
Targeted High-Throughput Sequencing Enables the Detection of Single Nucleotide Variations in CRISPR/Cas9 Gene-Edited Organisms
 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Sample Preparation
2.3. ddPCR Assays
2.4. Conventional PCR Assays
2.5. Library Preparation, Sequencing, and Data Analysis
3. Results
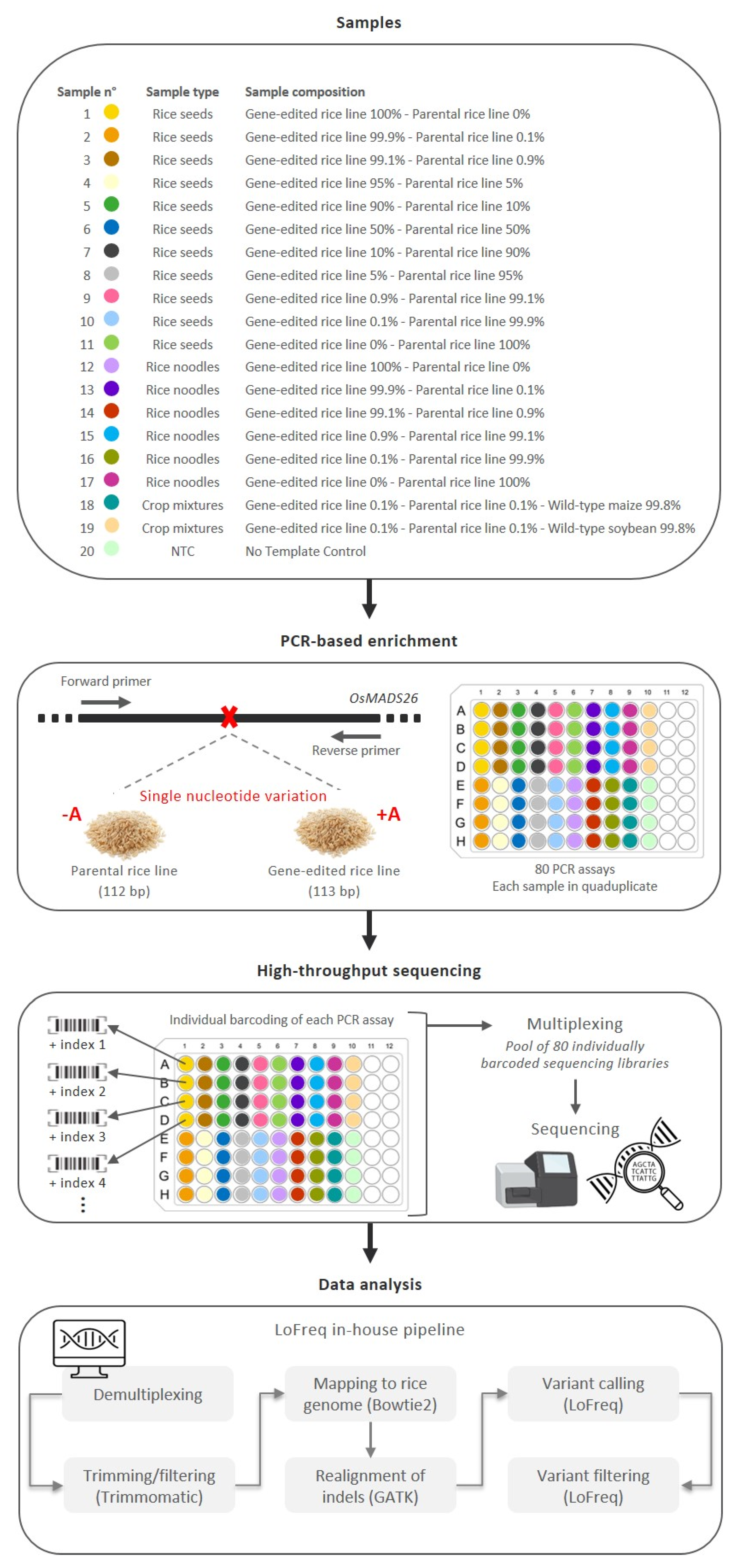
3.1. Development of a Workflow for Targeted High-Throughput Sequencing
3.2. Assessment of Sensitivity
3.3. Assessment of Applicability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ISAAA. Available online: https://www.isaaa.org/kc/cropbiotechupdate/article/default.asp?ID=19024 (accessed on 23 October 2022).
