
Optimization, Validation, and Application of Cleanup-Coupled Liquid Chromatography–Tandem Mass Spectrometry for the Simultaneous Analyses of 35 Mycotoxins and Their Derivatives in Cereals


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Standard Solution Preparation
2.3. Blank Matrix and Sample Collection
2.4. UPLC-MS/MS Analysis
2.5. Sample Preparation
2.5.1. Extraction Procedure (Step 1)
2.5.2. Sample Cleanup with Dispersive SPE Sorbent (Step 2)
2.6. Method Validation
2.7. Measurement Uncertainty
2.8. Stability
2.9. Application to Real Samples
2.10. Statistical Analysis
3. Results and Discussion
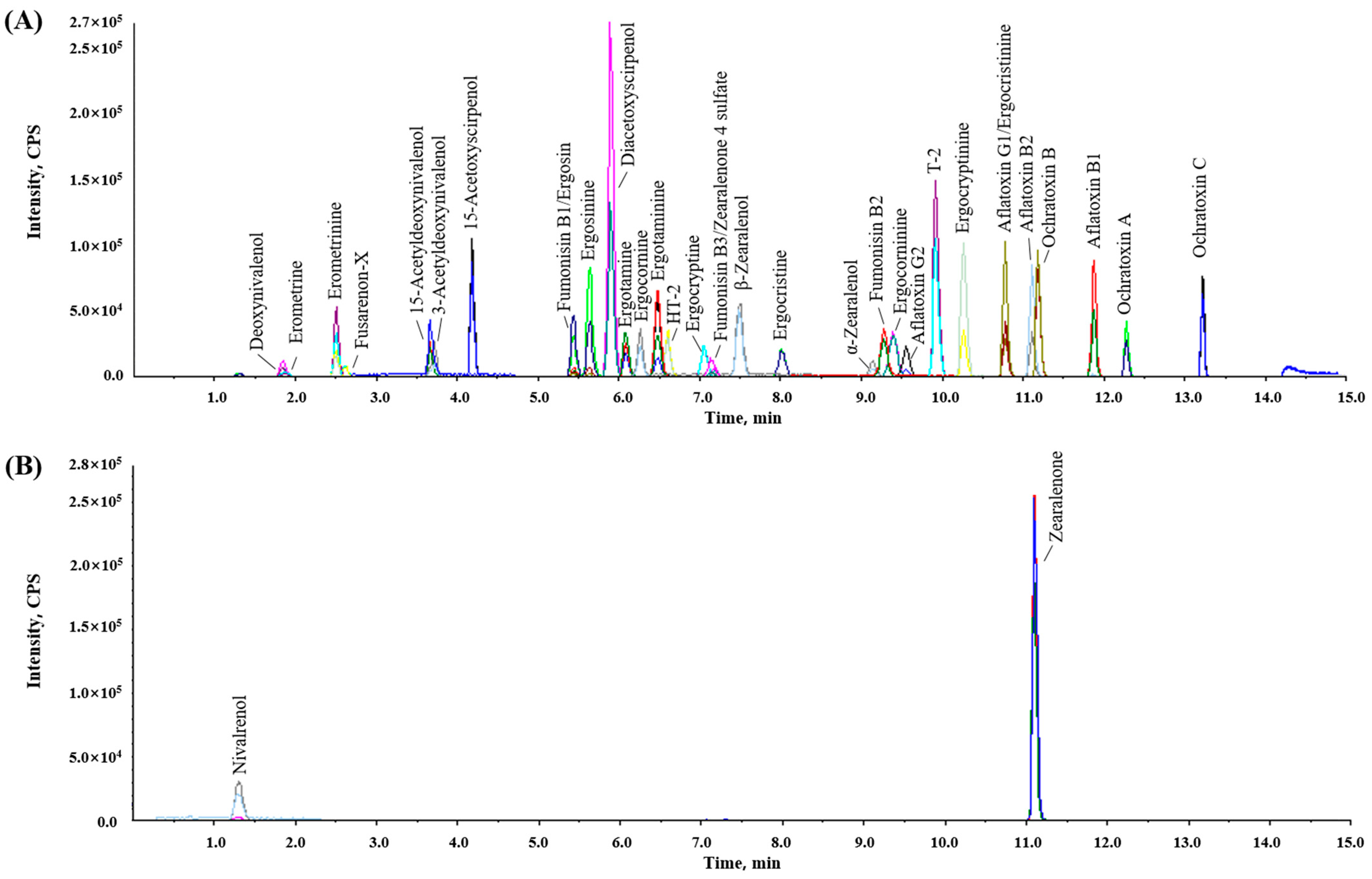
3.1. Optimization of UPLC–MS/MS Analysis Conditions
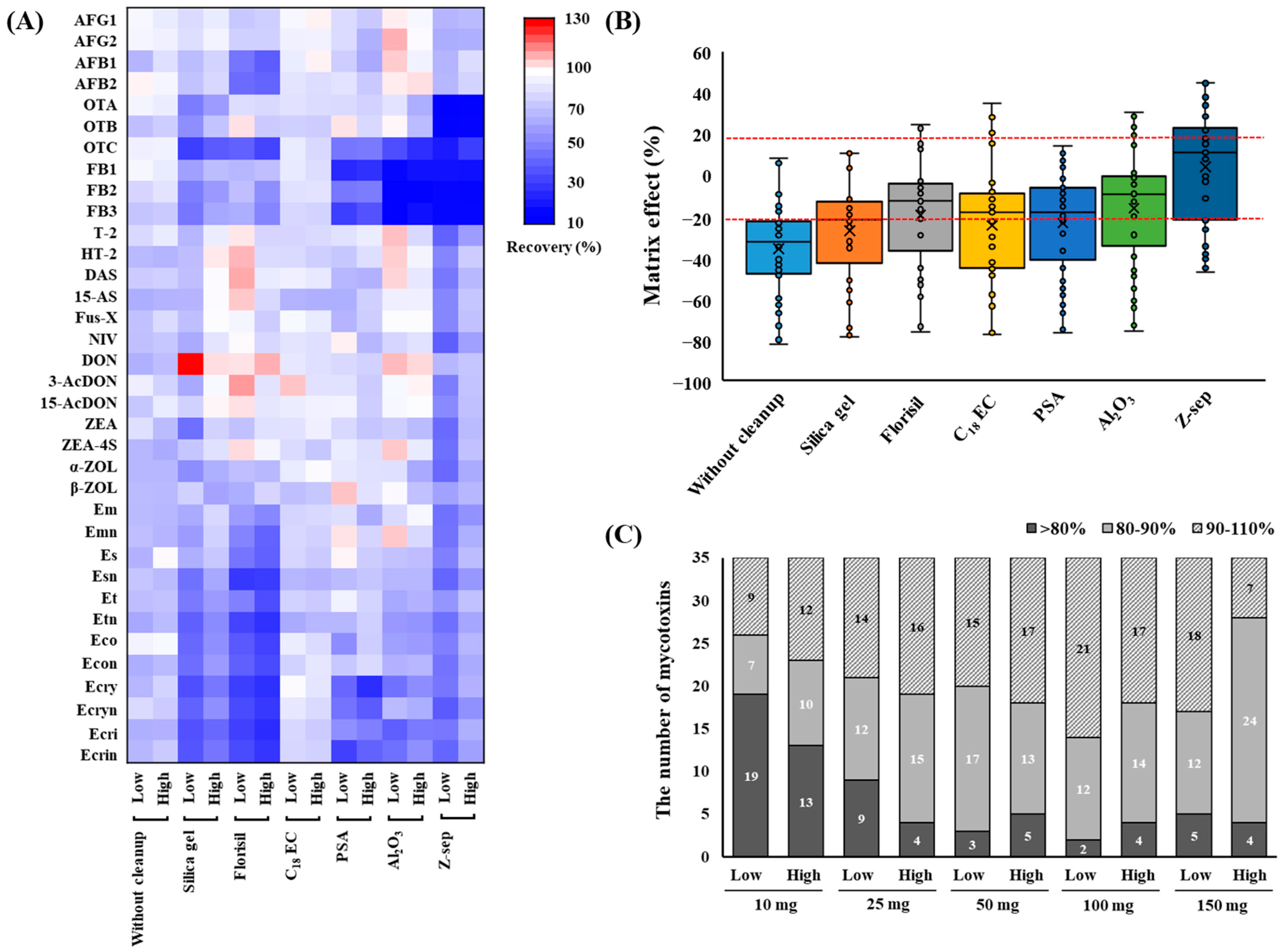
3.2. Optimization of the Cleanup Procedure
3.3. Analytical Method Validation
3.3.1. Linearity and Detectability
3.3.2. Accuracy and Precision
3.3.3. Matrix Effect
3.4. Estimation of Measurement of Uncertainty
3.5. Stability of Mycotoxins and Their Derivatives
3.6. Application to the Analysis of Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khaneghah, A.M.; Fakhri, Y.; Gahruie, H.H.; Niakousari, M.; Sant’Ana, A.S. Mycotoxins in cereal-based products during 24 years (1983–2017): A global systematic review. Trends Food Sci. Technol. 2019, 91, 95–105. [Google Scholar] [CrossRef]
- Leslie, J.F.; Moretti, A.; Mesterhazy, A.; Ameye, M.; Audenaert, K.; Singh, P.K.; Logrieco, A.F. Key global actions for mycotoxin management in wheat and other small grains. Toxins 2021, 13, 725. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Salleh, B.; Saad, B.; Abbas, H.; Abel, C.; Shier, W. An overview of mycotoxin contamination in foods and its implications for human health. Toxin Rev. 2010, 29, 3–26. [Google Scholar] [CrossRef]
- Kujawa, M.; IARC. Monographs on the evaluation of carcinogenic risks to humans. In Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins; World Health Organization: Geneva, Switzerland, 1993. [Google Scholar]
- Szabó-Fodor, J.; Szabó, A.; Kócsó, D.; Marosi, K.; Bóta, B.; Kachlek, M.; Mézes, M.; Balogh, K.; Kövér, G.; Nagy, I.; et al. Interaction between the three frequently co-occurring Fusarium mycotoxins in rats. J. Anim. Physiol. Anim. Nutr. 2019, 103, 370–382. [Google Scholar] [CrossRef]
- Klarić, M.Š. Adverse effects of combined mycotoxins. Arh. Hig. Rad. Toksikol. 2012, 63, 519–530. [Google Scholar] [CrossRef]
- Xu, W.; Han, X.; Li, F. Co-occurrence of multi-mycotoxins in wheat grains harvested in Anhui province, China. Food Control 2019, 96, 180–185. [Google Scholar] [CrossRef]
