
Unveiling the Microbial Signatures of Arabica Coffee Cherries: Insights into Ripeness Specific Diversity, Functional Traits, and Implications for Quality and Safety





Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, Processing, and on Site Natural Fermentation
2.2. Shotgun Metagenomic Sequencing
2.2.1. DNA Extraction and Library Construction
2.2.2. Sequencing and Data Processing
2.2.3. Metagenome De Novo Assembly, Gene Prediction, and Functional Annotation
2.3. Taxonomic Annotation and Data Analysis
2.4. Visualization and Statistical Significance Tests
2.5. Physicochemical Parameters Estimation
3. Results and Discussion
3.1. Metagenome Sequencing General Data
3.2. Bacterial Community Composition
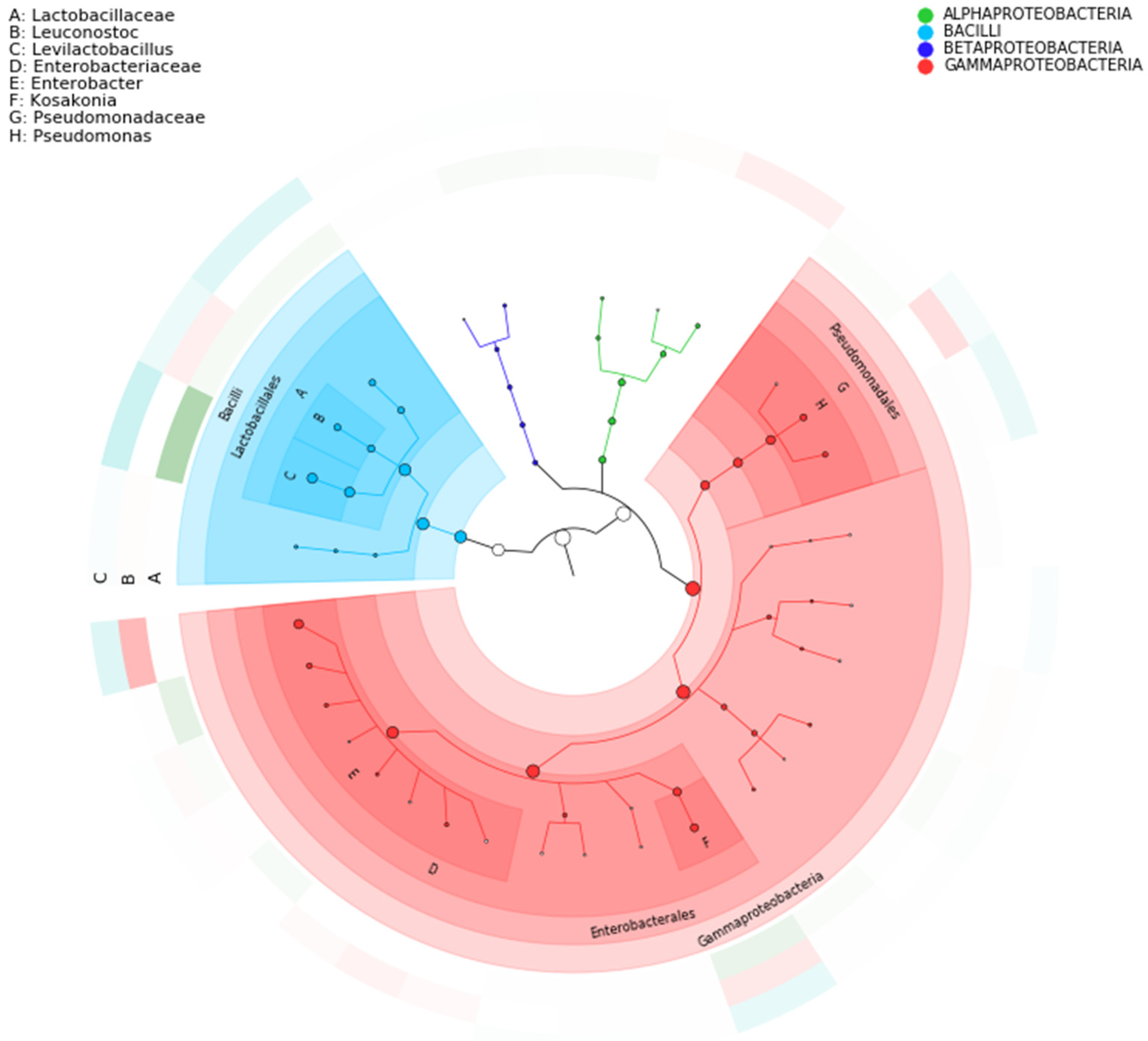
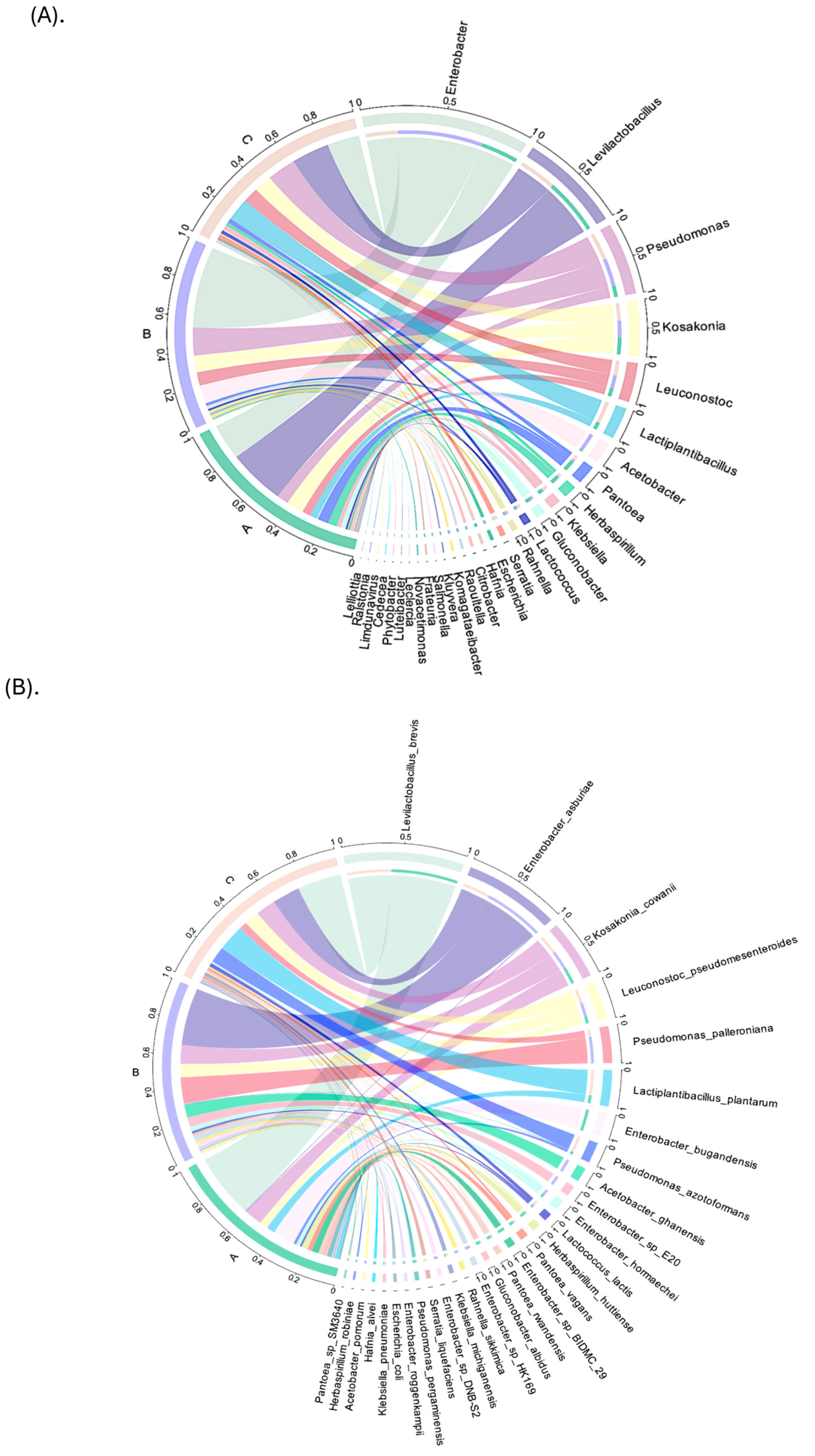
3.3. Taxonomic Annotation
3.4. Gene Prediction and Functional Analysis
3.5. EggNOG and COG Annotation Profile
3.6. KEGG Annotation Profile
3.7. CAZy and SwissProt Annotation Profile
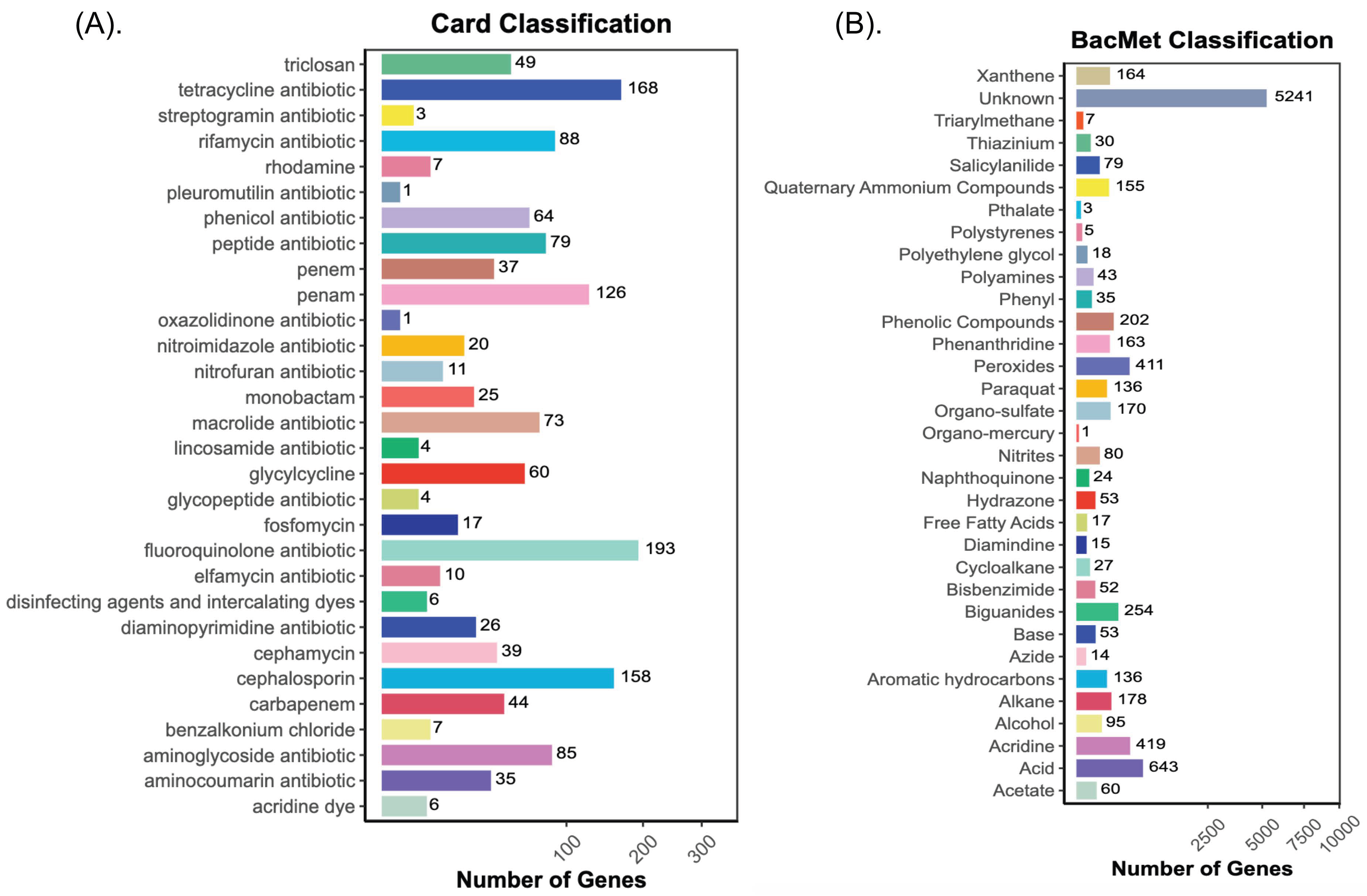
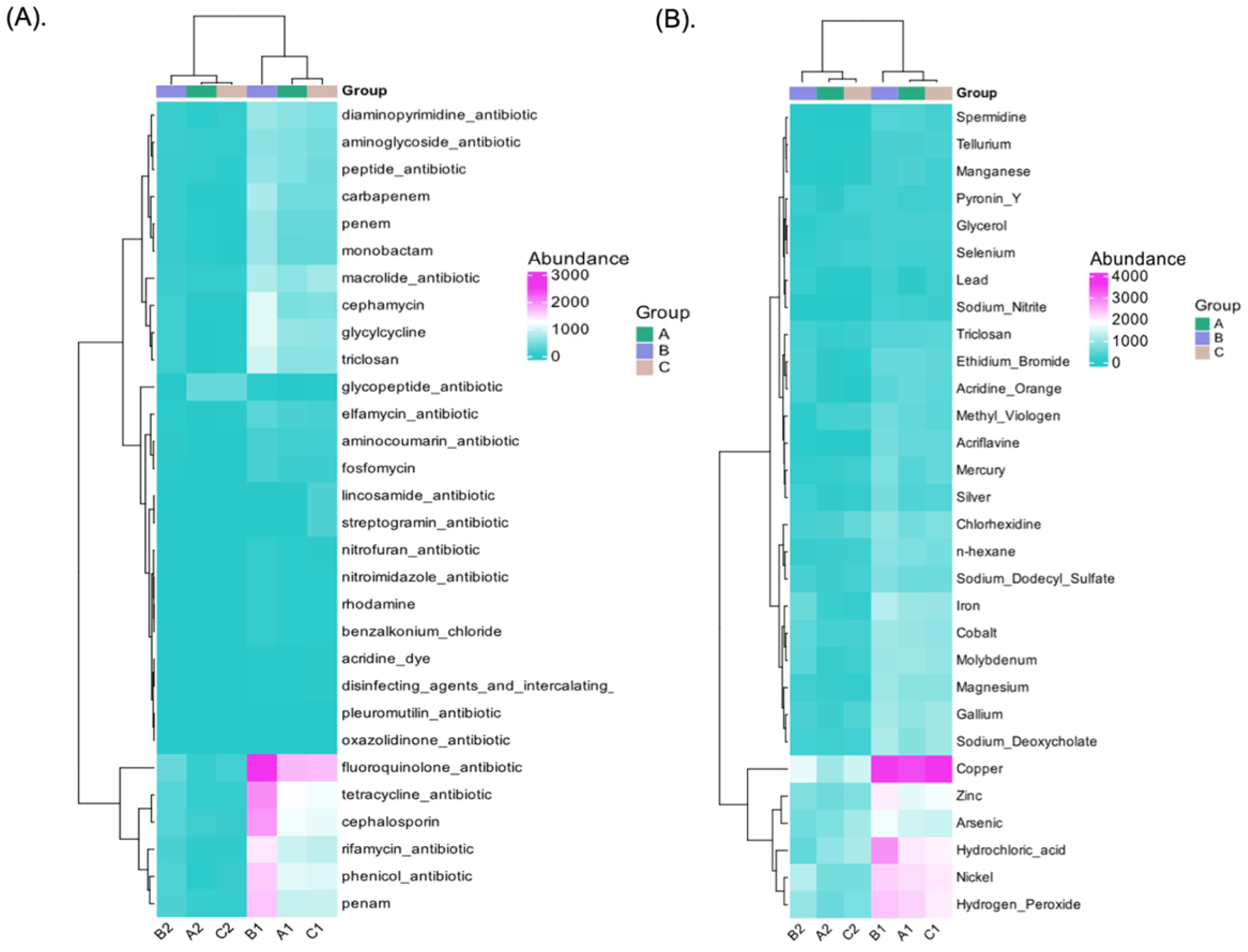
3.8. Antibiotic, Biocide, and Metal-Resistant Gene Annotation
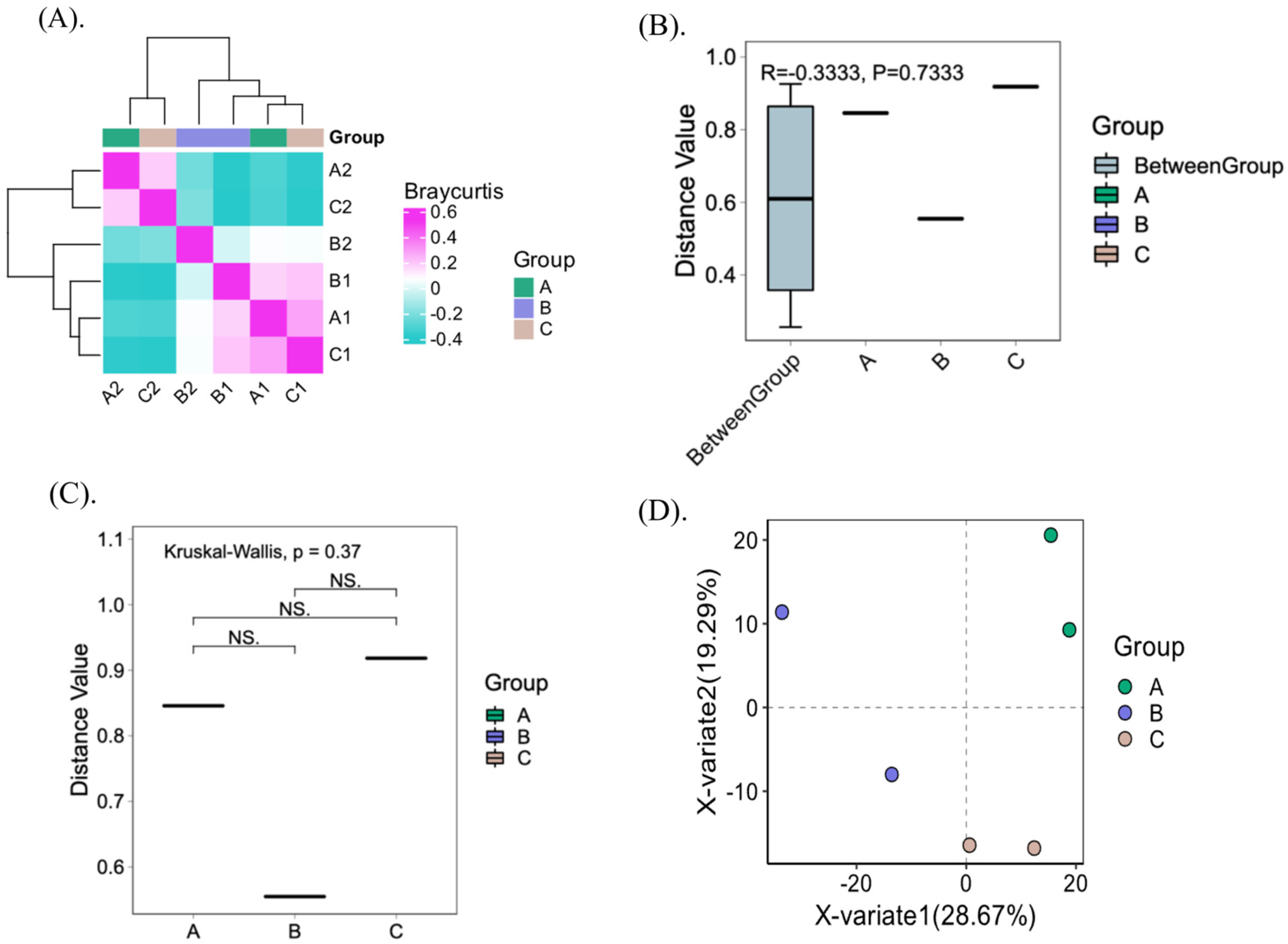
3.9. Physicochemical Properties and Microbiome Diversity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, B.; Furtado, A.; Henry, R.J. The coffee bean transcriptome explains the accumulation of the major bean components through ripening. Sci. Rep. 2018, 8, 11414. [Google Scholar] [CrossRef] [PubMed]
- Mihai, R.A.; Ortiz-Pillajo, D.C.; Iturralde-Proaño, K.M.; Vinueza-Pullotasig, M.Y.; Sisa-Tolagasí, L.A.; Villares-Ledesma, M.L.; Melo-Heras, E.J.; Cubi-Insuaste, N.S.; Catana, R.D. Comprehensive Assessment of Coffee Varieties (Coffea arabica L. Coffea canephora L.) from Coastal, Andean, and Amazonian Regions of Ecuador; A Holistic Evaluation of Metabolism, Antioxidant Capacity and Sensory Attributes. Horticulturae 2024, 10, 200. [Google Scholar] [CrossRef]
- De Beenhouwer, M.; Aerts, R.; Honnay, O. A global meta-analysis of the biodiversity and ecosystem service benefits of coffee and cacao agroforestry. Agric. Ecosyst. Environ. 2013, 175, 1–7. [Google Scholar] [CrossRef]
