


The Microbial Diversity and Traceability Analysis of Raw Milk from Buffalo Farms at Different Management Ranks in Guangxi Province

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. 16S rRNA Gene Sequencing
3. Results
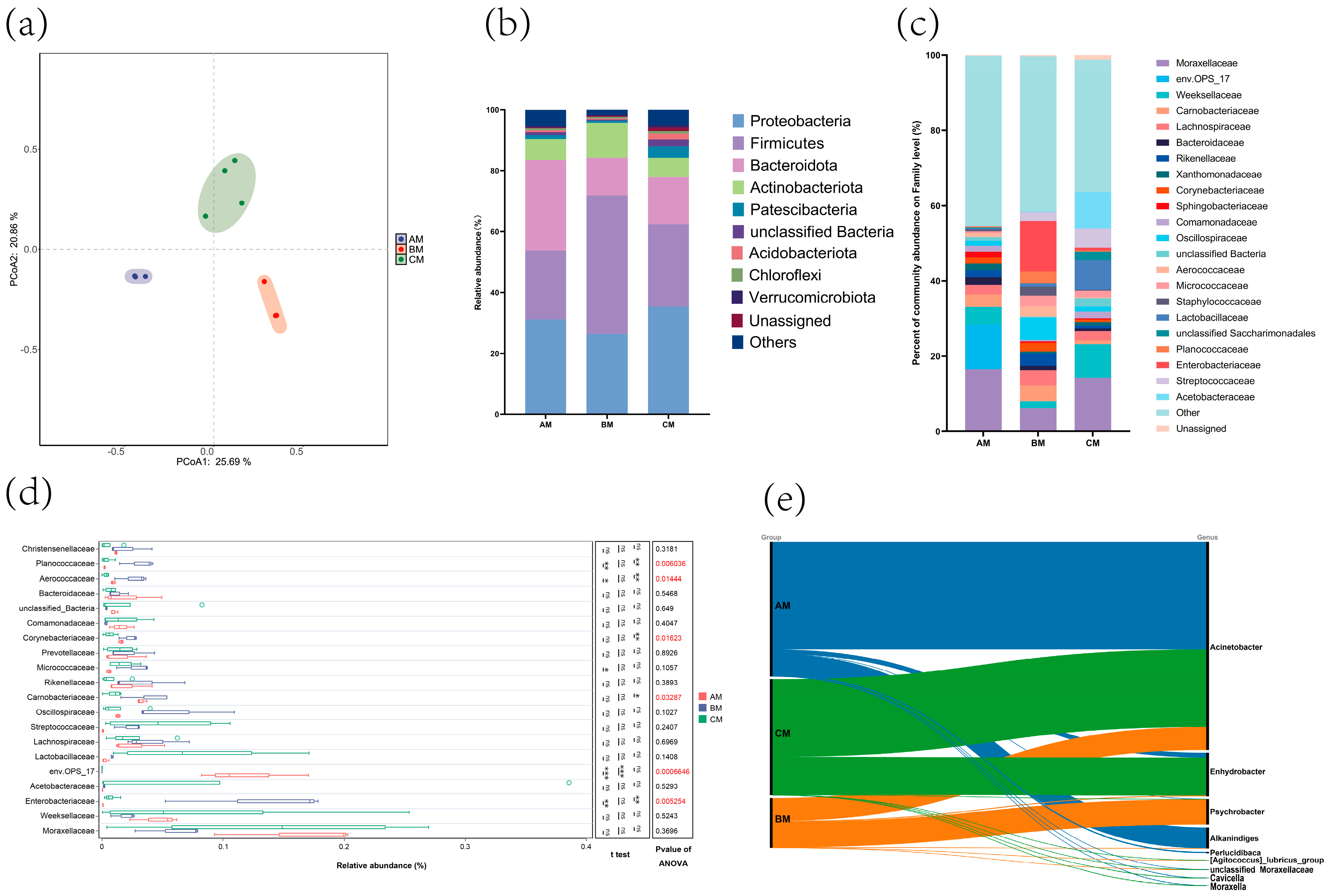
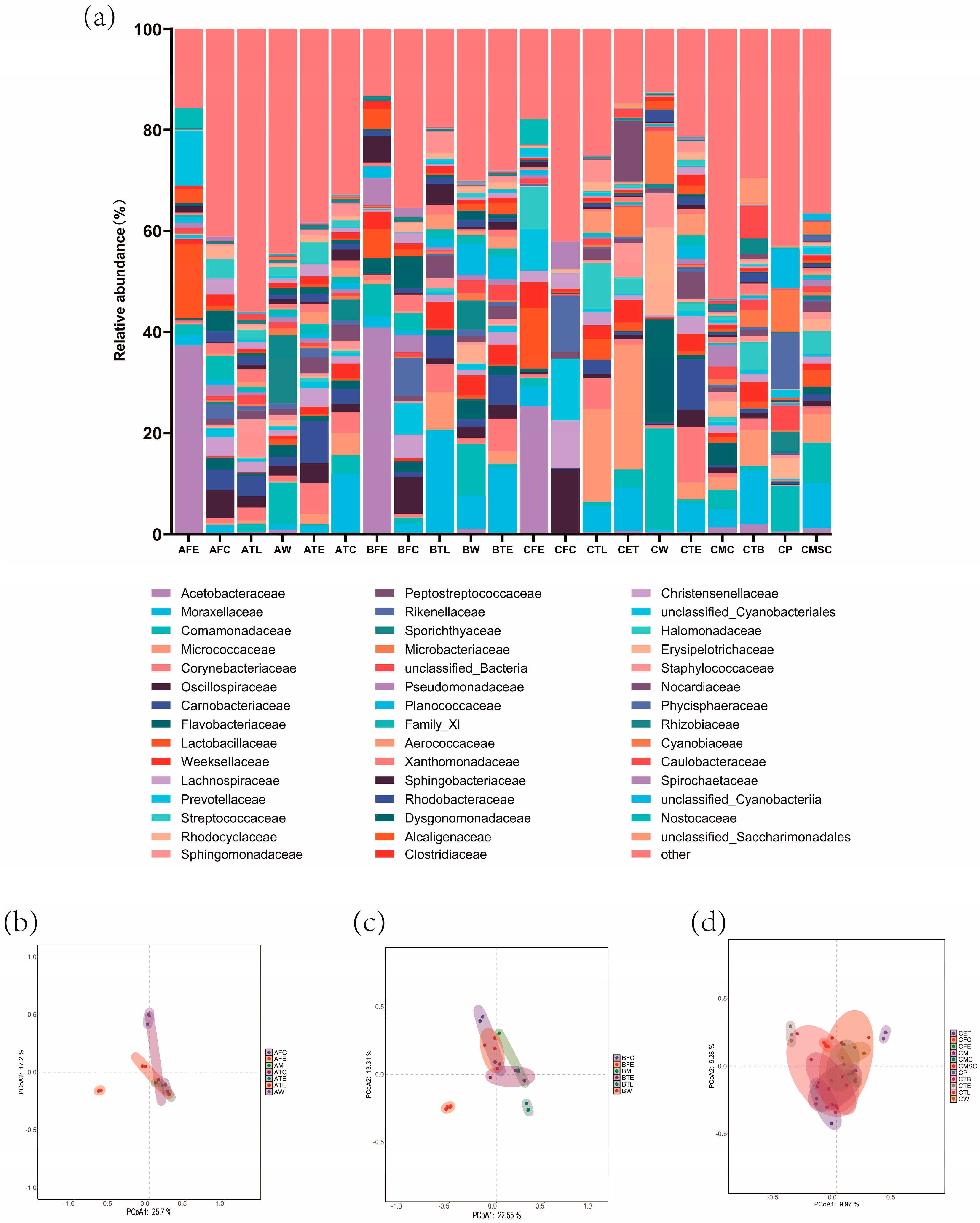
3.1. Effect of Management Rank on the Microbial Composition of Canned Raw Milk from Buffalo Farms
3.2. Analysis of Environmental Microbial Hazards in Buffalo Farms at Different Ranks of Management
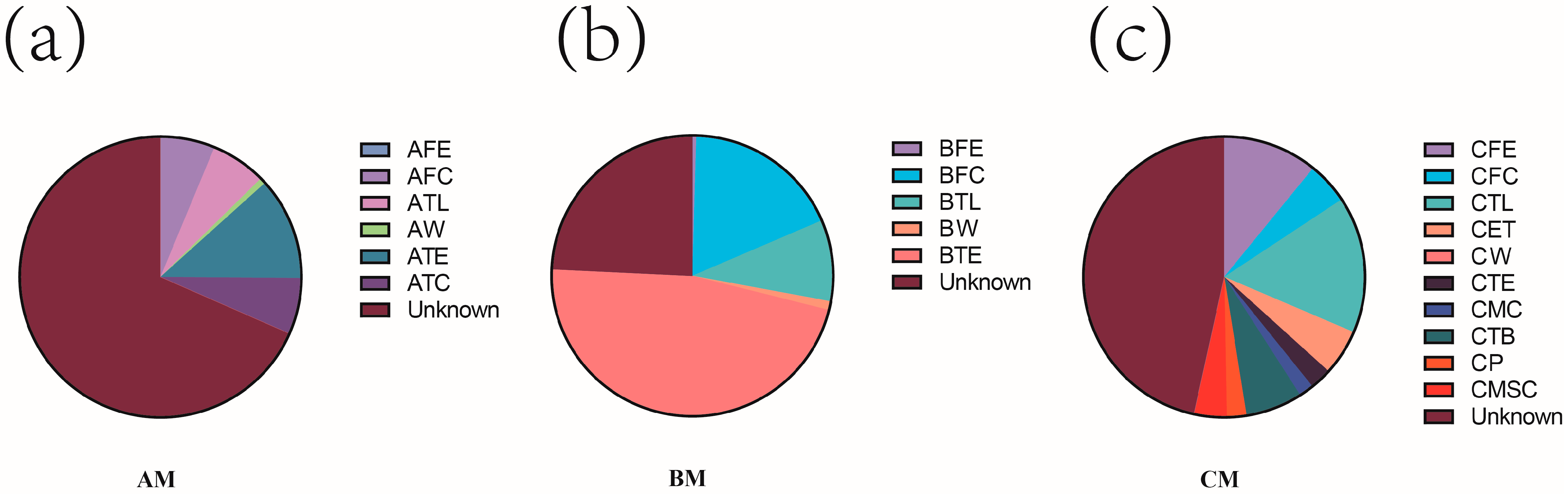
3.3. Further Assessment of the Contribution of Environmental Sample Sources to the Prediction of the Microbiota of Canned Raw Buffalo Milk Based on the FEAST Traceability Model
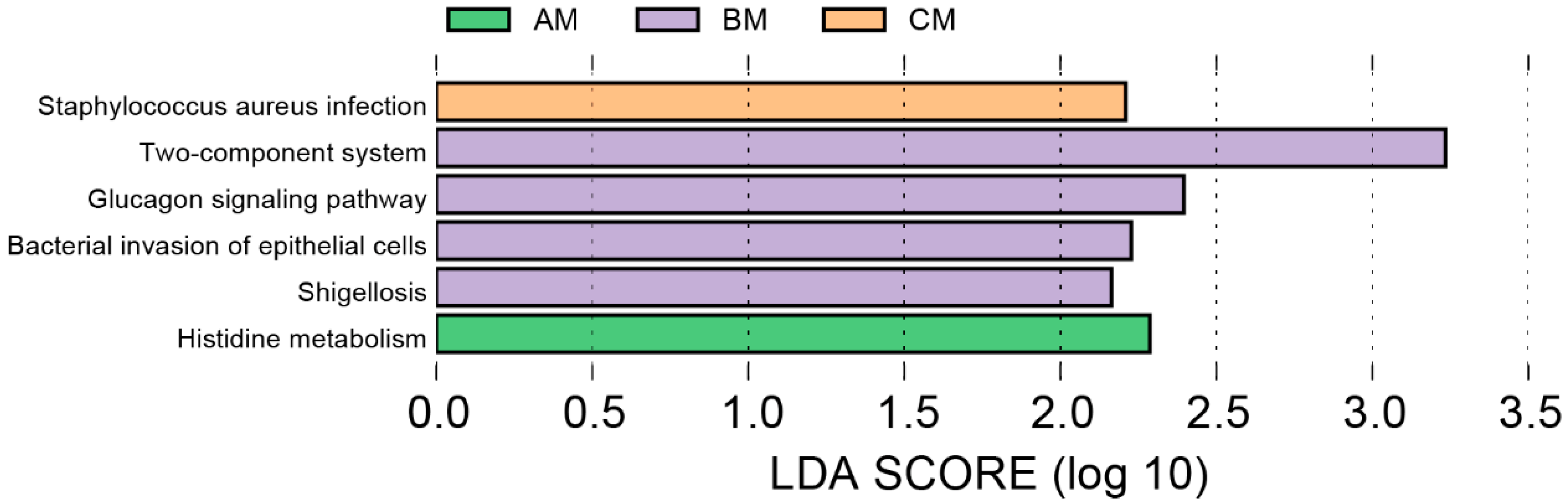
3.4. Functional Differences of Microbiome in Buffalo Milk at Different Ranks of Management
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vargas-Ramella, M.; Pateiro, M.; Maggiolino, A.; Faccia, M.; Franco, D.; De Palo, P.; Lorenzo, J.M. Buffalo Milk as a Source of Probiotic Functional Products. Microorganisms 2021, 9, 2303. [Google Scholar] [CrossRef] [PubMed]