- Stanciu, O.; Juan, C.; Miere, D.; Loghin, F.; Mañes, J. Occurrence and co-occurrence of Fusarium mycotoxins in wheat grains and wheat flour from Romania. Food Control 2017, 73, 147–155. [Google Scholar] [CrossRef]
- Woo, S.Y.; Lee, S.Y.; Park, S.B.; Chun, H.S. Simultaneous determination of 17 regulated and non-regulated Fusarium mycotoxins co-occurring in foodstuffs by UPLC-MS/MS with solid-phase extraction. Food Chem. 2024, 438, 137624. [Google Scholar] [CrossRef]
- Liao, C.D.; Wong, J.W.; Zhang, K.; Hayward, D.G.; Lee, N.S.; Trucksess, M.W. Multi-mycotoxin analysis of finished grain and nut products using high-performance liquid chromatography–triple-quadrupole mass spectrometry. J. Agric. Food Chem. 2013, 61, 4771–4782. [Google Scholar] [CrossRef]
- Liang, S.H.; York, J.L.; Konschnik, J.D.; Majer, H.; Steimling, J.A. Simultaneous determination of alternaria toxins, ergot alkaloid epimers, and other major mycotoxins in various food matrixes by LC-MS/MS. J. AOAC Int. 2023, 106, 333–340. [Google Scholar] [CrossRef]
- Arroyo-Manzanares, N.; De Ruyck, K.; Uka, V.; Gámiz-Gracia, L.; García-Campaña, A.M.; De Saeger, S.; Diana Di Mavungu, J. In-house validation of a rapid and efficient procedure for simultaneous determination of ergot alkaloids and other mycotoxins in wheat and maize. Anal. Bioanal. Chem. 2018, 410, 5567–5581. [Google Scholar] [CrossRef] [PubMed]
- Silva, Â.; Mateus, A.R.S.; Barros, S.C.; Silva, A.S. Ergot Alkaloids on Cereals and Seeds: Analytical Methods, Occurrence, and Future Perspectives. Molecules 2023, 28, 7233. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Stránská, M.; Václavíková, M.; Elliott, C.T.; Black, C.; Meneely, J.; Krska, R. Advanced LC–MS-based methods to study the co-occurrence and metabolization of multiple mycotoxins in cereals and cereal-based food. Anal. Bioanal. Chem. 2018, 410, 801–825. [Google Scholar] [CrossRef] [PubMed]
- Pascale, M.; De Girolamo, A.; Lippolis, V.; Stroka, J.; Mol, H.G.; Lattanzio, V.M. Performance evaluation of LC-MS methods for multimycotoxin determination. J. AOAC Int. 2019, 102, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- González-Jartín, J.M.; Rodriguez-Canas, I.; Alfonso, A.; Sainz, M.J.; Vieytes, M.R.; Gomes, A.; Botana, L.M. Multianalyte method for the determination of regulated, emerging and modified mycotoxins in milk: QuEChERS extraction followed by UHPLC–MS/MS analysis. Food Chem. 2021, 356, 129647. [Google Scholar] [CrossRef]
