
Application of Rice Husk-Derived SBA-15 Bifunctionalized with C18 and Sulfonic Groups for Solid-Phase Extraction of Tropane, Pyrrolizidine, and Opium Alkaloids in Gluten-Free Bread

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Solvents and Materials
2.2. Synthesis and Modification of RH-Derived SBA-15 Silica
2.2.1. Extraction of Silica from RH
2.2.2. Synthesis and Functionalization of RH-Derived SBA-15 Silica
2.3. Characterization of Mesoporous Silica
2.4. Preparation of Gluten-Free Bread Samples
2.5. Extraction by SLE and Purification by SPE with RH-SBA-15-SO3H-C18 of Alkaloids from Gluten-Free Bread Samples
2.6. Chromatographic Conditions
2.7. Method Validation
3. Results
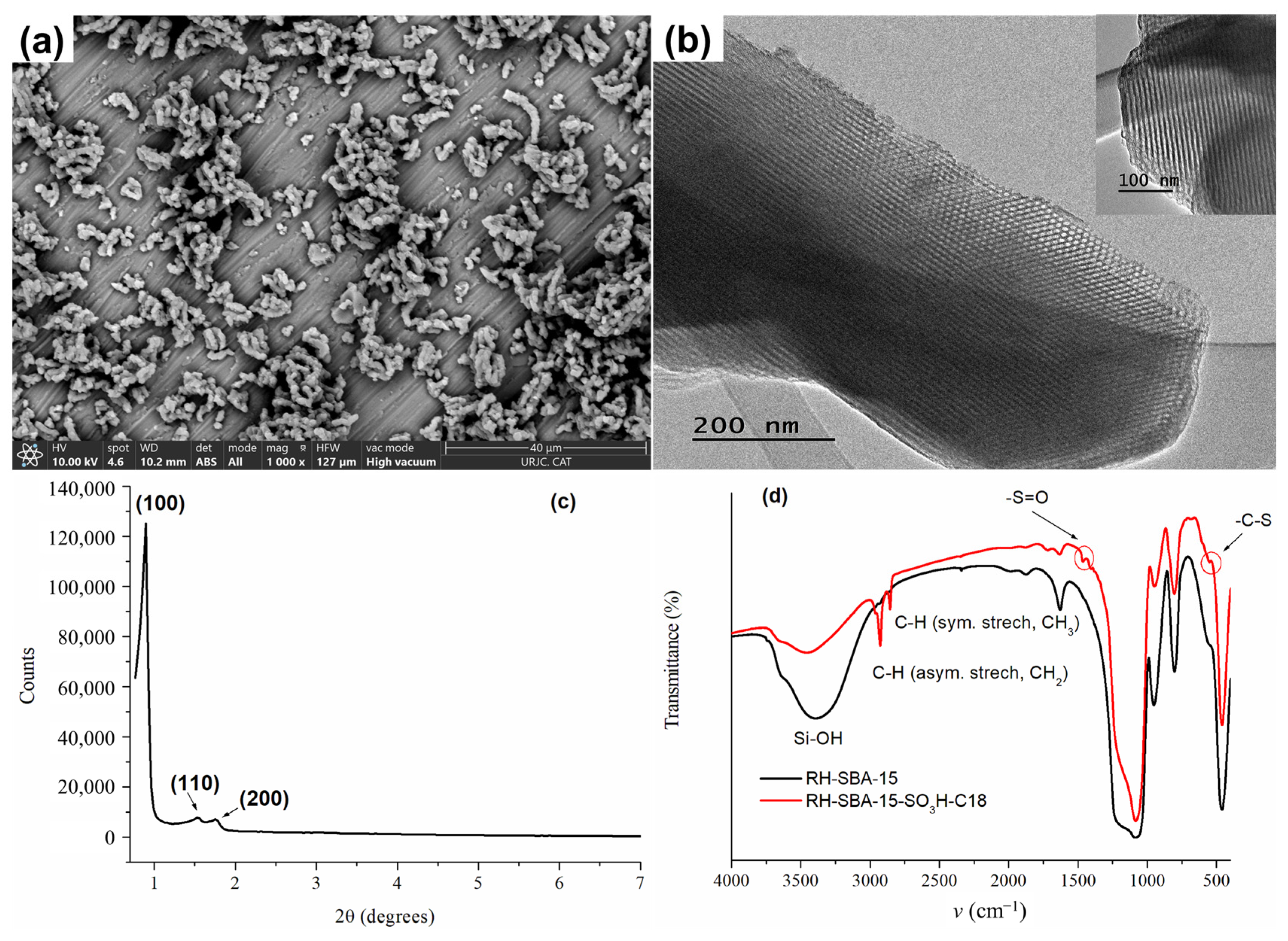
3.1. Characterization of Mesoporous Silicas
3.2. Evaluation of the Adsorption Capacity of RH-SBA-15-SO3H-C18 as SPE Sorbent
3.3. Optimization of the Extraction and Purification of Alkaloids from Gluten-Free Bread Samples
3.4. Method Validation
3.5. Application of the Validated Method to the Analysis of Gluten-Free Bread
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- EFSA Panel on Contaminants in the Food Chain, (CONTAM). Scientific Opinion on Pyrrolizidine alkaloids in food and feed. EFSA J. 2011, 9, 2406. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the risks for public health related to the presence of opium alkaloids in poppy seeds. EFSA J. 2011, 9, 2405. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain, (CONTAM). Scientific Opinion on Tropane alkaloids in food and feed. EFSA J. 2013, 11, 3386. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain, (CONTAM). Update of the Scientific Opinion on opium alkaloids in poppy seeds. EFSA J. 2018, 16, 5243. [Google Scholar] [CrossRef]
- Casado, N.; Casado-Hidalgo, G.; González-Gómez, L.; Morante-Zarcero, S.; Sierra, I. Insight into the Impact of Food Processing and Culinary Preparations on the Stability and Content of Plant Alkaloids Considered as Natural Food Contaminants. Appl. Sci. 2023, 13, 1704. [Google Scholar] [CrossRef]