- The Cuenca Dispatch. 2024. Available online: https://thecuencadispatch.com/ecuadorian-coffee-production-is-in-decline-and-now-supplies-only-50-of-national-consumption/#google_vignette (accessed on 9 June 2024).
- Torres Castillo, N.E.; Melchor Martínez, E.M.; Ochoa Sierra, J.S.; Ramirez-Mendoza, R.A.; Parra Saldívar, R.; Iqba, H.M.N. Impact of climate change and early development of coffee rust: An overview of control strategies to preserve organic cultivars in Mexico. Sci. Total Environ. 2020, 738, 140–225. [Google Scholar] [CrossRef]
- Venegas Sánchez, S.; Orellana Bueno, D.; Pérez Jara, P. La realidad ecuatoriana en la producción de café. RECIMUNDO 2018, 2, 72–91. [Google Scholar] [CrossRef]
- Cheng, B.; Furtado, A.; Smyth, H.E.; Henry, R.J. Influence of genotype and environment on coffee quality. Trends Food Sci. Technol. 2016, 57 Pt A, 20–30. [Google Scholar] [CrossRef]
- Leong, K.H.; Chen, Y.S.; Pan, S.F.; Chen, J.J.; Wu, H.C.; Chang, Y.C.; Yanagida, F. Diversity of lactic acid bacteria associated with fresh coffee cherries in Taiwan. Curr. Microbiol. 2014, 68, 440–447. [Google Scholar] [CrossRef]
- Ivamoto, S.T.; Reis, O.; Domingues, D.S.; Dos Santos, T.B.; de Oliveira, F.F.; Pot, D.; Leroy, T.; Vieira, L.G.; Carazzolle, M.F.; Pereira, G.A.; et al. Transcriptome analysis of leaves, flowers and fruits perisperm of Coffea arabica L. reveals the differential expression of genes involved in raffinose biosynthesis. PLoS ONE 2017, 12, e0169595. [Google Scholar] [CrossRef]
- De Melo Pereira, G.V.; de Carvalho Neto, D.P.; Magalhães Júnior, A.I.; Vásquez, Z.S.; Medeiros, A.B.P.; Vandenberghe, L.P.S.; Soccol, C.R. Exploring the impacts of postharvest processing on the aroma formation of coffee beans—A review. Food Chem. 2019, 272, 441–452. [Google Scholar] [CrossRef]
- Krajangsang, S.; Seephin, P.; Tantayotai, P.; Mahingsapun, R.; Meeampun, Y.; Panyachanakul, T.; Samosorn, S.; Dolsophon, K.; Jiamjariyatam, R.; Lorliam, W.; et al. New approach for screening of microorganisms from Arabica coffee processing for their ability to improve Arabica coffee flavor. 3 Biotech 2022, 12, 143. [Google Scholar] [CrossRef]