- Grohmann, L.; Keilwagen, J.; Duensing, N.; Dagand, E.; Hartung, F.; Wilhelm, R.; Bendiek, J.; Sprink, T. Detection and Identi-fication of Genome Editing in Plants: Challenges and Opportunities. Front. Plant Sci. 2019, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Chhalliyil, P.; Ilves, H.; Kazakov, S.A.; Howard, S.J.; Johnston, B.H.; Fagan, J.A. Real-Time Quantitative PCR Method Specific for Detection and Quantification of the First Commercialized Genome-Edited Plant. Foods 2020, 9, 1245. [Google Scholar] [CrossRef] [PubMed]
- Menz, J.; Modrzejewski, D.; Hartung, F.; Wilhelm, R.; Sprink, T. Genome Edited Crops Touch the Market: A View on the Global Development and Regulatory Environment. Front. Plant Sci. 2020, 11, 586027. [Google Scholar] [CrossRef]
- Tyagi, S.; Kumar, R.; Das, A.; Won, S.Y.; Shukla, P. CRISPR-Cas9 system: A genome-editing tool with endless possibilities. J. Biotechnol. 2022, 10, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hussain, A.; Manghwar, H.; Xie, K.; Xie, S.; Zhao, S.; Larkin, R.M.; Qing, P.; Jin, S.; Ding, F. Genome editing with the CRISPR-Cas system: An art, ethics and global regulatory perspective. Plant Biotechnol. J. 2020, 18, 1651–1669. [Google Scholar] [CrossRef]
- Ribarits, A.; Eckerstorfer, M.; Simon, S.; Stepanek, W. Genome-Edited Plants: Opportunities and Challenges for an Anticipatory Detection and Identification Framework. Foods 2021, 10, 430. [Google Scholar] [CrossRef]
- Directive 2001/18/EC of the European Parliament and of the Council of 12 March 2001 on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC. Off. J. Eur. Commun. 2001, L106, 1–38.
- Regulation (EC) No 1829/2003 of the European Parliament and of the Council of 22 September 2003 on genetically modified food and feed. Off. J. Eur. Union 2003, L268, 1–23.
- Regulation (EC) No 1830/2003 of the European Parliament and of the Council of 22 September 2003 concerning the traceability and labelling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending Directive 2001/18/EC. Off. J. Eur. Union 2003, L268, 24–28.
- European Court of Justice C-528/16-Judgement of 25 July 2018 on New Mutagenesis Techniques. Available online: http://curia.europa.eu/juris/document/document.jsf?text=&docid=204387&pageIndex=0&doclang=EN&mode=lst&dir=&occ=first&part=1&cid=138460 (accessed on 23 October 2022).