- Casado, N.; Gañán, J.; Morante-Zarcero, S.; Sierra, I. Recent food alerts and analytical advances related to the contamination of tropane and pyrrolizidine alkaloids in food. Front. Chem. Biol. 2024, 3, 1360027. [Google Scholar] [CrossRef]
- EU 2020/2040; Commission Regulation (EU) 2020/2040 of 11 December 2020 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Pyrrolizidine Alkaloids in Certain Foodstuffs (Text with EEA Relevance). European Union: Brussels, Belgium, 2020. Available online: http://data.europa.eu/eli/reg/2020/2040/oj (accessed on 9 October 2024).
- EU 2021/1408; Commission Regulation (EU) 2021/1408 of 27 August 2021 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Tropane Alkaloids in Certain Foodstuffs (Text with EEA Relevance). European Union: Brussels, Belgium, 2021. Available online: http://data.europa.eu/eli/reg/2021/1408/oj (accessed on 9 October 2024).
- EU 2021/2142; Commission Regulation (EU) 2021/2142 of 3 December 2021 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Opium Alkaloids in Certain Foodstuffs (Text with EEA Relevance). European Union: Brussels, Belgium, 2021. Available online: http://data.europa.eu/eli/reg/2021/2142/oj (accessed on 9 October 2024).
- EU 2023/915; Commission Regulation (EU) 2023/915 of 25 April 2023 on Maximum Levels for Certain Contaminants in Food and Repealing Regulation (EC) No 1881/2006 (Text with EEA Relevance). European Union: Brussels, Belgium, 2023. Available online: http://data.europa.eu/eli/reg/2023/915/oj (accessed on 9 October 2024).
- EU 2023/2783; Commission Implementing Regulation (EU) 2023/2783 of 14 December 2023 Laying Down the Methods of Sampling and Analysis for the Control of the Levels of Plant Toxins in Food and Repealing Regulation (EU) 2015/705 (Text with EEA Relevance). European Union: Brussels, Belgium, 2023. Available online: http://data.europa.eu/eli/reg_impl/2023/2783/oj (accessed on 9 October 2024).