- Kim, D.B.; Jung, Y.S.; Nam, T.G.; Lee, S.H.; Yoo, M.Y. Simultaneous determination of trichothecene mycotoxins in cereals by LC-MS/MS. Food Sci. Biotechnol. 2022, 31, 165–174. [Google Scholar] [CrossRef]
- Eurachem. EURACHEM/CITAC Guide: Quantifying Uncertainty in Analytical Measurement; Ellison, S.L.R., Williams, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Berger, U.; Oehme, M.; Kuhn, F. Quantitative determination and structure elucidation of type A-and B-trichothecenes by HPLC/ion trap multiple mass spectrometry. J. Agric. Food Chem. 1999, 47, 4240–4245. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Smedsgaard, J. Fungal metabolite screening: Database of 474 mycotoxins and fungal metabolites for dereplication by standardized liquid chromatography-UV-mass spectrometry methodology. J. Chromatogr. A 2003, 1002, 111–136. [Google Scholar] [CrossRef]
- Lehner, S.M.; Neumann, N.K.N.; Sulyok, M.; Lemmens, M.; Krska, R.; Schuhmacher, R. Evaluation of LC-high-resolution FT-Orbitrap MS for the quantification of selected mycotoxins and the simultaneous screening of fungal metabolites in food. Food Addit. Contam. Part A 2011, 28, 1457–1468. [Google Scholar] [CrossRef]
- Guan, X.; Feng, Y.; Suo, D.; Xiao, Z.; Wang, S.; Liang, Y.; Fan, X. Simultaneous Determination of 11 Mycotoxins in maize via multiple-impurity adsorption combined with liquid chromatography–tandem mass spectrometry. Foods 2022, 11, 3624. [Google Scholar] [CrossRef]
- Hu, Z.B.; Qi, P.; Wang, N.; Zhou, Q.Q.; Lin, Z.H.; Chen, Y.Z.; Li, C.Y. Simultaneous determination of multiclass illegal dyes with different acidic–basic properties in foodstuffs by LC-MS/MS via polarity switching mode. Food Chem. 2020, 309, 125745. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Zhou, Q.Q.; Lin, Z.H.; Cai, W.y.; Mao, X.W.; Jiang, J.J. Qualitative screening and quantitative determination of multiclass water-soluble synthetic dyes in foodstuffs by liquid chromatography coupled to quadrupole Orbitrap mass spectrometry. Food Chem. 2021, 360, 129948. [Google Scholar] [CrossRef]
- Łozowicka, B.; Mojsak, P.; Kaczyński, P.; Konecki, R.; Borusiewicz, A. The fate of spirotetramat and dissipation metabolites in Apiaceae and Brassicaceae leaf-root and soil system under greenhouse conditions estimated by modified QuEChERS/LC–MS/MS. Sci. Total Environ. 2017, 603, 178–184. [Google Scholar] [CrossRef]
