysates with Health Benefits. Curr. Opin. Food Sci. 2022, 48, 100914. [Google Scholar] [CrossRef]
- Regazzo, D.; Mollé, D.; Gabai, G.; Tomé, D.; Dupont, D.; Leonil, J.; Boutrou, R. The (193–209) 17-Residues Peptide of Bovine β-Casein Is Transported through Caco-2 Monolayer. Mol. Nutr. Food Res. 2010, 54, 1428–1435. [Google Scholar] [CrossRef]
- Grootaert, C.; Jacobs, G.; Matthijs, B.; Pitart, J.; Baggerman, G.; Possemiers, S.; Van der Saag, H.; Smagghe, G.; Van Camp, J.; Voorspoels, S. Quantification of Egg Ovalbumin Hydrolysate-Derived Anti-Hypertensive Peptides in an in Vitro Model Combining Luminal Digestion with Intestinal Caco-2 Cell Transport. Food Res. Int. 2017, 99, 531–541. [Google Scholar] [CrossRef]
- Kong, S.; Zhang, Y.H.; Zhang, W. Regulation of Intestinal Epithelial Cells Properties and Functions by Amino Acids. BioMed Res. Int. 2018, 2018, 2819154. [Google Scholar] [CrossRef]
- Takeda, J.; Park, H.-Y.; Kunitake, Y.; Yoshiura, K.; Matsui, T. Theaflavins, Dimeric Catechins, Inhibit Peptide Transport across Caco-2 Cell Monolayers via down-Regulation of AMP-Activated Protein Kinase-Mediated Peptide Transporter PEPT1. Food Chem. 2013, 138, 2140–2145. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Liu, S.; Xie, X.-N.; Tan, Z.-R. Regulation Profile of the Intestinal Peptide Transporter 1 (PepT1). Drug Des. Dev. Ther. 2017, 11, 3511–3517. [Google Scholar] [CrossRef]
- Ozorio, L.; Mellinger-Silva, C.; Cabral, L.M.; Jardin, J.; Boudry, G.; Dupont, D. The Influence of Peptidases in Intestinal Brush Border Membranes on the Absorption of Oligopeptides from Whey Protein Hydrolysate: An Ex Vivo Study Using an Ussing Chamber. Foods 2020, 9, 1415. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Hong, H.; Wu, J.; Yan, X. Bioavailability of Bioactive Peptides Derived from Food Proteins across the Intestinal Epithelial Membrane: A Review. Trends Food Sci. Technol. 2019, 86, 399–411. [Google Scholar] [CrossRef]
- Castro, P.; Madureira, R.; Sarmento, B.; Pintado, M. Tissue-Based in Vitro and Ex Vivo Models for Buccal Permeability Studies. In Concepts and Models for Drug Permeability Studies; Elsevier: Amsterdam, The Netherlands, 2016; pp. 189–202. [Google Scholar]
- Vermeirssen, V.; Camp, J.V.; Verstraete, W. Bioavailability of Angiotensin I Converting Enzyme Inhibitory Peptides. Br. J. Nutr. 2004, 92, 357–366. [Google Scholar] [CrossRef]
- Deacon, C.F.; Nauck, M.A.; Toft-Nielsen, M.; Pridal, L.; Willms, B.; Holst, J.J. Both Subcutaneously and Intravenously Administered Glucagon-like Peptide I Are Rapidly Degraded from the NH2-Terminus in Type II Diabetic Patients and in Healthy Subjects. Diabetes 1995, 44, 1126–1131. [Google Scholar]
- Shimizu, M.; Tsunogai, M.; Arai, S. Transepithelial Transport of Oligopeptides in the Human Intestinal Cell, Caco-2. Peptides 1997, 18, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Adibi, S. Intestinal Phase of Protein Assimilation in Man. Am. J. Clin. Nutr. 1976, 29, 205–215. [Google Scholar] [CrossRef]
- Chabance, B.; Marteau, P.; Rambaud, J.C.; Migliore-Samour, D.; Boynard, M.; Perrotin, P.; Guillet, R.; Jollès, P.; Fiat, A.M. Casein Peptide Release and Passage to the Blood in Humans during Digestion of Milk or Yogurt. Biochimie 1998, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Lambers, T.T.; Gloerich, J.; van Hoffen, E.; Alkema, W.; Hondmann, D.H.; van Tol, E.A.F. Clustering Analyses in Peptidomics Revealed That Peptide Profiles of Infant Formulae Are Descriptive. Food Sci. Nutr. 2015, 3, 81–90. [Google Scholar] [CrossRef]
