1. Introduction
Starch, a polysaccharide abundant in nature, is positioned as a renewable biomaterial with low environmental impact [
1]. Ranked as the second most abundant after cellulose, this natural component finds applications in various sectors [
2]. Its characteristics as a thickener, adhesive, and gelling agent make it a key ingredient in the food and textile industries [
3]. Among these properties, its ability to form thermoreversible gels stands out, which makes it ideal for the encapsulation and controlled release of bioactive compounds in pharmaceutical and food applications. In addition, its ability to interact with other materials, such as proteins and lipids, broadens its applications in the food industry, improving the stability of emulsions and foams. The filmogenic properties of starch make it an ideal material for the creation of edible films and biodegradable coatings, which is beneficial in food applications.
However, native starch has limitations that restrict its potential applications [
4]. To overcome the limitations of native starch, its modification is performed through chemical, physical, and enzymatic methods [
3]. These techniques allow for the adjustment of starch properties, such as particle size, stress resistance, thermal properties, and retrogradation behavior [
5].
The amylose and amylopectin contents in starch are crucial when starch is subjected to chemical, biological, or physical modifications due to their impact on the final properties of the modified starch. Amylose, being linear (more ordered and crystalline structures), tends to form stronger gels with higher retrogradability [
6], which can influence the texture and stability of food products. On the other hand, amylopectin, being branched (less crystalline structures), contributes to the formation of more flexible gels and is less prone to retrogradation. When modifications are made to starch, the relative contents of amylose and amylopectin can affect the swelling capacity, solubility [
7], crystallinity, granule size, chemical nature, and arrangement of polymers within the granule [
8]. Therefore, understanding and knowing the proportions of amylose and amylopectin are essential to adjusting the properties of modified starch according to the specific needs of different applications.
Chemical modification introduces functional groups into the starch molecule, resulting in highly functional derivatives with improved properties [
9]. These modifications can technically improve aspects such as morphology, crystalline structure, thermal behavior, and even particle size reduction [
4,
10].
Its high solubility, thermal stability, non-toxicity, and low cost have led to the use of nanostarch in numerous applications, including drug delivery, cosmetics, textiles, food, enhanced oil recovery (EOR), and tissue engineering [
9,
11,
12]. Of particular note is the effectiveness of nanostarch in inducing the stabilization of Pickering emulsions without the need for surfactants [
9]. These applications take advantage of the special functions that can be achieved by modifications in the structure and properties of native starch [
13].
Acid hydrolysis, a commonly used method to obtain nanostarch with high crystallinity and stability, is often combined with other techniques such as ultrasound, nanoprecipitation, milling, and crosslinking to optimize starch properties [
14,
15]. The efficiency depends on several factors during the hydrolysis process, such as the type and concentration of acid used, the reaction time and temperature, and the source of the starch itself [
16].
Enzymatic modification offers another approach that directly affects starch properties by creating a new structure [
13]. Factors such as enzyme concentration, incubation times, enzyme mixture, reaction temperature, and pH can significantly influence the degree of hydrolysis and various functional properties [
15,
17]. These factors can also influence water-holding capacity and stickiness, paving the way for “clean-label” starches with unique gelling properties [
18]. Enzymatic processing, combined with techniques such as nanoprecipitation, high-pressure homogenization, and ultrasonication [
2,
19], finds applications in the production of Pickering emulsions, films, and nanocomposites [
2,
20]. In addition, modified starches exhibit reinforcing properties in biocomposites, making them valuable not only for their functionality but also for their renewable and biodegradable nature [
1].
Recent research has explored the antisolvent precipitation method to produce biopolymeric nanoparticles from native starches [
21,
22]. This rapid method typically uses sonication, gamma irradiation, mild combinations of alkaline hydrolysis, and antisolvents, such as acetone or ethanol, added to the starch solution [
14,
23,
24,
25]. Some studies suggest that the dropwise addition of ethanol to a weak solution of gelatinized starch while stirring promotes the formation of smaller nanoparticles [
26].
In short, starch, with its exceptional properties and the possibility of obtaining nanoparticles and/or microparticles with even more versatile characteristics, is positioned as a biopolymer with great potential for the development of new materials and technologies in various sectors. In particular, biopolymeric nanoparticles often exhibit higher solubility and stability in water compared to their unmodified counterparts [
21].