- ENGL. Evaluation of the Scientific Publication “A Real-Time Quantitative PCR Method Specific for Detection and Quantification of the First Commercialized Genome-Edited Plant” P. Chhalliyil et al. in: Foods 2020, 9, 1245. Available online: https://gmo-crl.jrc.ec.europa.eu/ENGL/docs/ENGL%20Evaluation%20of%20the%20scientific%20publication%2002-10-2020.pdf (accessed on 22 October 2022). [CrossRef]
- Peng, C.; Wang, H.; Xu, X.; Wang, X.; Chen, X.; Wei, W.; Lai, Y.; Liu, G.; Godwin, I.D.; Li, J.; et al. High throughput detection and screening of plants modified by gene editing using quantitative real-time polymerase chain reaction. Plant J. 2018, 95, 557–567. [Google Scholar] [CrossRef]
- Peng, C.; Zheng, M.; Ding, L.; Chen, X.; Wang, X.; Feng, X.; Wang, J.; Xu, J. Accurate Detection and Evaluation of the Gene-Editing Frequency in Plants Using Droplet Digital PCR. Front. Plant Sci. 2020, 11, 610790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, J.; Zhao, S.; Yan, X.; Si, N.; Gao, H.; Li, Y.; Zhai, S.; Xiao, F.; Wu, G.; et al. An Editing-Site-Specific PCR Method for Detection and Quantification of CAO1-Edited Rice. Foods 2021, 10, 1209. [Google Scholar] [CrossRef] [PubMed]
- Fraiture, M.A.; Guiderdoni, E.; Meunier, A.C.; Papazova, N.; Roosens, N.H.C. ddPCR strategy to detect a gene-edited plant carrying a single variation point: Technical feasibility and interpretation issues. Food Control 2022, 137, 108904. [Google Scholar] [CrossRef]
- ENGL. Detection of Food and Feed Plant Products Obtained by New Mutagenesis Techniques. Available online: https://gmo-crl.jrc.ec.europa.eu/doc/JRC116289-GE-report-ENGL.pdf (accessed on 23 October 2022).
- Shillito, R.D.; Whitt, S.; Ross, M.; Ghavami, F.; De Vleesschauwer, D.; D’Halluin, K.; Van Hoecke, A.; Meulewaeter, F. Detection of genome edits in plants—From editing to seed. In Vitro Cell. Dev. Biol. Plant 2021, 57, 595–608. [Google Scholar] [CrossRef]
- Sturme, M.H.J.; van der Berg, J.P.; Bouwman, L.M.S.; De Schrijver, A.; de Maagd, R.A.; Kleter, G.A.; Battaglia-de Wilde, E. Occurrence and Nature of Off-Target Modifications by CRISPR-Cas Genome Editing in Plants. ACS Agric. Sci. Technol. 2022, 2, 192–201. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Herman, P.; Taverniers, I.; De Loose, M.; Deforce, D.; Roosens, N.H.C. Current and new approaches in gmo detection: Challenges and solutions. Biomed. Res. Int. 2015, 2015, 392872. [Google Scholar] [CrossRef]
- Whale, A.S.; Huggett, J.F.; Tzonev, S. Fundamentals of multiplexing with digital PCR. Biomol. Detect. Quantif. 2016, 10, 15–23. [Google Scholar] [CrossRef]
- Grohmann, L.; Barbante, A.; Eriksson, R.; Gatto, F.; Georgieva, T.; Huber, I.; Hulin, J.; Köppel, R.; Marchesi, U.; Marmin, L.; et al. Guidance Document on Multiplex Real-Time PCR Methods; EUR 30708 EN; Publications Office of the European Union: Luxembourg, 2021. [Google Scholar]
- Shirasawa, K.; Kuwata, C.; Watanabe, M.; Fukami, M.; Hirakawa, H.; Isobe, S. Target Amplicon Sequencing for Genotyping Genome-Wide Single Nucleotide Polymorphisms Identified by Whole-Genome Resequencing in Peanut. Plant Genome 2016, 9, 3. [Google Scholar] [CrossRef]
- Onda, Y.; Takahagi, K.; Shimizu, M.; Inoue, K.; Mochida, K. Multiplex PCR Targeted Amplicon Sequencing (MTA-Seq): Simple, Flexible, and Versatile SNP Genotyping by Highly Multiplexed PCR Amplicon Sequencing. Front. Plant Sci. 2018, 9, 201. [Google Scholar] [CrossRef]
- Jo, J.; Kim, Y.; Kim, G.W.; Kwon, J.K.; Kang, B.C. Development of a Panel of Genotyping-in-Thousands by Sequencing in Capsicum. Front. Plant Sci. 2021, 12, 769473. [Google Scholar] [CrossRef] [PubMed]
- Kovalic, D.; Garnaat, C.; Guo, L.; Yan, Y.; Groat, J.; Silvanovich, A.; Ralston, L.; Huang, M.; Tian, Q.; Christian, A.; et al. The use of next generation sequencing and junction sequence analysis bioinformatics to achieve molecular characterization of crops improved through modern biotechnology. Plant Genome 2022, 5, 149–163. [Google Scholar] [CrossRef]