- Todhanakasem, T.; Van Tai, N.; Pornpukdeewattana, S.; Charoenrat, T.; Young, B.M.; Wattanachaisaereekul, S. The relationship between microbial communities in coffee fermentation and aroma with metabolite attributes of finished products. Foods 2024, 13, 2332. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.N.; Do, H.D.K.; Loan Trinh, K.T.; Lee, N.Y. Metagenomics: An effective approach for exploring microbial diversity and functions. Foods 2023, 12, 2140. [Google Scholar] [CrossRef] [PubMed]
- Pothakos, V.; De Vuyst, L.; Zhang, S.J.; De Bruyn, F.; Verce, M.; Torres, J.; Callanan, M.; Moccand, C.; Weckx, S. Temporal shotgun metagenomics of an Ecuadorian coffee fermentation process highlights the predominance of lactic acid bacteria. Curr. Res. Biotechnol. 2020, 2, 1–15. [Google Scholar] [CrossRef]
- Haile, M.; Kang, W.H. The role of microbes in coffee fermentation and their impact on coffee quality. J. Food Qual. 2019, 2019, 4836709. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Freilich, S.; Bartuv, R.; Zhimo, V.Y.; Kumar, A.; Biasi, A.; Salim, S.; Feygenberg, O.; Burchard, E.; Dardick, C.; et al. Global analysis of the apple fruit microbiome: Are all apples the same? Environ. Microbiol. 2021, 23, 6038–6055. [Google Scholar] [CrossRef]
- Balcha, E.S.; Gómez, F.; Gemeda, M.T.; Bekele, F.B.; Abera, S.; Cavalazzi, B.; Woldesemayat, A.A. Shotgun metagenomics-guided prediction reveals the metal tolerance and antibiotic resistance of microbes in poly-extreme environments in the Danakil Depression, Afar Region. Antibiotics 2023, 12, 1697. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A mapreduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014, 42, D737–D743. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Poux, S.; Arighi, C.N.; Magrane, M.; Bateman, A.; Wei, C.H.; Lu, Z.; Boutet, E.; Bye-A-Jee, H.; Famiglietti, M.L.; Roechert, B.; et al. On expert curation and scalability: UniProtKB/Swiss-Prot as a case study. Bioinformatics 2017, 33, 3454–3460. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotecnol. 