- Alahmad, W.; Kaya, S.I.; Cetinkaya, A.; Varanusupakul, P.; Ozkan, S. Green chemistry methods for food analysis: Overview of sample preparation and determination. Adv. Sam. Prep. 2023, 5, 100053. [Google Scholar] [CrossRef]
- Jagirani, M.S.; Soylak, M. Green sorbents for the solid phase extraction of trace species. Curr. Opin. Green Sustain. Chem. 2024, 47, 100899. [Google Scholar] [CrossRef]
- United Nations (UN). Departament of Economic and Social Affairs. Development. Available online: https://sdgs.un.org/goals (accessed on 9 October 2024).
- Locatelli, M.; Kabir, A.; Perrucci, M.; Ulusoy, S.; Ulusoy, H.I.; Ali, I. Green profile tools: Current status and future perspectives. Adv. Sam. Prep. 2023, 6, 100068. [Google Scholar] [CrossRef]
- Werner, J.; Frankowski, R.; Grześkowiak, T.; Zgoła-Grześkowiak, A. Green sorbents in sample preparation techniques—Naturally occurring materials and biowastes. TrAC Trends Anal. Chem. 2024, 176, 117772. [Google Scholar] [CrossRef]
- Della, V.P.; Kühn, I.; Hotza, D. Rice husk ash as an alternate source for active silica production. Mater. Lett. 2002, 57, 818–821. [Google Scholar] [CrossRef]
- Steven, S.; Restiawaty, E.; Bindar, Y. Routes for energy and bio-silica production from rice husk: A comprehensive review and emerging prospect. Renew. Sust. Energy. Rev. 2021, 149, 111329. [Google Scholar] [CrossRef]
- Castaneda, F.N.; Prince, D.L.; Peirano, S.R.; Giovannoni, S.; Echevarria, R.N.; Keunchkarian, S.; Reta, M. New sorbents for sample pretreatment: Development and applications. TrAC Trends Anal. Chem. 2024, 180, 117924. [Google Scholar] [CrossRef]
- Pode, R. Potential applications of rice husk ash waste from rice husk biomass power plant. Renew. Sust. Energ. Rev. 2016, 53, 1468–1485. [Google Scholar] [CrossRef]
- Kresge, C.T.; Roth, W.J. The discovery of mesoporous molecular sieves from the twenty year perspective. Chem. Soc. Rev. 2013, 42, 3663–3670. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, X.; Feng, J.; Sun, M. Recent Advances of Ordered Mesoporous Silica Materials for Solid-Phase Extraction. J. Chromatogr. A 2022, 1675, 463157. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef]
- Vanichvattanadecha, C.; Singhapong, W.; Jaroenworaluck, A. Different sources of silicon precursors influencing on surface characteristics and pore morphologies of mesoporous silica nanoparticles. Appl. Surf. Sci. 2020, 513, 145568. [Google Scholar] [CrossRef]