- Pizzutti, I.R.; de Kok, A.; Scholten, J.; Righi, L.W.; Cardoso, C.D.; Rohers, G.N.; da Silva, R.C. Development, optimization and validation of a multimethod for the determination of 36 mycotoxins in wines by liquid chromatography–tandem mass spectrometry. Talanta 2014, 129, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, J.; Ma, L.; Wang, F. Development and validation of a simple and fast method for simultaneous determination of aflatoxin B1 and sterigmatocystin in grains. Food Chem. 2017, 221, 11–17. [Google Scholar] [CrossRef]
- Mateus, A.R.S.; Barros, S.; Pena, A.; Silva, A.S. Development and Validation of QuEChERS Followed by UHPLC-ToF-MS Method for Determination of Multi-Mycotoxins in Pistachio Nuts. Molecules 2021, 26, 5754. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EU) 2023/915 of 25 April 2023 on maximum levels for certain contaminants in food and repealing Regulation (EC) No 1881/2006 (Text with EEA relevance). Off. J. Eur. Union 2023, 119, 103–157. [Google Scholar]
- Kim, H.Y.; Baek, E.J.; Shin, B.G.; Kim, H.J.; Kim, J.E. In-house validation of an efficient and rapid procedure for the simultaneous determination and monitoring of 23 mycotoxins in grains in Korea. Toxins 2022, 14, 457. [Google Scholar] [CrossRef] [PubMed]
- De Colli, L.; Elliott, C.; Finnan, J.; Grant, J.; Arendt, E.K.; McCormick, S.P.; Danaher, M. Determination of 42 mycotoxins in oats using a mechanically assisted QuEChERS sample preparation and UHPLC-MS/MS detection. J. Chromatogr. B 2020, 1150, 122187. [Google Scholar] [CrossRef]
- Shi, H.; Yu, P. Correlation patterns prevalence, and co-occurrence of ergot alkaloids in cool-season adapted cereal grains revealed with molecular spectroscopy and LC-MS/MS equipped HPLC system. Food Chem. 2022, 393, 133322. [Google Scholar] [CrossRef]
- Varga, E.; Fodor, P.; Soros, C. Multi-mycotoxin LC-MS/MS method validation and its application to fifty-four wheat flours in Hungary. Food Addit. Contam. Part A 2021, 38, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Covarelli, L.; Beccari, G.; Colasante, V.; Mañes, J. Simultaneous analysis of twenty-six mycotoxins in durum wheat grain from Italy. Food Control 2016, 62, 322–329. [Google Scholar] [CrossRef]
- European Commission. Report on the Relationship Between Analytical Results, Measurement Uncertainty, Recovery Factors and the Provisions of EU Food and Feed Legislation, with Particular Reference to Community Legislation Concerning. 2004. Available online: https://food.ec.europa.eu/system/files/2016-10/cs_contaminants_sampling_analysis-report_2004_en.pdf (accessed on 14 October 2024).