2021, 39, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. Peer J. Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Chao, A. Non-parametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Tenea, G.N.; Reyes, P. Bacterial community changes in strawberry fruits (Fragaria × ananassa variety “Monterey”) from farm field to retail market stands, an indicator of postharvest contamination. Front. Microbiol. 2024, 15, 1348316. [Google Scholar] [CrossRef]
- Stat, M.; Pochon, X.; Franklin, E.C.; Bruno, J.F.; Casey, K.S.; Selig, E.R.; Gates, R.D. The distribution of the thermally tolerant symbiont lineage (Symbiodinium clade D) in corals from Hawaii: Correlations with host and the history of ocean thermal stress. Ecol. Evol. 2013, 3, 1317–1329. [Google Scholar] [CrossRef]
- Whittaker, R.H. Vegetation of the Siskiyou Mountains, Oregon and California. Ecol. Monogr. 1960, 30, 279–338. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J.; Chen, D.G. Statistical Analysis of Microbiome Data with R; Springer Nature: Singapore, 2018; pp. XXIII, 505. [Google Scholar] [CrossRef]
- Association of Official Analytical Chemists (AOAC). Official Methods of Analysis of AOAC International, 17th ed.; AOAC: Gaithersburg, MD, USA, 2003. [Google Scholar]
- Tang, J.; Liu, Y.; Lin, B.; Zhu, H.; Jiang, W.; Yang, Q.; Chen, S. Effects of ultra-long fermentation time on the microbial community and flavor components of light-flavor Xiaoqu Baijiu based on fermentation tanks. World J. Microbiol. Biotechnol. 2021, 38, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, Y.; Tang, W.; Chen, H.; Gong, R. Insights into the Coloring Mechanism of Dark-Red and Yellow Fruits in Sweet Cherry through Transcriptome and Metabolome Analysis. Agronomy 2023, 13, 2397. [Google Scholar] [CrossRef]
- De Bruyn, F.; Zhang, S.J.; Pothakos, V.; Torres, J.; Lambot, C.; Moroni, A.V.; Callanan, M.; Sybesma, W.; Weckx, S.; De Vuyst, L. Exploring the Impacts of Postharvest Processing on the Microbiota and Metabolite Profiles during Green Coffee Bean Production. Appl. Environ. Microbiol. 2016, 83, e02398-16. [Google Scholar] [CrossRef]