- Wahler, D.; Schauser, L.; Bendiek, J.; Grohmann, L. Next generation sequencing as a tool for detailed molecular characterisation of genomic insertions and flanking regions in genetically modified plants: A pilot study using a rice event unauthorised in the EU. Food Anal. Methods 2013, 6, 1718–1727. [Google Scholar] [CrossRef]
- Liang, C.; van Dijk, J.P.; Scholtens, I.M.; Staats, M.; Prins, T.W.; Voorhuijzen, M.M.; da Silva, A.M.; Arisi, A.C.; den Dunnen, J.T.; Kok, E.J. Detecting authorized and unauthorized genetically modified organisms containing vip3A by real-time PCR and next-generation sequencing. ABC 2014, 406, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Holst-Jensen, A.; Spilsberg, B.; Arulandhu, A.J.; Kok, E.; Shi, J.; Zel, J. Application of whole genome shotgun sequencing for detection and characterization of genetically modified organisms and derived products. ABC 2016, 408, 4595–4614. [Google Scholar] [CrossRef] [PubMed]
- Willems, S.; Fraiture, M.A.; Deforce, D.; De Keersmaecker, S.; De Loose, M.; Ruttink, T.; Van Nieuwerburgh, F.; Roosens, N. Statistical framework for detection of genetically modified organisms based on Next Generation Sequencing. Food Chem. 2016, 192, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Fraiture, M.A.; Herman, P.; Papazova, N.; De Loose, M.; Ruttink, T.; Roosens, N.H.C. An integrated strategy combining DNA walking and NGS to detect GMO. Food Chem. 2017, 232, 351–358. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Herman, P.; De Loose, M.; Debode, F.; Roosens, N.H.C. How can we better detect unauthorized GMO in the food and feed chain. Trends Biotechnol. 2017, 35, 508–517. [Google Scholar] [CrossRef]
- Bogožalec Košir, A.; Arulandhu, A.J.; Voorhuijzen, M.M.; Xiao, H.; Hagelaar, R.; Staats, M.; Costessi, A.; Žel, J.; Kok, E.J.; van Dijk, J.P. ALF: A strategy for identification of unauthorized GMOs in complex mixtures by a GW-NGS method and dedicated bioinformatics analysis. Sci. Rep. 2017, 7, 14155. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Saltykova, A.; Hoffman, S.; Winand, R.; Deforce, D.; Vanneste, K.; De Keersmaecker, S.C.J.; Roosens, N.H.C. Nanopore sequencing technology: A new route for the fast detection of unauthorized GMO. Sci. Rep. 2018, 8, 7903. [Google Scholar] [CrossRef]
- Debode, F.; Hulin, J.; Charloteaux, B.; Coppieters, W.; Hanikenne, M.; Karim, L.; Berben, G. Detection and identification of transgenic events by next generation sequencing combined with enrichment technologies. Sci. Rep. 2019, 9, 15595. [Google Scholar] [CrossRef] [PubMed]
- Fraiture, M.A.; Ujhelyi, G.; Ovesná, J.; Van Geel, D.; De Keersmaecker, S.C.J.; Saltykova, A.; Papazova, N.; Roosens, N.H.C. MinION sequencing technology to characterize unauthorized GM petunia plants circulating on the European Union market. Sci. Rep. 2019, 9, 7141. [Google Scholar] [CrossRef] [PubMed]
- Boutigny, A.L.; Fioriti, F.; Rolland, M. Targeted MinION sequencing of transgenes. Sci. Rep. 2020, 10, 15144. [Google Scholar] [CrossRef] [PubMed]
- Fraiture, M.A.; Papazova, N.; Vanneste, K.; De Keersmaecker, S.C.J.; Roosens, N.H.C. GMO Detection and Identification Using Next-generation Sequencing in DNA Techniques to Verify Food Authenticity: Applications in Food Fraud; Burns, M., Foster, L., Walker, M., Eds.; Royal Society of Chemistry Publishing: Cambridge, UK, 2020; pp. 96–106. [Google Scholar]
- Saltykova, S.; Van Braekel, J.; Papazova, N.; Fraiture, M.A.; Deforce, D.; Vanneste, K.; De Keersmaecker, S.C.J.; Roosens, N.H.C. Detection and identification of authorized and unauthorized GMOs using high-throughput sequencing with the support of a sequence-based GMO database. Food Chem. Mol. Sci. 2022, 4, 100096. [Google Scholar] [CrossRef] [PubMed]
- ENGL. Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing. Available online: https://gmo-crl.jrc.ec.europa.eu/guidance-documents (accessed on 2 November 2022).