- Cherewyk, J.E.; Grusie-Ogilvie, T.J.; Parker, S.E.; Blakley, B.R.; Al-Dissi, A.N. The impact of storage temperature and time on ergot alkaloid concentrations. Toxins 2023, 15, 497. [Google Scholar] [CrossRef] [PubMed]
- Miguel Alfonso, R.A.; Yael Yvette, B.H.; Irma Martha, M.D.; Cyndia Azucena, G.A.; Briscia Socorro, B.V.; Jos’e Francisco, H.M.; Aurora Elizabeth, R.G. Genotoxic effects of the ochratoxin A (OTA), its main metabolite (OTα) per se and in combination with fumonisin B1 in HepG2 cells and human lymphocytes. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2022, 878, 503482. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Analyte a | Formula | RT (min) | Adduct | Precursor ion (m/z) | Production (m/z) b | DP (V) c | CE (V) d | CXP (V) e |
|---|---|---|---|---|---|---|---|---|
| AFG1 | C17H12O7 | 10.8 | [M+H]+ | 328.9 | 242.9/199.9 | 106 | 39/55 | 8/6 |
| AFG2 | C17H14O7 | 9.3 | [M+H]+ | 331.0 | 312.9/244.8 | 121 | 35/43 | 66 |
| AFB1 | C17H12O6 | 11.8 | [M+H]+ | 313.0 | 285.0/213.0 | 116 | 35/61 | 8/6 |
| AFB2 | C17H14O6 | 11.1 | [M+H]+ | 315.0 | 258.9/287.3 | 101 | 41/39 | 4/24 |
| OTA | C20H18ClNO6 | 12.3 | [M+H]+ | 404.0 | 238.8/220.9 | 81 | 35/51 | 6/6 |
| OTB | C20H19NO6 | 11.2 | [M+H]+ | 370.0 | 204.9/187.2 | 56 | 31/51 | 6/10 |
| OTC | C22H22ClNO6 | 13.2 | [M+H]+ | 432.0 | 239.2/358.2 | 111 | 41/25 | 8/12 |
| FB1 | C34H59NO15 | 5.2 | [M+H]+ | 722.3 | 334.2/352.1 | 126 | 57/51 | 10/10 |
| FB2 | C34H59NO14 | 8.8 | [M+H]+ | 706.2 | 336.4/688.2 | 141 | 51/41 | 12/20 |
| FB3 | C34H59NO14 | 6.9 | [M+H]+ | 706.3 | 336.1/81.2 | 111 | 51/111 | 10/8 |
| T-2 | C24H34O9 | 9.9 | [M+NH4]+ | 484.2 | 305.2/215.1 | 71 | 21/25 | 12/6 |
| HT-2 | C22H32O8 | 6.6 | [M+NH4]+ | 442.2 | 263.1/215.1 | 6 | 19/19 | 8/6 |
| DAS | C19H26O7 | 5.9 | [M+H]+ | 384.1 | 307.2/247.2 | 1 | 17/19 | 10/10 |
| 15-AS | C17H24O6 | 4.2 | [M+NH4]+ | 342.1 | 265.4/107.2 | 16 | 13/25 | 10/14 |
| Fus-X | C17H22O8 | 2.6 | [M+H]+ | 355.1 | 247.2/175.3 | 66 | 15/27 | 10/12 |
| NIV | C15H20O7 | 1.3 | [M+HCOO]− | 357.0 | 281.0/310.9 | −5 | −18/−14 | −9/−11 |
| DON | C15H20O6 | 1.8 | [M+H]+ | 297.1 | 249.2/203.2 | 76 | 17/21 | 10/6 |
| 3-AcDON | C17H22O7 | 3.7 | [M+H]+ | 339.1 | 231.3/203.1 | 71 | 13/17 | 10/8 |
| 15-AcDON | C17H22O7 | 3.6 | [M+H]+ | 339.1 | 321.1/261.1 | 16 | 13/25 | 14/8 |
| ZEA | C18H22O5 | 11.0 | [M-H]− | 317.0 | 174.9/131.0 | −105 | −34/−38 | −9/−7 |
| ZEA-4S | C18H25NO8S | 7.2 | [M+H]+ | 416.1 | 319.2/187.2 | 66 | 17/41 | 10/6 |
| α-ZOL | C18H24O5 | 9.3 | [M+H]+ | 321.2 | 303.0/285.0 | 51 | 11/17 | 12/8 |
| β-ZOL | C18H24O5 | 7.5 | [M+H]+ | 321.1 | 303.1/285.4 | 56 | 11/17 | 8/8 |