- De Oliveira Junqueira, A.C.; de Melo Pereira, G.V.; Coral Medina, J.D.; Alvear, M.C.R.; Rosero, R.; de Carvalho Neto, D.P.; Enriquez, H.G.; Soccol, C.R. First description of bacterial and fungal communities in Colombian coffee beans fermentation analyzed using Illumina-based amplicon sequencing. Sci. Rep. 2019, 9, 8794. [Google Scholar] [CrossRef]
- Mahatmanto, T.; Sunarharum, W.B.; Putri, F.A.; Susanto, C.A.; Davian, A.O.; Murdiyatmo, U. The microbiology of arabica and robusta coffee cherries: A comparative study of indigenous bacteria with presumptive impact on coffee quality. FEMS Microbiol. Lett. 2023, 370, fnad024. [Google Scholar] [CrossRef]
- Zhang, Y.; Peng, S.; Ren, Y.; Yao, T.; Chu, H.; Gao, Y.; Tian, X. First report of Pseudomonas palleroniana causing potato soft rot in China. Plant Dis. 2022, 107, 553. [Google Scholar] [CrossRef]
- Cleenwerck, I.; Camu, N.; Engelbeen, K.; De Winter, T.; Vandemeulebroecke, K.; De Vos, P.; De Vuyst, L. Acetobacter ghanensis sp. nov.; a novel acetic acid bacterium isolated from traditional heap fermentations of Ghanaian cocoa beans. Int. J. System. Evol. Microbiol. 2007, 57 Pt 7, 1647–1652. [Google Scholar] [CrossRef]
- Evangelista, S.R.; Miguel, M.G.; Silva, C.F.; Pinheiro, A.C.; Schwan, R.F. Microbiological diversity associated with the spontaneous wet method of coffee fermentation. Int. J. Food Microbiol. 2015, 210, 102–112. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, B.; Zheng, T.; Zhao, C.; Shen, X.; Wang, X.; Qiu, M.; Fan, J. Integrating metabolomics and proteomics technologies provides insights into the flavor precursor changes at different maturity stages of Arabica coffee cherries. Foods 2023, 12, 1432. [Google Scholar] [CrossRef]
- Liang, T.; Jiang, T.; Liang, Z.; Zhang, N.; Dong, B.; Wu, Q.; Gu, B. Carbohydrate-active enzyme profiles of Lactiplantibacillus plantarum strain 84-3 contribute to flavor formation in fermented dairy and vegetable products. Food Chem. 2023, 20, 101036. [Google Scholar] [CrossRef]
- Szabó, M.; Kiss, J.; Olasz, F. Functional Organization of the Inverted Repeats of IS30. J. Bacteriol. 2010, 192, 3414–3423. [Google Scholar] [CrossRef] [PubMed]