- Kamari, S.; Ghorbani, F. Extraction of highly pure silica from rice husk as an agricultural by-product and its application in the production of magnetic mesoporous silica MCM–41. Biomass Conv. Bioref. 2021, 11, 3001–3009. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Y.; Wang, R.; Chen, Z.; Luo, X.; Wang, L.; Zhao, X.; Zhang, C.; Yu, P. Removal of aflatoxin B1 from aqueous solution using amino-grafted magnetic mesoporous silica prepared from rice husk. Food Chem. 2022, 389, 132987. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Shi, Y.; Cao, J. Recent advances and applications of novel advanced materials in solid-phase microextraction for natural products. TrAC Trends Anal. Chem. 2024, 178, 117858. [Google Scholar] [CrossRef]
- Liou, T.; Wang, S.; Lin, Y.; Yang, S. Sustainable utilization of rice husk waste for preparation of ordered nanostructured mesoporous silica and mesoporous carbon: Characterization and adsorption performance. Colloids Surf. A Physicochem. Eng. Asp. 2022, 636, 128150. [Google Scholar] [CrossRef]
- González-Gómez, L.; Gañán, J.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Sulfonic Acid-Functionalized SBA-15 as Strong Cation-Exchange Sorbent for Solid-Phase Extraction of Atropine and Scopolamine in Gluten-Free Grains and Flours. Foods 2020, 9, 1854. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Aceituno, L.; Casado, N.; Arriero-Romo, E.; Morante-Zarcero, S.; Lázaro, A.; Sierra, I. Development of Gluten-Free Bread Based on Maize and Buckwheat and Enriched with Aromatic Herbs and Spices. Appl. Sci. 2024, 14, 3348. [Google Scholar] [CrossRef]
- Vera-Baquero, F.L.; Casado, N.; Morante-Zarcero, S.; Sierra, I. Improving the food safety of bakery products by simultaneously monitoring the occurrence of pyrrolizidine, tropane and opium alkaloids. Food Chem. 2024, 460, 140769. [Google Scholar] [CrossRef]
- European Commission SANTE. European Commission SANTE 11312/2021. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. 2021. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/SANTE_11312_2021.pdf (accessed on 9 October 2024).
- Tietje, A.B. (Ed.) Q2(R1) ICH Guidelines (Internation Council for Harmonisation, 2005). In Handbook of Transnational Economic Governance Regimes; Brill: Leiden, The Netherlands, 2010; pp. 1041–1053. [Google Scholar] [CrossRef]
- Priego-Capote, F. 6—Solid–liquid extraction techniques. In Analytical Sample Preparation with Nano- and Other High-Performance Materials; Lucena, R., Cárdenas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 111–130. [Google Scholar] [CrossRef]
- Dzuman, Z.; Jonatova, P.; Stranska-Zachariasova, M.; Prusova, N.; Brabenec, O.; Novakova, A.; Fenclova, M.; Hajslova, J. Development of a new LC-MS method for accurate and sensitive determination of 33 pyrrolizidine and 21 tropane alkaloids in plant-based food matrices. Anal. Bioanal. Chem. 2020, 412, 7155–7167. [Google Scholar] [CrossRef]
- Meos, A.; Saks, L.; Raal, A. Content of alkaloids in ornamental Papaver somniferum L. cultivars growing in Estonia. Proc. Est. Acad. Sci. 2017, 66, 34–39. [Google Scholar] [CrossRef]