- Mejares, C.T.; Huppertz, T.; Chandrapala, J. Thermal processing of buffalo milk—A review. Int. Dairy J. 2022, 129, 105311. [Google Scholar] [CrossRef]
- Hau, E.H.; Li, L.; Mah, S.H. Bioactive peptides from livestock milk and casein as alternative functional foods. Food Saf. Health 2024, 1–5. [Google Scholar] [CrossRef]
- Albenzio, M.; Santillo, A.; Caroprese, M.; Marino, R.; Centoducati, P.; Sevi, A. Effect of different ventilation regimens on ewes’ milk and Canestrato Pugliese cheese quality in summer. J. Dairy Res. 2005, 72, 447–455. [Google Scholar] [CrossRef]
- Huijps, K.; Hogeveen, H.; Lam, T.J.G.M.; Oude Lansink, A.G.J.M. Costs and efficacy of management measures to improve udder health on Dutch dairy farms. J. Dairy Sci. 2010, 93, 115–124. [Google Scholar] [CrossRef]
- Quintana, Á.R.; Seseña, S.; Garzón, A.; Arias, R. Factors Affecting Levels of Airborne Bacteria in Dairy Farms: A Review. Animals 2020, 10, 526. [Google Scholar] [CrossRef]
- Fusco, V.; Chieffi, D.; Fanelli, F.; Logrieco, A.F.; Cho, G.-S.; Kabisch, J.; Böhnlein, C.; Franz, C.M.A.P. Microbial quality and safety of milk and milk products in the 21st century. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2013–2049. [Google Scholar] [CrossRef]
- Vissers, M.; Driehuis, F. On-farm hygienic milk production. In Milk Processing and Quality Management; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2008; pp. 1–22. [Google Scholar]
- Du, B.; Meng, L.; Liu, H.; Zheng, N.; Zhang, Y.; Guo, X.; Zhao, S.; Li, F.; Wang, J. Impacts of milking and housing environment on milk microbiota. Animals 2020, 10, 2339. [Google Scholar] [CrossRef]
- Parente, E.; Ricciardi, A.; Zotta, T. The microbiota of dairy milk: A review. Int. Dairy J. 2020, 107, 104714. [Google Scholar] [CrossRef]
- Billington, C.; Kingsbury, J.M.; Rivas, L. Metagenomics Approaches for Improving Food Safety: A Review. J. Food Prot. 2022, 85, 448–464. [Google Scholar] [CrossRef]