- Corrochano, A.R.; Cal, R.; Kennedy, K.; Wall, A.; Murphy, N.; Trajkovic, S.; O’Callaghan, S.; Adelfio, A.; Khaldi, N. Characterising the Efficacy and Bioavailability of Bioactive Peptides Identified for Attenuating Muscle Atrophy within a Vicia Faba-Derived Functional Ingredient. Curr. Res. Food Sci. 2021, 4, 224–232. [Google Scholar] [CrossRef]
- Dallas, D.C.; Guerrero, A.; Parker, E.A.; Robinson, R.C.; Gan, J.; German, J.B.; Barile, D.; Lebrilla, C.B. Current Peptidomics: Applications, Purification, Identification, Quantification, and Functional Analysis. Proteomics 2015, 15, 1026–1038. [Google Scholar] [CrossRef]
- Arnold, S.L.; Stevison, F.; Isoherranen, N. Impact of Sample Matrix on Accuracy of Peptide Quantification: Assessment of Calibrator and Internal Standard Selection and Method Validation. Anal. Chem. 2016, 88, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Warwood, S.; Byron, A.; Humphries, M.J.; Knight, D. The Effect of Peptide Adsorption on Signal Linearity and a Simple Approach to Improve Reliability of Quantification. J. Proteom. 2013, 85, 160–164. [Google Scholar] [CrossRef]
- Chauhan, S. Using Peptidomics and Machine Learning to Assess Effects of Drying Processes on the Peptide Profile within a Functional Ingredient. Processes 2021, 9, 425. [Google Scholar] [CrossRef]
- Sun, W.; Wu, S.; Wang, X.; Zheng, D.; Gao, Y. A Systematical Analysis of Tryptic Peptide Identification with Reverse Phase Liquid Chromatography and Electrospray Ion Trap Mass Spectrometry. Genom. Proteom. Bioinform. 2004, 2, 174–183. [Google Scholar] [CrossRef]
- Foreman, R.E.; George, A.L.; Reimann, F.; Gribble, F.M.; Kay, R.G. Peptidomics: A Review of Clinical Applications and Methodologies. J. Proteome Res. 2021, 20, 3782–3797. [Google Scholar] [CrossRef]
- van der Kloet, F.M.; Bobeldijk, I.; Verheij, E.R.; Jellema, R.H. Analytical Error Reduction Using Single Point Calibration for Accurate and Precise Metabolomic Phenotyping. J. Proteome Res. 2009, 8, 5132–5141. [Google Scholar] [CrossRef] [PubMed]
- Mesmin, C.; Dubois, M.; Becher, F.; Fenaille, F.; Ezan, E. Liquid Chromatography/Tandem Mass Spectrometry Assay for the Absolute Quantification of the Expected Circulating Apelin Peptides in Human Plasma. Rapid Commun. Mass Spectrom. 2010, 24, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Kandi, S.; Savaryn, J.P.; Ji, Q.C.; Jenkins, G.J. Use of In-Sample Calibration Curve Approach for Quantification of Peptides with High-Resolution Mass Spectrometry. Rapid Commun. Mass Spectrom. 2022, 36, e9377. [Google Scholar] [CrossRef]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef]
- Cal, R.; Davis, H.; Kerr, A.; Wall, A.; Molloy, B.; Chauhan, S.; Trajkovic, S.; Holyer, I.; Adelfio, A.; Khaldi, N. Preclinical Evaluation of a Food-Derived Functional Ingredient to Address Skeletal Muscle Atrophy. Nutrients 2020, 12, 2274. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Valenzo, O.M.M.; Williams, D.R. Prediction of Peptides Retention Behavior in Reversed-Phase Liquid Chromatography Based on Their Hydrophobicity. J. Sep. Sci. 2023, 46, 2200743. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]
- Krieger, J.; Xin, L.; Shan, B. Multi-User, High-Throughput PEAKS Online Workflow, for Large Scale DIA and DDA Proteomic Analysis. J. Biomol. Tech. 2020, 31, S22–S23. [Google Scholar]
- Fernández-Costa, C.; Martínez-Bartolomé, S.; McClatchy, D.B.; Saviola, A.J.; Yu, N.-K.; Yates, J.R.I. Impact of the Identification Strategy on the Reproducibility of the DDA and DIA Results. J. Proteome Res. 2020, 19, 3153–3161. [Google Scholar] [CrossRef]
- Muggeo, V.M.R. Estimating Regression Models with Unknown Break-Points. Stat. Med. 2003, 22, 3055–3071. [Google Scholar] [CrossRef]
- Muggeo, V.M.R. Segmented: An R Package to Fit Regression Models with Broken-Line Relationships. R News 2008, 8, 20–25. [Google Scholar]