- Regulation (EC) No 619/2011 of 24 June 2011 laying down the methods of sampling and analysis for the official control of feed as regards presence of genetically modified material for which an authorisation procedure is pending or the authorisation of which has expired. Off. J. Eur. Union 2011, L166, 9–15.
- Takabatake, R.; Egi, E.; Soga, K.; Narushima, J.; Yoshiba, S.; Shibata, N.; Nakamura, K.; Kondo, K.; Kishine, M.; Mano, J.; et al. Development and Interlaboratory Validation of a Novel Reproducible Qualitative Method for GM Soybeans Using Comparative Cq-Based Analysis for the Revised Non-GMO Labeling System in Japan. Anal. Chem. 2022, 94, 13447–13454. [Google Scholar] [CrossRef] [PubMed]
- Mansueto, L.; Fuentes, R.R.; Borja, F.N.; Detras, J.; Abriol-Santos, J.M.; Chebotarov, D.; Sanciangco, M.; Palis, K.; Copetti, D.; Poliakov, A.; et al. Rice SNP-seek database update: New SNPs, indels, and queries. Nucleic Acids Res. 2017, 45, D1075–D1081. [Google Scholar] [CrossRef]
- ISO 21571:2005. Foodstuffs—Methods of Analysis for the Detection of Genetically Modified Organisms and Derived Products—Nucleic acid Extraction. ISO: Genève, Switzerland, 2015.
- EURL. Sampling and DNA extraction of cotton seeds. Report from the Validation of the “CTAB/Genomic-tip 20” method for DNA extraction from ground cotton seeds. Available online: http://gmo-crl.jrc.ec.europa.eu/summaries/281-3006%20Cotton_DNAExtr.pdf (accessed on 7 October 2022).
- Fraiture, M.A.; Herman, P.; Taverniers, I.; De Loose, M.; Deforce, D.; Roosens, N.H.C. An innovative and integrated approach based on DNA walking to identify unauthorised GMOs. Food Chem. 2014, 147, 60–69. [Google Scholar] [CrossRef]
- Broeders, S.; Fraiture, M.A.; Vandermassen, E.; Delvoye, M.; Barbau-Piednoir, E.; Lievens, A.; Roosens, N.H.C. New qualitative trait-specific SYBR®Green qPCR methods to expand the panel of GMO screening methods used in the CoSYPS. Eur. Food Res. Technol. 2015, 241, 275–287. [Google Scholar] [CrossRef]
- Arumuganathan, K.; Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Leitch, I.J.; Johnston, E.; Pellicer, J.; Hidalgo, O.; Bennett, M.D. The Plant DNA C-values database (release 7.1): An updated online repository of plant genome size data for comparative studies. New Phytol. 2019, 226, 301–305. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2013, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Picard Toolkit. Broad Institute, GitHub Repository. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 12 March 2022).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2014, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Khong, N.G.; Pati, P.K.; Richaud, F.; Parizot, B.; Bidzinski, P.; Mai, C.D.; Bes, M.; Bourrié, I.; Meynard, D.; Beeckman, T.; et al. OsMADS26 negatively regulates resistance to pathogens and drought tolerance in rice. Plant Physiol. 2015, 169, 2935–2949. [Google Scholar] [CrossRef]