- Sieradzki, E.T.; Nuccio, E.E.; Pett-Ridge, J.; Firestone, M.K. Expression of macromolecular organic nitrogen degrading enzymes identifies potential mediators of soil organic N availability to an annual grass. ISME J. 2023, 17, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.P.; Cu, X.; Xu, K.; Peng, J.H.; Liu, H.R.; Zhao, R.T.; Wang, Z.; Wang, T.; Xu, Z.S. The effect of glutathione biosynthesis of Streptococcus thermophilus ST-1 on cocultured Lactobacillus delbrueckii ssp. bulgaricus ATCC11842. J. Dairy Sci. 2023, 106, 884–896. [Google Scholar] [CrossRef]
- Neves, A.L.A.; Yu, J.; Suzuki, Y.; Baez-Magana, M.; Arutyunova, E.; O’Hara, E.; McAllister, T.; Ominski, K.H.; Lemieux, M.J.; Guan, L.L. Accelerated discovery of novel glycoside hydrolases using targeted functional profiling and selective pressure on the rumen microbiome. Microbiome 2021, 9, 229. [Google Scholar] [CrossRef]
- You, L.; Yang, C.; Jin, H.; Kwok, L.-Y.; Sun, Z.; Zhang, H. Metagenomic features of traditional fermented milk products. LWT-Food Sci. Technol. 2022, 155, 112945. [Google Scholar] [CrossRef]
- Tingley, J.P.; Low, K.E.; Xing, X.; Abbott, D.W. Combined whole cell wall analysis and streamlined in silico carbohydrate-active enzyme discovery to improve biocatalytic conversion of agricultural crop residues. Biotechnol. Biofuels 2021, 14, 16. [Google Scholar] [CrossRef]
- Avallone, S.; Brillouet, J.M.; Guyot, B.; Olguin, E.; Guiraud, J.P. Involvement of pectolytic micro-organisms in coffee fermentation. Int. J. Food Sci. Technol. 2002, 37, 191–198. [Google Scholar] [CrossRef]
- Waters, L.S.; Sandoval, M.; Storz, G. The Escherichia coli MntR miniregulon includes genes encoding a small protein and an efflux pump required for manganese homeostasis. J. Bacteriol. 2011, 193, 5887–5897. [Google Scholar] [CrossRef]
- O’Connell, K.P.; Gustafson, A.M.; Lehmann, M.D.; Thomashow, M.F. Identification of cold shock gene loci in Sinorhizobium meliloti by using a luxAB reporter transposon. Appl. Environ. Microbiol. 2000, 66, 401–405. [Google Scholar] [CrossRef]