This study aims to produce nanostructures and microstructures of native potato starches modified by acidic, enzymatic, and ethanolic precipitation treatments to characterize their physicochemical, structural, and thermal properties, which will allow us to evaluate their potential use in various food applications.
2. Materials and Methods
2.1. Materials and Reagents
Four native potato varieties, cultivated in the Cusco region of Peru, were utilized in the investigation: two sweet varieties, Aq’hu Pukucho (APE) (S: 14°44′36.5″; W: 71°26′22.9″; altitude 3925 m) and Yurakk Kkachun Wakkachi (YKW) (S: 14°05′19.8″; W: 71°17′55.8″; altitude 3475 m), and two bitter varieties, Yurac Anca (YA) (S: 14°52′19.8″; W: 71°31′48.4″; altitude 4001 m) and Huarmi Mallco (HM) (S: 14°05′19.8″; W: 71°17′55.8″; altitude 3475 m). These varieties may exhibit variations in starch composition, potentially influencing their behavior during modification. Starch extraction was conducted via the wet method, with the resulting samples utilized for various modification treatments. Reagents included α-amylase enzyme (Amylase Liquid IFCC, Quimica Clinica Aplicada S.A., Amposta, Spain), orthophosphoric acid with a minimum purity of 85% (Merck KGaA, Darmstadt, Germany, CAS 7664-38-2), and ethanol with 96% purity (Alkofarma, Lima, Peru). All solutions were prepared using distilled water.
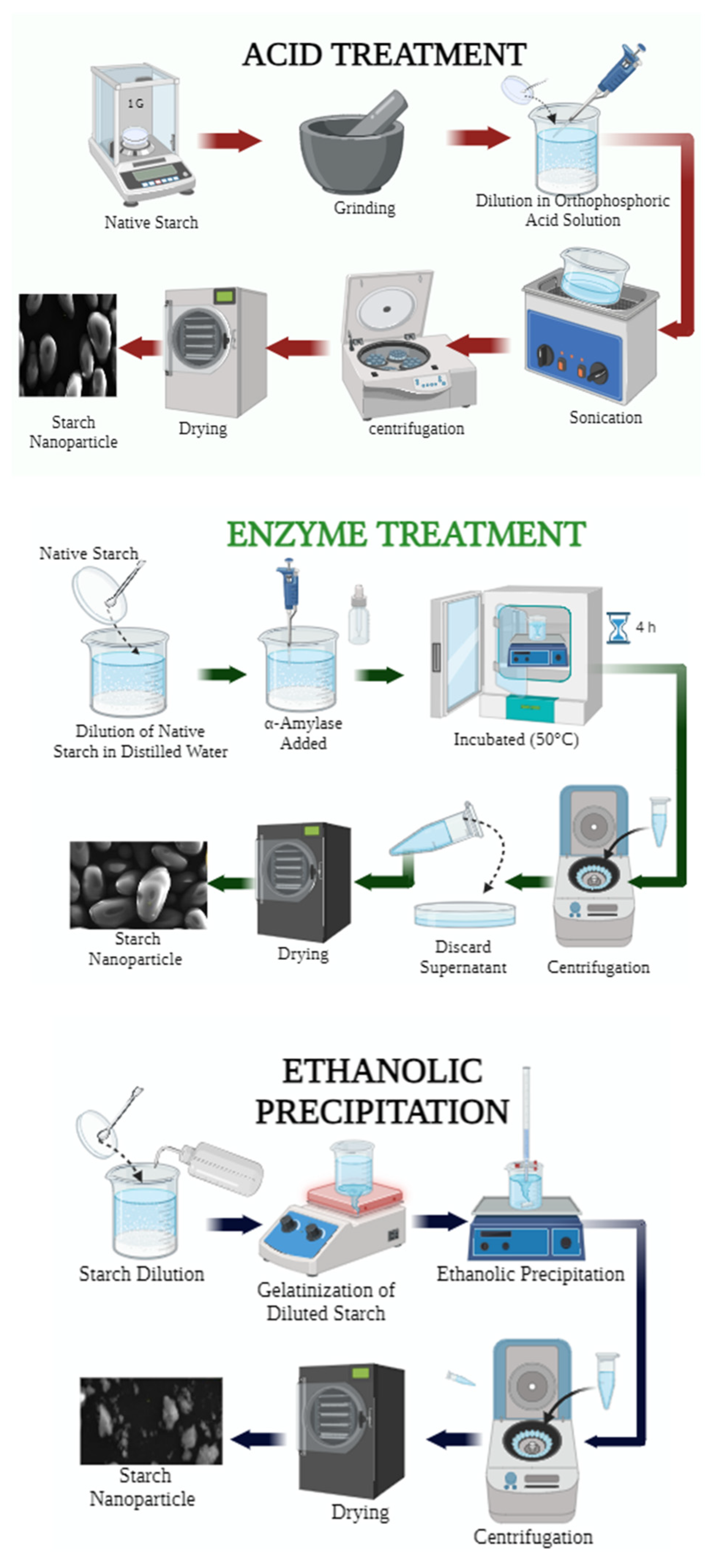
2.2. Acid Treatment
The method outlined by Ahmad et al. [
23] was employed with certain adaptations. Five grams of starch were dispersed in 50 mL of orthophosphoric acid (1 M). The solution underwent ultrasonication at 40 Hz (Biobase equipment, model UC-08 Myanmar, Silicon Valley, CA, USA) for 30 min at 40 °C. Subsequently, the supernatant was separated via centrifugation at 10,000×
g for 10 min, and the resulting sediment was washed eight times with distilled water under continuous stirring until reaching a neutral pH. The resultant product was vacuum dried (10 mBar) at 20 °C for 4 h using a vacuum oven (BINDER, model VD56, Tuttingen, Germany), followed by sieving through a UTEST electric sieve (model UTG-0416, Lima, Peru) with a mesh size of 63 μm. Finally, the samples were packaged for subsequent characterization.
2.3. Enzymatic Treatment
This procedure was conducted following the protocol proposed by Dukare et al. [