- Gillette, M.A.; Carr, S.A. Quantitative Analysis of Peptides and Proteins in Biomedicine by Targeted Mass Spectrometry. Nat. Methods 2013, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel Reaction Monitoring Using Quadrupole-Orbitrap Mass Spectrometer: Principle and Applications. Proteomics 2016, 16, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Bezstarosti, K.; Van der Wal, L.; Doff, W.A.S.; Demmers, J.A.A. Parallel Reaction Monitoring Targeted Mass Spectrometry as a Fast and Sensitive Alternative to Antibody-Based Protein Detection. Front. Anal. Sci. 2024, 4, 1397810. [Google Scholar] [CrossRef]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef]
- Barkovits, K.; Pacharra, S.; Pfeiffer, K.; Steinbach, S.; Eisenacher, M.; Marcus, K.; Uszkoreit, J. Reproducibility, Specificity and Accuracy of Relative Quantification Using Spectral Library-Based Data-Independent Acquisition. Mol. Cell. Proteom. 2020, 19, 181–197. [Google Scholar] [CrossRef]
- Hoffman, M.A.; Fang, B.; Haura, E.B.; Rix, U.; Koomen, J.M. Comparison of Quantitative Mass Spectrometry Platforms for Monitoring Kinase ATP Probe Uptake in Lung Cancer. J. Proteome Res. 2018, 17, 63–75. [Google Scholar] [CrossRef]
- Klaproth-Andrade, D.; Hingerl, J.; Bruns, Y.; Smith, N.H.; Träuble, J.; Wilhelm, M.; Gagneur, J. Deep Learning-Driven Fragment Ion Series Classification Enables Highly Precise and Sensitive de Novo Peptide Sequencing. Nat. Commun. 2024, 15, 151. [Google Scholar] [CrossRef]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted Proteomic Quantification on Quadrupole-Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2012, 11, 1709–1723. [Google Scholar] [CrossRef]
- van Bentum, M.; Selbach, M. An Introduction to Advanced Targeted Acquisition Methods. Mol. Cell. Proteom. MCP 2021, 20, 100165. [Google Scholar] [CrossRef] [PubMed]
- Gilani, G.S.; Xiao, C.; Lee, N. Need for Accurate and Standardized Determination of Amino Acids and Bioactive Peptides for Evaluating Protein Quality and Potential Health Effects of Foods and Dietary Supplements. J. AOAC Int. 2008, 91, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Rutherfurd-Markwick, K.J. Food Proteins as a Source of Bioactive Peptides with Diverse Functions. Br. J. Nutr. 2012, 108, S149–S157. [Google Scholar] [CrossRef]
- Mamone, G.; Picariello, G.; Caira, S.; Addeo, F.; Ferranti, P. Analysis of Food Proteins and Peptides by Mass Spectrometry-Based Techniques. J. Chromatogr. A 2009, 1216, 7130–7142. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide of Interest | Spiked Internal Standard | ||
|---|---|---|---|
| Peptide Sequence | Molecular Weight (Da) | Peptide Sequence | Molecular Weight (Da) |
| HLPSYSPSP | 984.4785 | HLPSYSPSP[Label:13C(5)15N(1)] | 990.4923 |
| HLPSYSPSPQ | 1112.5371 | HLP[Label:13C(5)15N(1)]SYSPSPQ | 1118.5509 |
| TIKIPAGT | 800.4876 | TI[Label:13C(6)15N(2)]KIPAGT | 808.4876 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manguy, J.; Papoutsidakis, G.I.; Doyle, B.; Trajkovic, S. Quantification of Peptides in Food Hydrolysate from Vicia faba. Foods 2025, 14, 1180. https://doi.org/10.3390/foods14071180
Manguy J, Papoutsidakis GI, Doyle B, Trajkovic S. Quantification of Peptides in Food Hydrolysate from Vicia faba. Foods. 2025; 14(7):1180. https://doi.org/10.3390/foods14071180
Chicago/Turabian StyleManguy, Jean, Georgios I. Papoutsidakis, Ben Doyle, and Sanja Trajkovic. 2025. "Quantification of Peptides in Food Hydrolysate from Vicia faba" Foods 14, no. 7: 1180. https://doi.org/10.3390/foods14071180
APA StyleManguy, J., Papoutsidakis, G. I., Doyle, B., & Trajkovic, S. (2025). Quantification of Peptides in Food Hydrolysate from Vicia faba. Foods, 14(7), 1180. https://doi.org/10.3390/foods14071180