- Zhang, D.; Peng, Y.; Chan, C.L.; On, H.; Wai, H.K.; Shekhawat, S.S.; Gupta, A.B.; Varshney, A.K.; Chuanchuen, R.; Zhou, X.; et al. Metagenomic survey reveals more diverse and abundant antibiotic resistance genes in municipal wastewater than hospital wastewater. Front. Microbiol. 2021, 12, 712843. [Google Scholar] [CrossRef]
- Dean, R.J.; Shimmield, T.M.; Black, K.D. Copper, zinc and cadmium in marine cage fish farm sediments: An extensive survey. Environ. Pollut. 2007, 145, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Tom-Petersen, A.; Nybroe, O. Copper amendment of agricultural soil selects for bacterial antibiotic resistance in the field. Lett. Appl. Microbiol. 2005, 40, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, Q.; Jin, M.; Huang, L.; Hui, D.; Sardans, J.; Peñuelas, J.; O’Connor, P.; Zhu, Y.; Yang, X.; et al. From grasslands to genes: Exploring the major microbial drivers of antibiotic-resistance in microhabitats under persistent overgrazing. Microbiome 2024, 12, 245. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coffee Variety/Group Annotation | Cherries Ripe Stage | Sample | Contig Number | Assembly Length (bp) | N50 (bp) | N90 (bp) | Max Length (bp) | Min Length (bp) | Average Length (bp) |
|---|---|---|---|---|---|---|---|---|---|
| C. arabica L. var. Typica/group A | immature green | A1 | 66,231 | 138,086,378 | 9238 | 621 | 981,927 | 300 | 2084 |
| mature red | A2 | 53,051 | 100,746,497 | 7152 | 590 | 399,572 | 300 | 1899 | |
| C. arabica L. var. Yellow Caturra/group B | immature green | B1 | 28,951 | 69,111,399 | 6264 | 855 | 825,737 | 300 | 2387 |
| mature yellow | B2 | 62,057 | 90,049,394 | 3197 | 485 | 896,691 | 300 | 1451 | |
| C. arabica L. var. Red Caturra/group C | immature green | C1 | 29,237 | 104,006,429 | 21,010 | 1197 | 1,033,477 | 300 | 3557 |
| mature red | C2 | 61,270 | 83,508,992 | 4249 | 411 | 723,331 | 300 | 1362 |
| Coffee Variety/Group | Cherries Ripe | Sample | Kingdom | Phylum | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|---|---|---|
| C. arabica L. var. Typica/group A | immature green | A1 | 4 | 31 | 62 | 139 | 301 | 921 | 3407 |