- Chen, H.; Bai, X.; Li, Y.; Jing, L.; Chen, R.; Teng, Y. Source identification of antibiotic resistance genes in a peri-urban river using novel crAssphage marker genes and metagenomic signatures. Water Res. 2019, 167, 115098. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.; Rivera, B.; Gerba, C.P. Chapter 14—Microbial Source Tracking. In Environmental Microbiology, 3rd ed.; Pepper, I.L., Gerba, C.P., Gentry, T.J., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 309–317. [Google Scholar] [CrossRef]
- Li, L.G.; Yin, X.; Zhang, T. Tracking antibiotic resistance gene pollution from different sources using machine-learning classification. Microbiome 2018, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Yin, G.; Liu, M.; Chen, C.; Jiang, Y.; Hou, L.; Zheng, Y. A systematic review of antibiotics and antibiotic resistance genes in estuarine and coastal environments. Sci. Total Environ. 2021, 777, 146009. [Google Scholar] [CrossRef] [PubMed]
- Shenhav, L.; Thompson, M.; Joseph, T.A.; Briscoe, L.; Furman, O.; Bogumil, D.; Mizrahi, I.; Pe’er, I.; Halperin, E. FEAST: Fast expectation-maximization for microbial source tracking. Nat. Methods 2019, 16, 627–632. [Google Scholar] [CrossRef]
- Li, L.; Miao, W.; Li, Z.; Huang, L.; Hau, E.; Khan, M.F.; Liu, Q.; Zeng, Q.; Cui, K. Meta-Genomic Analysis of Different Bacteria and Their Genomes Found in Raw Buffalo Milk Obtained in Various Farms Using Different Milking Methods. Genes 2024, 15, 1081. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Y.X.; Li, X. USEARCH 12: Open-source software for sequencing analysis in bioinformatics and microbiome. iMeta 2024, 3, e236. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Khleborodova, A.; Gamboa-Tuz, S.D.; Ramos, M.; Segata, N.; Waldron, L.; Oh, S. Lefser: Implementation of metagenomic biomarker discovery tool, LEfSe, in R. Bioinformatics 2024, btae707. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- CRecommended Mastitis Control Program. National Mastitis Council: Madison, WI, USA, 2016; pp. 1–2. Available online: https://www.nmconline.org/wp-content/uploads/2016/08/RECOMMENDED-MASTITIS-CONTROL-PROGRAM-International.pdf (accessed on 16 December 2024).