| Em | C19H23N3O2 | 1.9 | [M+H]+ | 326.1 | 206.9/222.9 | 76 | 59/33 | 6/6 |
| Emn | C19H23N3O2 | 2.5 | [M+H]+ | 325.8 | 208.0/207.1 | 131 | 39/59 | 6/6 |
| Es | C30H37N5O5 | 5.4 | [M+H]+ | 548.0 | 223.0/208.1 | 81 | 45/59 | 6/6 |
| Esn | C30H37N5O5 | 5.6 | [M+H]+ | 547.8 | 222.9/152.0 | 131 | 45/165 | 6/4 |
| Et | C33H35N5O5 | 6.1 | [M+H]+ | 582.0 | 223.0/207.9 | 91 | 47/65 | 6/6 |
| Etn | C33H35N5O5 | 6.5 | [M+H]+ | 582.1 | 223.0/208.1 | 86 | 47/69 | 6/6 |
| Eco | C31H39N5O5 | 6.3 | [M+H]+ | 562.0 | 268.0/223.0 | 96 | 37/51 | 8/6 |
| Econ | C31H39N5O5 | 9.4 | [M+H]+ | 562.4 | 223.0/227.0 | 1 | 51/39 | 6/8 |
| Ecry | C32H41N5O5 | 7.1 | [M+H]+ | 576.0 | 267.9/208.0 | 106 | 37/67 | 8/6 |
| Ecryn | C32H41N5O5 | 10.3 | [M+H]+ | 576.1 | 222.9/305.3 | 6 | 51/39 | 6/10 |
| Ecri | C35H39N5O5 | 7.6 | [M+H]+ | 610.1 | 208.0/223.0 | 96 | 65/55 | 6/6 |
| Ecrin | C35H39N5O5 | 10.5 | [M+H]+ | 609.9 | 223.0/305.2 | 1 | 55/39 | 6/10 |
| Analyte a | Calibration Range (μg/kg) | Matrix-Matched Calibration (R2) | Matrix Effect (%) | LOD b (μg/kg) | LOQ c (μg/kg) | U d (%) | Intra-Day (n = 5) | Inter-Day (n = 15) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low e | Medium f | High g | Low | Medium | High | ||||||||||
| RE (%) h | RSDr | RE (%) | RSDr | RE (%) | RSDr | RSDR | RSDR | RSDR | |||||||
| AFG1 | 0.25–10 | 0.9995 | −48.23 | 0.14 | 0.43 | 10.90 | 93.33 | 3.83 | 91.86 | 4.60 | 91.24 | 2.48 | 8.65 | 4.65 | 5.04 |
| AFG2 | 0.25–10 | 0.9996 | −24.36 | 0.06 | 0.17 | 10.64 | 92.77 | 5.84 | 87.58 | 4.29 | 85.92 | 1.61 | 6.52 | 4.34 | 5.36 |
| AFB1 | 0.25–10 | 0.9997 | −64.61 | 0.04 | 0.13 | 11.33 | 101.43 | 2.30 | 98.97 | 1.21 | 95.24 | 2.21 | 5.84 | 7.54 | 6.71 |
| AFB2 | 0.25–10 | 0.9999 | −64.06 | 0.02 | 0.06 | 11.43 | 103.16 | 6.21 | 104.02 | 5.91 | 101.1 | 3.79 | 10.42 | 9.27 | 8.94 |
| OTA | 0.25–10 | 0.9997 | −48.63 | 0.03 | 0.08 | 10.47 | 97.15 | 5.94 | 91.80 | 0.88 | 88.93 | 4.56 | 7.77 | 5.34 | 6.52 |
| OTB | 0.25–10 | 0.9998 | −8.14 | 0.01 | 0.04 | 11.11 | 97.10 | 5.00 | 94.17 | 4.22 | 91.06 | 2.88 | 4.62 | 3.57 | 5.11 |
| OTC | 0.25–10 | 0.9998 | 17.31 | 0.04 | 0.12 | 9.49 | 85.01 | 1.00 | 84.83 | 2.21 | 86.00 | 1.91 | 2.59 | 3.07 | 3.24 |
| FB1 | 0.25–10 | 0.9999 | 31.57 | 0.10 | 0.30 | 10.95 | 92.21 | 7.48 | 89.56 | 2.08 | 88.32 | 1.76 | 6.46 | 13.77 | 4.63 |
| FB2 | 0.25–10 | 0.9998 | 27.32 | 0.03 | 0.09 | 10.05 | 87.88 | 3.69 | 85.55 | 3.39 | 80.39 | 3.53 | 4.63 | 5.34 | 6.18 |
| FB3 | 0.25–10 | 0.9995 | 13.53 | 0.37 | 1.12 | 10.55 | 98.04 | 5.44 | 89.67 | 3.09 | 88.74 | 3.14 | 6.68 | 8.84 | 4.06 |
| T-2 | 1.25–50 | 0.9996 | −2.62 | 0.15 | 0.45 | 5.55 | 94.21 | 4.30 | 91.61 | 2.95 | 89.43 | 1.92 | 5.42 | 3.73 | 4.18 |