| immature red | A2 | 4 | 32 | 79 | 161 | 341 | 982 | 2910 | |
| C. arabica L. var. Yellow Caturra/group B | immature green | B1 | 4 | 32 | 56 | 110 | 227 | 591 | 2097 |
| immature yellow | B2 | 4 | 39 | 76 | 157 | 324 | 902 | 2855 | |
| C. arabica L. var. Red Caturra/group C | immature green | C1 | 4 | 32 | 67 | 145 | 296 | 936 | 3550 |
| immature red | C2 | 4 | 25 | 45 | 92 | 198 | 555 | 1870 |
| Parameter | TSS | pH Cherries Juice | Acidity Cherries Juice (%) | pH Fermented Cherries | Shannon Diversity |
|---|---|---|---|---|---|
| TSS | 1 | −0.216 | 0.756 | −0.942 | −0.435 |
| pH Cherries Juice | −0.216 | 1 | 0.171 | 0.082 | −0.183 |
| Acidity Cherries Juice (%) | 0.756 | 0.171 | 1 | −0.853 | −0.366 |
| pH Fermented Cherries | −0.942 | 0.082 | −0.853 | 1 | 0.623 |
| Shannon Diversity | −0.435 | −0.183 | −0.366 | 0.623 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tenea, G.N.; Cifuentes, V.; Reyes, P.; Cevallos-Vallejos, M. Unveiling the Microbial Signatures of Arabica Coffee Cherries: Insights into Ripeness Specific Diversity, Functional Traits, and Implications for Quality and Safety. Foods 2025, 14, 614. https://doi.org/10.3390/foods14040614
Tenea GN, Cifuentes V, Reyes P, Cevallos-Vallejos M. Unveiling the Microbial Signatures of Arabica Coffee Cherries: Insights into Ripeness Specific Diversity, Functional Traits, and Implications for Quality and Safety. Foods. 2025; 14(4):614. https://doi.org/10.3390/foods14040614
Chicago/Turabian StyleTenea, Gabriela N., Victor Cifuentes, Pamela Reyes, and Marcelo Cevallos-Vallejos. 2025. "Unveiling the Microbial Signatures of Arabica Coffee Cherries: Insights into Ripeness Specific Diversity, Functional Traits, and Implications for Quality and Safety" Foods 14, no. 4: 614. https://doi.org/10.3390/foods14040614
APA StyleTenea, G. N., Cifuentes, V., Reyes, P., & Cevallos-Vallejos, M. (2025). Unveiling the Microbial Signatures of Arabica Coffee Cherries: Insights into Ripeness Specific Diversity, Functional Traits, and Implications for Quality and Safety. Foods, 14(4), 614. https://doi.org/10.3390/foods14040614