1] with certain modifications. A solution was prepared containing 1 g of starch and 100 mL of distilled water. Subsequently, 50 µL of α-amylase enzyme (A = buffer/enzyme and B = buffer/substrate) was added and mixed using a magnetic stirrer (CAT, model M6, Staufen, Germany). The reaction temperature was maintained at 50 °C using an incubator (BINDER, model DIN 12880, Tuttlingen, Germany) for 4 h with continuous stirring (BINDER, model VD56, Tuttingen, Germany). The resulting sample was cooled to room temperature and centrifuged (Centurión, 1.C2024, Chichester, UK) at 4000 rpm for 10 min to separate the supernatant. The obtained samples were then vacuum dried (10 mBar) (BINDER, model VD56, Tuttingen, Germany) at 20 °C for 4 h and ground using a Retsch PM100 planetary ball mill (Haan, Germany). Finally, the material was sieved through a 63 μm mesh and packed for the evaluation of its physicochemical and structural properties.
2.4. Ethanolic Precipitation
The method described by Qin et al. [
27] was followed, involving the gelatinization of starch followed by precipitation with ethanol (96% purity, Alkofarma, Lima, Peru). Two grams of starch were dispersed in 200 mL of distilled water and continuously stirred (CAT, Modelo M6, Staufen, Alemania) at 900 rpm at 80 °C for 30 min. The gelatinized starch was then cooled to room temperature and precipitated using 800 mL of 96% ethanol. Ethanol was added dropwise upon contact with the gelatinized starch while stirring at 700 rpm, gradually increasing the volume of alcohol until precipitation was complete. The precipitation process was maintained under agitation at 150 rpm for 18 h. Subsequently, the samples were centrifuged at (Centurión, 1.C2024, Chichester, UK) 3500 rpm for 10 min (twice) to remove the supernatant. Finally, the samples were vacuum dried (10 mBar) (BINDER, model VD56, Tuttlingen, Germany) at 20 °C for 4 h (
Figure 1).
2.5. Color Analysis
The color assessment of the modified starch samples was conducted using a Konica Minolta benchtop colorimeter, model CR-5 (Tokyo, Japan). Colorimetric measurements were employed to ascertain lightness (L*, where 0 represents black and 100 represents white), hue angle (h*, Equation (1)), and chroma (C*, Equation (2)) by the standards set by the International Commission on Illumination (CIE). The yellowness index (YI) and the whiteness index (WI) were determined using Equations (3) and (4), respectively [
28].