| HT-2 | 1.25–50 | 0.9994 | −21.50 | 0.41 | 1.23 | 5.23 | 107.16 | 1.83 | 98.61 | 2.81 | 95.88 | 4.00 | 4.55 | 6.31 | 4.82 |
| DAS | 1.25–50 | 0.9999 | 15.39 | 0.12 | 0.37 | 5.55 | 98.12 | 1.85 | 96.44 | 1.92 | 94.06 | 1.70 | 4.03 | 2.51 | 3.10 |
| 15-AS | 1.25–50 | 0.9998 | −18.12 | 0.14 | 0.43 | 4.74 | 100.73 | 1.82 | 95.56 | 2.87 | 95.13 | 1.98 | 3.52 | 4.51 | 3.44 |
| Fus-X | 1.25–50 | 0.9996 | −35.65 | 0.76 | 2.31 | 5.48 | 95.72 | 8.06 | 95.13 | 5.08 | 95.51 | 3.26 | 9.10 | 5.77 | 5.85 |
| NIV | 2.5–100 | 0.9992 | −76.67 | 7.50 | 22.73 | 1.62 | 98.89 | 7.26 | 91.72 | 4.38 | 84.98 | 1.79 | 9.51 | 6.97 | 6.20 |
| DON | 1.25–50 | 0.9989 | −29.86 | 3.38 | 10.23 | 5.27 | 85.78 | 4.77 | 88.42 | 10.41 | 95.75 | 10.12 | 10.16 | 11.40 | 9.56 |
| 3-AcDON | 1.25–50 | 0.9993 | −76.84 | 2.85 | 8.65 | 5.36 | 99.22 | 4.53 | 100.19 | 4.33 | 96.90 | 5.24 | 7.26 | 10.99 | 9.17 |
| 15-AcDON | 1.25–50 | 0.9990 | −75.69 | 2.54 | 7.70 | 5.01 | 97.52 | 8.26 | 89.89 | 4.23 | 96.53 | 5.68 | 9.42 | 4.91 | 7.71 |
| ZEA | 1.25–50 | 0.9997 | −59.09 | 0.39 | 1.19 | 5.37 | 93.44 | 4.10 | 88.21 | 1.09 | 84.35 | 5.80 | 8.43 | 8.30 | 10.37 |
| ZEA-4S | 1.25–50 | 0.9995 | −63.46 | 0.20 | 0.59 | 4.94 | 98.20 | 8.60 | 95.41 | 3.10 | 90.34 | 5.39 | 8.52 | 6.76 | 11.70 |
| α-ZOL | 1.25–50 | 0.9967 | −9.72 | 0.84 | 2.53 | 4.94 | 82.60 | 5.55 | 86.26 | 2.27 | 92.53 | 2.45 | 12.13 | 7.33 | 4.46 |
| β-ZOL | 1.25–50 | 0.9976 | −0.28 | 2.11 | 6.41 | 4.94 | 88.56 | 6.10 | 91.54 | 4.20 | 85.28 | 1.05 | 12.20 | 9.34 | 8.46 |
| Em | 0.25–10 | 0.9988 | −20.22 | 0.39 | 1.17 | 10.72 | 88.50 | 6.82 | 98.62 | 8.93 | 87.33 | 7.40 | 12.02 | 11.03 | 9.03 |
| Emn | 0.25–10 | 0.9999 | −16.70 | 0.10 | 0.29 | 10.03 | 86.94 | 7.77 | 88.28 | 3.75 | 88.43 | 2.54 | 9.38 | 5.65 | 3.96 |
| Es | 0.25–10 | 0.9997 | −27.19 | 0.01 | 0.03 | 10.73 | 98.10 | 5.42 | 100.17 | 4.64 | 97.99 | 4.51 | 3.82 | 5.06 | 4.45 |
| Esn | 0.25–10 | 0.9996 | −11.26 | 0.04 | 0.12 | 9.88 | 93.86 | 7.36 | 86.83 | 4.82 | 80.55 | 4.31 | 10.86 | 10.78 | 8.29 |
| Et | 0.25–10 | 0.9997 | −12.52 | 0.12 | 0.37 | 9.88 | 97.93 | 6.09 | 94.95 | 7.67 | 95.41 | 7.94 | 6.70 | 8.29 | 6.51 |
| Etn | 0.25–10 | 0.9998 | −21.30 | 0.02 | 0.06 | 9.88 | 85.43 | 5.34 | 84.65 | 2.50 | 84.93 | 3.54 | 9.33 | 4.89 | 5.86 |
| Eco | 0.25–10 | 0.9998 | −14.68 | 0.02 | 0.05 | 9.05 | 97.83 | 6.34 | 92.81 | 7.45 | 84.61 | 5.93 | 14.20 | 9.32 | 10.79 |
| Econ | 0.25–10 | 0.9997 | −10.92 | 0.07 | 0.22 | 9.52 | 86.42 | 2.07 | 84.85 | 1.99 | 81.07 | 3.93 | 4.26 | 3.16 | 4.37 |
| Ecry | 0.25–10 | 0.9994 | −30.01 | 0.10 | 0.29 | 9.92 | 92.42 | 8.01 | 87.63 | 5.27 | 83.09 | 6.76 | 10.08 | 5.59 | 11.40 |