- Napolitano, F.; De Rosa, G.; Chay-Canul, A.; Álvarez-Macías, A.; Pereira, A.M.F.; Bragaglio, A.; Mora-Medina, P.; Rodríguez-González, D.; García-Herrera, R.; Hernández-Ávalos, I.; et al. The Challenge of Global Warming in Water Buffalo Farming: Physiological and Behavioral Aspects and Strategies to Face Heat Stress. Animals 2023, 13, 3103. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG database. In Proceedings of the ‘In Silico’ Simulation of Biological Processes: Novartis Foundation Symposium 247; Novartis Foundation: Basel, Switzerland, 2002; pp. 91–103. [Google Scholar]
- Cheng, L.; Wei, M.; Guo, G.; Hu, Q.; Li, B.; Jiang, Y.; Hu, Z. Effects of feeding mode on the formation and stability of aerobic granular sludge under combined antibiotic stress. Chem. Eng. J. 2023, 475, 145996. [Google Scholar] [CrossRef]
- Rakitin, A.L.; Begmatov, S.; Beletsky, A.V.; Philippov, D.A.; Kadnikov, V.V.; Mardanov, A.V.; Dedysh, S.N.; Ravin, N.V. Highly Distinct Microbial Communities in Elevated Strings and Submerged Flarks in the Boreal Aapa-Type Mire. Microorganisms 2022, 10, 170. [Google Scholar] [CrossRef]
- Saeed, M.M. INFLUENCE OF MILK AND RAW MILK AS CARRIERS IN DISEASE TRANSMISSION BETWEEN HUMANS AND ANIMALS. Eur. Sci. Methodical J. 2024, 2, 15–35. [Google Scholar]
- Paramasivam, R.; Gopal, D.R.; Dhandapani, R.; Subbarayalu, R.; Elangovan, M.P.; Prabhu, B.; Veerappan, V.; Nandheeswaran, A.; Paramasivam, S.; Muthupandian, S. Is AMR in Dairy Products a Threat to Human Health? An Updated Review on the Origin, Prevention, Treatment, and Economic Impacts of Subclinical Mastitis. Infect. Drug Resist. 2023, 16, 155–178. [Google Scholar] [CrossRef]
- Mladenović, K.G.; Grujović, M.Ž.; Kiš, M.; Furmeg, S.; Tkalec, V.J.; Stefanović, O.D.; Kocić-Tanackov, S.D. Enterobacteriaceae in food safety with an emphasis on raw milk and meat. Appl. Microbiol. Biotechnol. 2021, 105, 8615–8627. [Google Scholar] [CrossRef]
- AoDaohu; Karin, A.; Kiiru, G.P.; Naoki, N. Variations in milk, udder skin, and fecal microbiota and their relationships with blood metabolites and milk composition in dairy cows. Lett. Appl. Microbiol. 2024, 77, ovae014. [Google Scholar] [CrossRef]
- Wu, H.; Nguyen, Q.D.; Tran, T.T.M.; Tang, M.T.; Tsuruta, T.; Nishino, N. Rumen fluid, feces, milk, water, feed, airborne dust, and bedding microbiota in dairy farms managed by automatic milking systems. Anim. Sci. J. 2019, 90, 445–452. [Google Scholar] [CrossRef]
- Luziatelli, F.; Melini, F.; Ficca, A.G.; Melini, V.; Nardilli, F.; Ruzzi, M. Core microbiome and bacterial diversity of the Italian Mediterranean river buffalo milk. Appl. Microbiol. Biotechnol. 2023, 107, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Wu, H.; Nishino, N. An investigation of seasonal variations in the microbiota of milk, feces, bedding, and airborne dust. Asian-Australas. J. Anim. Sci. 2020, 33, 1858–1865. [Google Scholar] [CrossRef] [PubMed]