2.6. Amylose and Amylopectin
The determination of amylose content was carried out utilizing a modified protocol based on the methodology proposed by Galicia et al. [
29]. A 20 mg sample of starch was placed in a 50 mL screw-capped tube, into which 0.2 mL of 95% ethanol and 1.8 mL of 1 M sodium hydroxide were added to create a solution. The mixture stood for one day at room temperature, after which the volume was adjusted to 20 mL with deionized water and vigorously shaken for 30 min. Subsequently, a one-milliliter sample of the resulting solution was combined with two mL of 1 M acetic acid. Then, 0.4 mL of Lugol’s solution was added, and the volume was adjusted to 20 mL with deionized water and allowed to stand for 20 min. A portion of the solution was carefully withdrawn and transferred to a cuvette for absorbance measurement at a wavelength of 620 nm using a UV-Vis spectrophotometer. A standard curve was prepared using a 1 mg/mL amylose solution in deionized water to facilitate accurate quantification of the amylose content in the samples. The absorbance data were fitted to a linear equation, and the percentage of amylose was determined according to Equation (5).
where M is the amylose (mg) obtained from the fitted equation, d is the dilution factor, and f is the mass of starch.
The difference between total starch and amylose represents the amylopectin content.
2.7. Particle Size
Next, 0.10 g of ethanolic precipitation-modified starch was suspended in 100 mL of ultrapure water and constantly stirred at 200 rpm for 24 h. Subsequently, the samples underwent ultrasound for 10 min at room temperature using a Sonics model VCX 750 sonicator (Newtown, CT, USA). An aliquot was extracted in a polystyrene cell, and the particle size distribution was determined by dynamic light scattering (DLS) using a Malvern brand instrument, model Zetasizer ZSU3100 (Worcestershire, UK).
2.8. Zeta Potential (ζ)
Four milligrams of the modified starch sample were dispersed in 5 mL of ultrapure water, stirred at 1000 rpm for 5 min, and then subjected to ultrasound for 10 min. Subsequently, 2 mL was taken and placed in a polystyrene cell in an apparatus (Malvern, model Zetasizer ZSU3100, Worcestershire, UK). Measurements were conducted using a laser with a wavelength of 632.8 nm, a scattering angle of −14.14°, and an electric field strength of 10 V/cm, with direct reading.
2.9. Swelling Power (SP), Water Solubility Index (WSI), and Water Absorption Index (WAI)
The procedure was performed following the guidelines outlined by Gani et al. [
30], with certain modifications. A 0.2 g sample of modified starch (M
0) was dispersed in 10 mL of distilled water. The swelling power, water solubility index, and water absorption index were calculated at 60, 70, and 80 °C. After manual shaking for 30 min, the mixture was centrifuged at 900×
g for 30 min at room temperature using a centrifuge (Tom’s/Hampton-USA Science and Technology Corporation, model 033R-2). The supernatant was then carefully removed from the Petri dishes, and the swollen starch granules (M
1) were weighed. The supernatant was dried for four hours at a temperature of 90 °C until reaching constant weight (M
2). SP, WSI, and WAI values were calculated using Equations (6), (7), and (8), respectively.
2.10. Paste Properties
Paste property analysis was carried out using an Anton Paar rotational rheometer (Model MCR 702e, Graz, Austria) equipped with a starch cell attachment. Initially, the water content of the starch was determined, and a 10% w/w suspension was prepared and allowed to stand for 60 min. Subsequently, 20 g of the suspension was weighed and introduced to the starch cell. The experimental procedure consisted of an initial agitation at 600 rpm for 20 s, followed by a constant speed of 160 rpm throughout the test. This protocol included a 2 min stabilization phase at 50 °C, followed by heating at 6 °C/min to 95 °C, a 5 min isothermal hold at 95 °C, cooling at 6 °C/min to 50 °C, and finally an isothermal retention of 2 min at 50 °C. Parameters such as paste temperature, maximum viscosity, minimum viscosity, final viscosity, burst viscosity, and recovery viscosity were recorded using Reocompass© software V1.30.999.Release. To ensure the accuracy and reliability of the results, all measurements were performed in duplicate.