- Sproll, C.; Perz, R.C.; Lachenmeier, D.W. Optimized LC/MS/MS Analysis of Morphine and Codeine in Poppy Seed and Evaluation of Their Fate during Food Processing as a Basis for Risk Analysis. J. Agric. Food Chem. 2006, 54, 5292–5298. [Google Scholar] [CrossRef]
- Marín-Sáez, J.; Romero-González, R.; Garrido Frenich, A. Effect of tea making and boiling processes on the degradation of tropane alkaloids in tea and pasta samples contaminated with Solanaceae seeds and coca leaf. Food Chem. 2019, 287, 265–272. [Google Scholar] [CrossRef]
- Shetge, S.A.; Dzakovich, M.P.; Cooperstone, J.L.; Kleinmeier, D.; Redan, B.W. Concentrations of the opium alkaloids morphine, codeine, and thebaine in poppy seeds are reduced after thermal and washing treatments but are not affected when incorporated in a model baked product. J. Agric. Food Chem. 2020, 68, 5241–5248. [Google Scholar] [CrossRef] [PubMed]
- Carlin, M.G.; Dean, J.R.; Ames, J.M. Opium alkaloids in harvested and thermally processed poppy seeds. Front. Chem. 2020, 8, 737. [Google Scholar] [CrossRef]
- Shetge, S.A.; Redan, B.W. Assessment of dry heating, water rinsing, and baking on concentrations of the opium alkaloid noscapine in poppy seeds. ACS Food Sci. Technol. 2022, 2, 541–547. [Google Scholar] [CrossRef]
- Vera-Baquero, F.L.; Morante-Zarcero, S.; Sierra, I. Evaluation of Thermal Degradation of Tropane and Opium Alkaloids in Gluten-Free Corn Breadsticks Samples Contaminated with Stramonium Seeds and Baked with Poppy Seeds under Different Conditions. Foods 2022, 11, 2196. [Google Scholar] [CrossRef]
- González-Gómez, L.; Casado-Hidalgo, G.; Gañán, J.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Evaluating the stability of tropane and opium alkaloids during baking in homemade gluten-free poppy seed crackers. LWT 2024, 214, 117080. [Google Scholar] [CrossRef]
- Kaltner, F.; Rychlik, M.; Gareis, M.; Gottschalk, C. Occurrence and Risk Assessment of Pyrrolizidine Alkaloids in Spices and Culinary Herbs from Various Geographical Origins. Toxins 2020, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Letsyo, E.; Adams, Z.S.; Dzikunoo, J.; Asante-Donyinah, D. Uptake and accumulation of pyrrolizidine alkaloids in the tissues of maize (Zea mays L.) plants from the soil of a 4-year-old Chromolaena odorata dominated fallow farmland. Chemosphere 2021, 270, 128669. [Google Scholar] [CrossRef]
- García-Juan, A.; León, N.; Armenta, S.; Pardo, O. Development and Validation of an Analytical Method for the Simultaneous Determination of 12 Ergot, 2 Tropane, and 28 Pyrrolizidine Alkaloids in Cereal-Based Food by LC-MS/MS. Food Res. Int. 2023, 174, 113614. [Google Scholar] [CrossRef]