| Ecryn | 0.25–10 | 0.9987 | −45.35 | 0.46 | 1.40 | 9.50 | 85.10 | 6.13 | 81.65 | 3.13 | 84.94 | 2.78 | 7.97 | 4.89 | 8.25 |
| Ecri | 0.25–10 | 0.9998 | −9.55 | 0.01 | 0.02 | 10.07 | 82.86 | 5.54 | 81.34 | 2.21 | 83.73 | 6.38 | 9.28 | 12.65 | 12.66 |
| Ecrin | 0.25–10 | 0.9999 | −43.34 | 0.05 | 0.15 | 9.72 | 84.50 | 6.30 | 81.95 | 2.84 | 81.71 | 2.69 | 8.85 | 9.41 | 8.80 |
| Samples | OTA a | T-2 | HT-2 | DON | 3-AcDON | EAs | ZEA | Number of Detected Mycotoxins |
|---|---|---|---|---|---|---|---|---|
| S1 | – a | – | – | – | – | 609.63 | – | 1 |
| S2 | – | – | – | 204.85 | <LOQ b | 3.70 | 11.01 | 4 |
| S4 | – | <LOQ | 7.37 | – | – | 16.85 | – | 3 |
| S5 | – | <LOQ | 9.79 | 82.05 | – | 202.91 | <LOQ | 5 |
| S6 | – | 0.71 | 11.69 | 119.76 | – | 294.93 | – | 4 |
| S7 | – | <LOQ | 9.10 | 47.68 | – | 5.27 | – | 4 |
| S8 | – | – | – | – | <LOQ | 72.55 | – | 2 |
| S9 | 0.19 | – | – | 79.34 | 11.55 | 11.55 | – | 4 |
| S13 | – | – | – | 22.93 | – | – | – | 1 |
| S15 | <LOQ | – | – | 13.73 | – | 11.97 | – | 3 |
| S17 | 0.13 | 0.64 | <LOQ | <LOQ | – | – | – | 4 |
| S18 | – | – | – | 18.78 | – | – | – | 1 |
| S19 | – | – | – | – | – | 2.97 | – | 1 |
| S20 | – | – | – | – | – | 1.25 | – | 1 |
| S21 | – | – | – | <LOQ | – | 2.29 | – | 2 |
| S22 | – | – | – | – | – | 0.42 | – | 1 |
| S23 | <LOQ | – | – | <LOQ | – | 10.11 | – | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-B.; Nam, M.; Kim, Y.-S.; Kim, M.-S. Optimization, Validation, and Application of Cleanup-Coupled Liquid Chromatography–Tandem Mass Spectrometry for the Simultaneous Analyses of 35 Mycotoxins and Their Derivatives in Cereals. Foods 2024, 13, 3617. https://doi.org/10.3390/foods13223617
Kim D-B, Nam M, Kim Y-S, Kim M-S. Optimization, Validation, and Application of Cleanup-Coupled Liquid Chromatography–Tandem Mass Spectrometry for the Simultaneous Analyses of 35 Mycotoxins and Their Derivatives in Cereals. Foods. 2024; 13(22):3617. https://doi.org/10.3390/foods13223617
Chicago/Turabian StyleKim, Dan-Bi, Miso Nam, Yong-Suk Kim, and Min-Sun Kim. 2024. "Optimization, Validation, and Application of Cleanup-Coupled Liquid Chromatography–Tandem Mass Spectrometry for the Simultaneous Analyses of 35 Mycotoxins and Their Derivatives in Cereals" Foods 13, no. 22: 3617. https://doi.org/10.3390/foods13223617
APA StyleKim, D.-B., Nam, M., Kim, Y.-S., & Kim, M.-S. (2024). Optimization, Validation, and Application of Cleanup-Coupled Liquid Chromatography–Tandem Mass Spectrometry for the Simultaneous Analyses of 35 Mycotoxins and Their Derivatives in Cereals. Foods, 13(22), 3617. https://doi.org/10.3390/foods13223617