- Nisaa, A.A.; Oon, C.E.; Sreenivasan, S.; Balakrishnan, V.; Rajendran, D.; Tan, J.J.; Roslan, F.F.; Todorov, S.D.; Jeong, W.S.; Zhao, F.; et al. Vaginal Infections during Pregnancy Increase Breast Milk Microbiome Alpha Diversity and Alter Taxonomic Composition. Prev. Nutr. Food Sci. 2023, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, Z.; Avillan, J.J.; Call, D.R.; Davis, M.; Sischo, W.M.; Zhang, A. Dairy farm soil presents distinct microbiota and varied prevalence of antibiotic resistance across housing areas. Environ. Pollut. 2019, 254, 113058. [Google Scholar] [CrossRef]
- Yuan, L.; Sadiq, F.A.; Liu, T.-j.; Li, Y.; Gu, J.-s.; Yang, H.-y.; He, G.-q. Spoilage potential of psychrotrophic bacteria isolated from raw milk and the thermo-stability of their enzymes. J. Zhejiang Univ. Sci. B 2018, 19, 630. [Google Scholar] [CrossRef]
- Grace, D.; Wu, F.; Havelaar, A.H. MILK Symposium review: Foodborne diseases from milk and milk products in developing countries—Review of causes and health and economic implications*. J. Dairy Sci. 2020, 103, 9715–9729. [Google Scholar] [CrossRef]
- Mohammad, A.-M.; Chowdhury, T.; Biswas, B.; Absar, N. Food poisoning and intoxication: A global leading concern for human health. In Food Safety and Preservation; Elsevier: Amsterdam, The Netherlands, 2018; pp. 307–352. [Google Scholar]
- Kerro Dego, O.; Vidlund, J. Staphylococcal mastitis in dairy cows. Front. Vet. Sci. 2024, 11, 1356259. [Google Scholar] [CrossRef]
- Ózsvári, L.; Ivanyos, D. The use of teat disinfectants and milking machine cleaning products in commercial Holstein-Friesian farms. Front. Vet. Sci. 2022, 9, 956843. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | AM | BM | CM | p Value |
|---|---|---|---|---|
| ASVs | 1528.3333 ± 185.4730 | 1782.3333 ± 123.8287 | 1859.2500 ± 1400.1788 | 0.918 |
| ACE | 1531.0378 ± 185.1027 | 1788.7559 ± 123.6023 | 1862.6068 ± 1398.7170 | 0.918 |
| Chao1 | 1529.3634 ± 185.5027 | 1784.2837 ± 123.4209 | 1860.0646 ± 1399.7299 | 0.918 |
| Simpson | 0.9761 ± 0.0107 | 0.9753 ± 0.0135 | 0.9408 ± 0.0575 | 0.518 |
| Shannon | 7.9736 ± 0.4333 | 8.4521 ± 0.5869 | 7.2540 ± 2.1636 | 0.661 |
| Good coverage | 0.9998 ± 0.000 | 0.9997 ± 0.0000 | 0.9998 ± 0.0001 | 0.098 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, W.; Wang, D.; Li, L.; Hau, E.; Zhang, J.; Shi, Z.; Huang, L.; Zeng, Q.; Cui, K. The Microbial Diversity and Traceability Analysis of Raw Milk from Buffalo Farms at Different Management Ranks in Guangxi Province. Foods 2024, 13, 4080. https://doi.org/10.3390/foods13244080
Miao W, Wang D, Li L, Hau E, Zhang J, Shi Z, Huang L, Zeng Q, Cui K. The Microbial Diversity and Traceability Analysis of Raw Milk from Buffalo Farms at Different Management Ranks in Guangxi Province. Foods. 2024; 13(24):4080. https://doi.org/10.3390/foods13244080
Chicago/Turabian StyleMiao, Wenhao, Dong Wang, Ling Li, Enghuan Hau, Jiaping Zhang, Zongce Shi, Li Huang, Qingkun Zeng, and Kuiqing Cui. 2024. "The Microbial Diversity and Traceability Analysis of Raw Milk from Buffalo Farms at Different Management Ranks in Guangxi Province" Foods 13, no. 24: 4080. https://doi.org/10.3390/foods13244080
APA StyleMiao, W., Wang, D., Li, L., Hau, E., Zhang, J., Shi, Z., Huang, L., Zeng, Q., & Cui, K. (2024). The Microbial Diversity and Traceability Analysis of Raw Milk from Buffalo Farms at Different Management Ranks in Guangxi Province. Foods, 13(24), 4080. https://doi.org/10.3390/foods13244080