- Bessaire, T.; Savoy, M.; Ernest, M.; Christinat, N.; Badoud, F.; Desmarchelier, A.; Carrères, B.; Chan, W.; Wang, X.; Delatour, T. Enhanced Surveillance of >1100 Pesticides and Natural Toxins in Food: Harnessing the Capabilities of LC-HRMS for Reliable Identification and Quantification. Foods 2024, 13, 3040. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bakery Products | Analytes | Sample Preparation | Analytical Technique | LOQ * | Recovery (%) | RSD * (%) | ME * (%) | Reference |
|---|---|---|---|---|---|---|---|---|
| Baking mixes | Morphine, codeine, papaverine and noscapine | SLE Sample: 10 g Extraction solvent: 30 mL methanol with 0.1% acetic acid | LC/MS/MS | 0.07–0.3 a mg/kg | N.S | 7.4–9.0 | N.S | [38] |
| Bread | Atropine, scopolamine and 15 other tropane alkaloids | SLE Sample: 3 g Extraction solvent: 30 mL methanol:water (2:1, v/v) with 0.5% acetic acid | LC-Orbitrap-MS | N.S | 75–101% | 1–13% | N.S | [39] |
| Muffin | Morphine, codeine and thebaine | SLE Sample: 15 g Extraction solvent: 45 mL acetonitrile/water (80/20; v/v) with 0.1% formic acid | UPLC−MS/MS | 0.6–2.3 pg | N.S | N.S | N.S | [40] |
| Muffins and poppy rolls (baking topping) | Morphine, codeine, thebaine, noscapine and papaverine | SLE Sample: 400 mg Extraction solvent: 1 mL chloroform: isopropanol (90:10, v/v) at pH 3.5 | HPLC−IT−MS | 10 ng/mL | N.S | 1.1–3.6 | N.S | [41] |
| Muffin batter | Noscapine | SLE Sample: 15 g Extraction solvent: 45 mL acetonitrile/water (80/20; v/v) with 0.1% formic acid | UPLC−MS/MS | 3.6 mg/kg | N.S | N.S | N.S | [42] |
| Gluten-Free Corn Breadsticks | Morphine, codeine, thebaine, papaverine, noscapine, atropine, scopolamine and anisodamine | SLE Sample: 2.5 g Extraction solvent: 20 mL of methanol with 0.1% acetic acid | HPLC-DAD | 1.01–8.16 mg/kg | 96–110 | 1–18 | 80–109 | [43] |
| Bakery products (bread, slice bread, bread toasts, breadsticks, crackers and biscuits) | 21 pyrrolizidine, 2 tropane and 6 opium alkaloids | SLE/SPE Sample: 0.5 g Extraction solvent: 5mL water with 1% HCl Purification: 150 mg Oasis MCX | UHPLC-IT-MS/MS | 0.38–5.43 µg/kg | 77–98 | 1–10 | −19–0 | [32] |
| Homemade gluten-free ground poppy seed crackers | Morphine, codeine, thebaine, papaverine, noscapine and oripavine | SLE/SPE Sample: 0.5 g Extraction solvent: 8 mL acidified water (pH 1.0 HCl) Purification: 50 mg SBA-15- SO3H-CN | UHPLC-TQ-MS/MS | 0.6–1.1 μg/kg for TAs 0.06–0.46 mg/kg for OAs | 79–107 | 2–17 | −80–15 | [44] |
| Bread | 21 pyrrolizidine, 2 tropane and 6 opium alkaloids | SLE/SPE Sample: 0.5 g Extraction solvent: 5 mL water with 0.2% formic acid Purification: 150 mg RH-SBA-15-SO3H-C18 | HPLC-IT-MS/MS | 0.38–5.43 µg/kg | 63–100 | 1–11 | −11–11 | This work |
| Analytes | Linear Range (µg/kg) | Matrix-Matched Calibration (R2) | Recovery (% ± SD) | Mean Recovery (% ± SD) | Repeatability Precision (RSD%) | Within-Laboratory Precision (RSD%) | MDL * (µg/kg) | MQL * (µg/kg) | ME (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low a | Medium b | High c | Low a | Medium b | High c | Low a | Medium b | High c | |||||||