- Van Poelvoorde, L.A.E.; Delcourt, T.; Coucke, W.; Herman, P.; De Keersmaecker, S.C.J.; Saelens, X.; Roosens, N.H.C.; Vanneste, K. Strategy and performance evaluation of low-frequency variant calling for SARS-CoV-2 using targeted deep Illumina sequencing. Front. Microbiol. 2021, 12, 747458. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Herman, P.; Taverniers, I.; De Loose, M.; Van Nieuwerburgh, F.; Deforce, D.; Roosens, N.H.C. Validation of a sensitive DNA walking strategy to characterise unauthorised GMOs using model food matrices mimicking common rice products. Food Chem. 2015, 173, 1259–1265. [Google Scholar] [CrossRef]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffron, S.; Van Der Vossen, J.; Tang, S.; Katase, M.; et al. The use of next generation sequencing for improving food safety: Translation into practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef]
- Zhu, H.; Misel, L.; Graham, M.; Robinson, M.L.; Liang, C. CT-Finder: A Web Service for CRISPR Optimal Target Prediction and Visualization. Sci Rep. 2016, 6, 25516. [Google Scholar] [CrossRef] [PubMed]
- Entine, J.; Felipe, M.S.S.; Groenewald, J.H.; Kershen, D.L.; Lema, M.; McHughen, A.; Nepomuceno, A.L.; Ohsawa, R.; Ordonio, R.L.; Parrott, W.A.; et al. Regulatory approaches for genome edited agricultural plants in select countries and jurisdictions around the world. Transgenic Res. 2021, 30, 551–584. [Google Scholar] [CrossRef] [PubMed]
- Zimny, T.; Sowa, S. Potential effects of asymmetric legal classification of gene edited plant products in international trade, from the perspective of the EU. EFB Bioecon. J. 2021, 1, 100016. [Google Scholar] [CrossRef]
- D’aes, J.; Fraiture, M.A.; Bogaerts, B.; De Keersmaecker, S.C.J.; Roosens, N.H.C.; Vanneste, K. Metagenomic Characterization of Multiple Genetically Modified Bacillus Contaminations in Commercial Microbial Fermentation Products. Life 2022, 12, 1971. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Rice Seed Sample Description | CRISPR/Cas9 Adenosine Insertion | ||||||
|---|---|---|---|---|---|---|---|
| Sample n° | GE Rice Line Content | Allele Frequency ~ | Depth of Coverage ~ | ||||
| Percentage | Copy Number | Detection | Percentage | StDev | Value | StDev | |
| 1 | 100 | 13,733.3 | + | 99.92 | 0.03 | 119,592 | 29,499 |
| 2 | 99.9 | 13,346.7 | + | 99.86 | 0.03 | 97,038 | 25,007 |
| 3 | 99.1 | 13,273.3 | + | 99.73 | 0.02 | 70,873 | 12,339 |
| 4 | 95 | 12,113.3 | + | 97.97 | 0.15 | 101,581 | 63,687 |
| 5 | 90 | 10,753.3 | + | 92.06 | 0.24 | 75,787 | 36,739 |
| 6 | 50 | 5640.0 | + | 53.80 | 0.57 | 71,791 | 19,099 |
| 7 | 10 | 1153.3 | + | 8.01 | 0.29 | 86,841 | 10,531 |
| 8 | 5 | 528.0 | + | 4.00 | 0.15 | 98,283 | 40,588 |
| 9 | 0.9 | 85.3 | + | 0.66 | 0.01 | 101,464 | 33,345 |
| 10 | 0.1 | 10.8 | + * | 0.13 * | 0.02 | 126,724 | 42,631 |
| 11 | 0 | 0 | − | 0.00 | 0.00 | 158,436 | 99,002 |
| Rice noodle Sample Description | CRISPR/Cas9 Adenosine Insertion | ||||||