2.11. X-ray Diffraction
X-ray diffraction measurements were performed on modified starch samples using an X-ray diffractometer, X’Pert Pro (PANalytical, Almelo, The Netherlands). CuKa-1 radiation at 40 mA and 40 kV was used. Data were obtained by scanning over a 2θ interval at room temperature, specifically between 5 and 50 °C. Two grams of sample were taken and adjusted to a moisture content of 4% prior to analysis. For analysis, the starch sample was placed in a PANalytical universal powder sample holder. Origin Pro v.2022 software was used to determine the crystalline and amorphous regions of the modified starches, successfully separating them. The relative crystallinity was then calculated by dividing the crystalline peak area by the total diffraction area obtained.
2.12. Thermal Analysis
The thermal stability of the modified starch samples was evaluated by two methods: thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). In TGA, the sample was loaded into an alumina crucible (Al2O3) and analyzed using a calorimetric analyzer (TA Instruments, TGA550, New Castle, DE, USA). Spectra were recorded using Trios V5.0.0.44616T software, with a temperature range of 20 to 600 °C, a heating rate of 10 °C/min, and a nitrogen flow rate of 50 mL/min. Differential scanning calorimetry (DSC) (DSC2500, TA Instruments, New Castle, DE, USA) was used to determine the thermal conversion parameters in a nitrogen atmosphere (50 mL/min). The modified starch samples were hermetically sealed in aluminum containers and heated from 20 to 200 °C at a heating rate of 5 °C/min. The initial DSC measurement was performed using an empty plate as a reference under the same conditions as for the sample measurement. The spectra were recorded using Trios V5.0.0.44616 software.
2.13. FT-IR Spectra
Tablets containing a 0.1% concentration of KBr-modified starch (IR grade, Darmstadt, Germany) were prepared and loaded into the transmission module of a Thermo Fisher Nicolet IS50 (Waltham, MA, USA) FT-IR (Fourier transform IR spectroscopy) spectrometer. The wavenumber range used was 4000 to 400 cm−1, with a resolution of 4 cm−1.
2.14. SEM Morphology
A Quanta model 200 scanning electron microscope (SEM, Thermo Fisher, Waltham, MA, USA) was used to analyze the modified starch samples. The starch granules were fixed directly onto a conductive carbon tape, ensuring stability during analysis. Each sample was introduced into the SEM vacuum chamber and measured at an accelerating voltage of 25 kV and 1000× magnification. The SEM software AutoScript 4 enabled image tracing and quantitative measurements of the morphology of the modified starch to be obtained.
2.15. Statistical Analysis
The results obtained were analyzed using the general linear model ANOVA with Tukey’s multiple comparisons test with a confidence level of 95% in the trial version of the Minitab software v.20. Tables were organized in Microsoft Excel, and figures were created with Origin Pro software.
4. Conclusions
Modified starch obtained from native potatoes is seen as a promising raw material for the manufacture of biocomposites for the food and pharmaceutical industries. Among the modification techniques employed, ethanol precipitation has stood out for its ability to significantly reduce the amylose content compared to other methods. On the other hand, enzymatic hydrolysis has been shown to maintain relatively high levels of amylose, suggesting its relevance for applications that demand a high content of this component. As for the changes generated by starch modification, ethanol precipitation induced a decrease in the water absorption index (WAI) in all the varieties studied, showing a marked interaction between the factors involved. Likewise, this technique has caused alterations in both the structure and properties of starch, with a notable reduction in viscosity measured by RVA. Despite this decrease in viscosity, an improvement in the ability of the modified starch to form a paste was observed, suggesting greater stability during gelatinization, both at high and low temperatures, as well as greater strength of the resulting gels. On the other hand, acid treatment generated a decrease in starch viscosity, although it maintained the typical characteristics of starch paste. In addition, both acid and enzymatic treatments produced modifications in the X-ray diffraction patterns in certain varieties, with a consequent decrease in the crystalline components. In contrast, ethanol precipitation reduced the amorphous region and led to an increase in the crystalline region in all the varieties studied. Acid treatment was found to confer higher thermal stability compared to enzymatic hydrolysis and ethanol precipitation, and exceptionally low gelatinization temperatures were detected by DSC. Also, ethanol precipitation altered the morphology of native starch, as demonstrated by scanning electron microscopy (SEM) images and nanometer particle sizes (DLS).
These characteristics suggest a broad application potential for modified starches, without the need for additional heat treatment.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}