| Atropine | 1.00–300 | y = 296,785x + 3106 (0.999) | 97 ± 1 | 100 ± 1 | 94 ± 2 | 97 ± 3 | 1 | 1 | 2 | 4 | 3 | 3 | 0.12 | 0.38 | 4 |
| Codeine | 2.00–3000 | y = 99,111x + 2106 (0.999) | 82 ± 8 | 85 ± 2 | 88 ± 7 | 85 ± 3 | 10 | 3 | 8 | 9 | 8 | 7 | 0.42 | 1.38 | −12 |
| Echimidine | 1.00–300 | y = 285,245x − 131,526 (0.999) | 94 ±2 | 83 ± 5 | 94 ± 4 | 90 ± 6 | 2 | 6 | 4 | 4 | 7 | 7 | 0.28 | 0.92 | 4 |
| Echimidine N-oxide | 2.00–300 | y = 454,369x − 17,066 (0.999) | 90 ± 4 | 91 ± 6 | 87 ± 6 | 90 ± 2 | 5 | 6 | 7 | 5 | 7 | 7 | 0.47 | 1.56 | −10 |
| Europine | 6.00–300 | y = 212,879x + 571,075 (0.999) | 86 ± 6 | 78 ± 4 | 82 ± 8 | 82 ± 4 | 7 | 6 | 9 | 9 | 5 | 10 | 1.63 | 5.43 | −12 |
| Europine N-oxide | 6.00–300 | y = 362,953x + 400,796 (0.999) | 98 ± 2 | 83 ± 2 | 82 ± 8 | 87 ± 9 | 2 | 2 | 10 | 5 | 8 | 8 | 1.56 | 5.20 | −3 |
| Heliotrine | 1.00–300 | y = 226,107x + 44,518 (0.999) | 87 ± 7 | 79 ± 7 | 98 ± 4 | 88 ± 9 | 8 | 9 | 4 | 8 | 6 | 5 | 0.26 | 0.87 | −2 |
| Heliotrine N-oxide | 1.00–300 | y = 280,627x + 33,815 (0.999) | 93 ± 2 | 88 ± 3 | 93 ± 7 | 91 ± 3 | 3 | 4 | 7 | 4 | 4 | 7 | 0.50 | 1.67 | 1 |
| Intermedine | 2.00–300 | y = 151,285x + 64,688 (0.999) | 87 ± 3 | 79 ± 6 | 89 ± 7 | 85 ± 5 | 3 | 8 | 8 | 2 | 10 | 7 | 0.52 | 1.72 | 11 |
| Intermedine N-oxide | 2.00–300 | y = 168,591x + 256,072 (0.999) | 93 ± 4 | 90 ± 8 | 88 ± 4 | 90 ± 3 | 5 | 9 | 5 | 5 | 8 | 6 | 0.52 | 1.73 | −5 |
| Lasiocarpine | 2.00–300 | y = 587,404x − 620,362 (0.999) | 94 ± 9 | 85 ± 7 | 95 ± 9 | 92 ± 6 | 10 | 8 | 9 | 4 | 4 | 8 | 0.37 | 1.22 | 4 |
| Lasiocarpine N-oxide | 2.00–300 | y = 586,066x − 123,946 (0.999) | 92 ± 3 | 93 ± 3 | 89 ± 5 | 91 ± 2 | 3 | 3 | 6 | 3 | 9 | 5 | 0.37 | 1.24 | −6 |
| Lycopsamine | 2.00–300 | y = 136,734x + 61,960 (0.999) | 96 ± 2 | 90 ± 5 | 89 ± 5 | 92 ± 4 | 2 | 6 | 6 | 5 | 9 | 6 | 0.43 | 1.44 | −7 |
| Lycopsamine N-oxide | 2.00–300 | y = 169,105x + 260,741 (0.999) | 93 ± 2 | 93 ± 4 | 90 ± 5 | 92 ± 2 | 2 | 5 | 6 | 3 | 10 | 5 | 0.39 | 1.30 | −9 |
| Morphine | 1.00–3000 | y = 79,799x + 459,065 (0.999) | 100 ± 4 | 73 ± 5 | 79 ± 3 | 84 ± 14 | 4 | 7 | 4 | 10 | 10 | 10 | 0.25 | 0.83 | −13 |
| Noscapine | 1.00–3000 | y = 5,666,667x – 5,668,012 (0.999) | 74 ± 4 | 74 ± 4 | 88 ± 9 | 79 ± 8 | 5 | 6 | 10 | 3 | 5 | 7 | 0.13 | 0.43 | −6 |
| Oripavine | 2.00–3000 | y = 25,775x + 33,223 (0.999) | 88 ± 7 | 85 ± 3 | 88 ± 3 | 87 ± 2 | 8 | 3 | 4 | 5 | 10 | 8 | 0.42 | 1.38 | 0 |
| Papaverine | 1.00–3000 | y = 894,000x + 609,973 (0.999) | 81 ± 4 | 77 ± 3 | 77 ± 4 | 78 ± 3 | 5 | 4 | 5 | 3 | 8 | 5 | 0.23 | 0.78 | 0 |
| Retrorsine | 1.00–300 | y = 145,873x + 54,022 (0.999) | 95 ± 5 | 77 ± 3 | 92 ± 3 | 88 ± 9 | 6 | 3 | 3 | 6 | 6 | 2 | 0.25 | 0.84 | −10 |
| Retrorsine N-oxide | 2.00–300 | y = 35,152x + 85,205 (0.999) | 92 ± 3 | 80 ± 4 | 91 ± 5 | 88 ± 7 | 4 | 6 | 5 | 4 | 9 | 6 | 0.52 | 1.73 | −4 |