|---|---|---|---|---|---|---|---|
| Sample n° | GE Rice Line Content | Allele Frequency ~ | Depth of Coverage ~ | ||||
| Percentage | Copy Number | Detection | Percentage | StDev | Value | StDev | |
| 12 | 100 | 13,610.0 | + | 99.85 | 0.06 | 120,711 | 60,941 |
| 13 | 99.9 | 13,596.4 | + | 99.68 | 0.15 | 47,261 | 27,646 |
| 14 | 99.1 | 13,487.0 | + | 99.05 | 0.16 | 69,972 | 26,283 |
| 15 | 0.9 | 122.5 | + | 1.03 | 0.03 | 62,408 | 16,122 |
| 16 | 0.1 | 13.6 | + | 0.24 | 0.04 | 68,359 | 8826 |
| 17 | 0 | 0 | − | 0.00 | 0.00 | 67,906 | 37,437 |
| (A) | |||||||
| Sample Composition | |||||||
| Sample n°18 | 0.1 % GE rice line (14 estimated haploid genome copies) | ||||||
| 0.1 % parental rice line (14 estimated haploid genome copies) | |||||||
| 99.8 % WT maize (113,972 estimated haploid genome copies) | |||||||
| Sample n°19 | 0.1 % GE rice line (14 estimated haploid genome copies) | ||||||
| 0.1 % parental rice line (14 estimated haploid genome copies) | |||||||
| 99.8 % WT soybean (113,972 estimated haploid genome copies) | |||||||
| (B) | |||||||
| Crop Mixture Sample Description | CRISPR/Cas9 Adenosine Insertion | ||||||
| Sample n° | GE Rice Line Content | Allele Frequency ~ | Depth of Coverage ~ | ||||
| Copy Number | Rice Ratio * | Detection | Percentage | StDev | Value | StDev | |
| 18 | 10.6 | 50 | + | 40.46 | 10.78 | 533 | 50 |
| 19 | 9.3 | 50 | + | 48.52 | 18.12 | 23,124 | 3181 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraiture, M.-A.; D’aes, J.; Guiderdoni, E.; Meunier, A.-C.; Delcourt, T.; Hoffman, S.; Vandermassen, E.; De Keersmaecker, S.C.J.; Vanneste, K.; Roosens, N.H.C. Targeted High-Throughput Sequencing Enables the Detection of Single Nucleotide Variations in CRISPR/Cas9 Gene-Edited Organisms. Foods 2023, 12, 455. https://doi.org/10.3390/foods12030455
Fraiture M-A, D’aes J, Guiderdoni E, Meunier A-C, Delcourt T, Hoffman S, Vandermassen E, De Keersmaecker SCJ, Vanneste K, Roosens NHC. Targeted High-Throughput Sequencing Enables the Detection of Single Nucleotide Variations in CRISPR/Cas9 Gene-Edited Organisms. Foods. 2023; 12(3):455. https://doi.org/10.3390/foods12030455
Chicago/Turabian StyleFraiture, Marie-Alice, Jolien D’aes, Emmanuel Guiderdoni, Anne-Cécile Meunier, Thomas Delcourt, Stefan Hoffman, Els Vandermassen, Sigrid C. J. De Keersmaecker, Kevin Vanneste, and Nancy H. C. Roosens. 2023. "Targeted High-Throughput Sequencing Enables the Detection of Single Nucleotide Variations in CRISPR/Cas9 Gene-Edited Organisms" Foods 12, no. 3: 455. https://doi.org/10.3390/foods12030455
APA StyleFraiture, M.-A., D’aes, J., Guiderdoni, E., Meunier, A.-C., Delcourt, T., Hoffman, S., Vandermassen, E., De Keersmaecker, S. C. J., Vanneste, K., & Roosens, N. H. C. (2023). Targeted High-Throughput Sequencing Enables the Detection of Single Nucleotide Variations in CRISPR/Cas9 Gene-Edited Organisms. Foods, 12(3), 455. https://doi.org/10.3390/foods12030455