| Scopolamine | 1.00–300 | y = 116,194x + 497,310 (0.999) | 99 ± 2 | 98 ± 3 | 90 ± 3 | 96 ± 5 | 2 | 3 | 3 | 2 | 1 | 4 | 0.18 | 0.59 | −9 |
| Senecionine | 2.00–300 | y = 349,644x − 400,100 (0.999) | 93 ± 1 | 89 ± 6 | 99 ± 2 | 94 ± 5 | 1 | 7 | 2 | 2 | 5 | 8 | 0.44 | 1.47 | −7 |
| Senecionine N-oxide | 2.00–300 | y = 51,515x + 39,822 (0.999) | 92 ± 6 | 76 ± 4 | 91 ± 4 | 87 ± 9 | 6 | 6 | 5 | 6 | 5 | 5 | 0.38 | 1.27 | −12 |
| Seneciphylline | 2.00–300 | y = 122,051x + 343,330 (0.999) | 82 ± 6 | 84 ± 7 | 93 ± 4 | 86 ± 6 | 8 | 9 | 4 | 7 | 5 | 3 | 0.37 | 1.24 | −2 |
| Seneciphylline N-oxide | 4.00–300 | y = 50,238x + 17,004 (0.999) | 85 ± 3 | 79 ± 6 | 93 ± 5 | 86 ± 7 | 4 | 7 | 6 | 6 | 7 | 7 | 1.04 | 3.46 | −9 |
| Senecivernine | 2.00–300 | y = 314,944x + 81,413 (0.999) | 94 ± 4 | 84 ± 6 | 90 ± 5 | 89 ± 5 | 4 | 7 | 5 | 6 | 5 | 5 | 0.37 | 1.23 | −3 |
| Senecivernine N-oxide | 2.00–300 | y = 64,911x − 7928 (0.999) | 84 ± 6 | 79 ± 6 | 89 ± 5 | 84 ± 5 | 7 | 8 | 6 | 7 | 8 | 6 | 0.43 | 1.44 | −5 |
| Senkirkine | 2.00–300 | y = 139,817x − 89,539 (1) | 91 ± 5 | 93 ± 7 | 92 ± 7 | 92 ± 1 | 5 | 7 | 7 | 10 | 8 | 8 | 0.39 | 1.31 | −6 |
| Thebaine | 2.00–3000 | y = 49,133x + 30,837 (0.999) | 81 ± 3 | 63 ± 7 | 67 ± 1 | 70 ± 10 | 3 | 11 | 2 | 5 | 9 | 5 | 0.52 | 1.74 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vera-Baquero, F.L.; Gañán, J.; Casado, N.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Application of Rice Husk-Derived SBA-15 Bifunctionalized with C18 and Sulfonic Groups for Solid-Phase Extraction of Tropane, Pyrrolizidine, and Opium Alkaloids in Gluten-Free Bread. Foods 2025, 14, 1156. https://doi.org/10.3390/foods14071156
Vera-Baquero FL, Gañán J, Casado N, Pérez-Quintanilla D, Morante-Zarcero S, Sierra I. Application of Rice Husk-Derived SBA-15 Bifunctionalized with C18 and Sulfonic Groups for Solid-Phase Extraction of Tropane, Pyrrolizidine, and Opium Alkaloids in Gluten-Free Bread. Foods. 2025; 14(7):1156. https://doi.org/10.3390/foods14071156
Chicago/Turabian StyleVera-Baquero, Fernando L., Judith Gañán, Natalia Casado, Damián Pérez-Quintanilla, Sonia Morante-Zarcero, and Isabel Sierra. 2025. "Application of Rice Husk-Derived SBA-15 Bifunctionalized with C18 and Sulfonic Groups for Solid-Phase Extraction of Tropane, Pyrrolizidine, and Opium Alkaloids in Gluten-Free Bread" Foods 14, no. 7: 1156. https://doi.org/10.3390/foods14071156
APA StyleVera-Baquero, F. L., Gañán, J., Casado, N., Pérez-Quintanilla, D., Morante-Zarcero, S., & Sierra, I. (2025). Application of Rice Husk-Derived SBA-15 Bifunctionalized with C18 and Sulfonic Groups for Solid-Phase Extraction of Tropane, Pyrrolizidine, and Opium Alkaloids in Gluten-Free Bread. Foods, 14(7), 1156. https://doi.org/10.3390/foods14071156