
Exploring the Role of Active Functional Microbiota in Flavor Generation by Integrated Metatranscriptomics and Metabolomics during Niulanshan Baijiu Fermentation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling of the Fermented Grains
2.2. Metatranscriptomic Sequencing
2.3. Metabolite Profiles by LC-MS Analysis
2.4. Metabolite Profiles by GC-MS Analysis
2.5. Metabolite Profiles by HS-SPME-GC-MS Analysis
2.6. Data Processing
2.7. Multivariate Statistical Analysis
3. Results
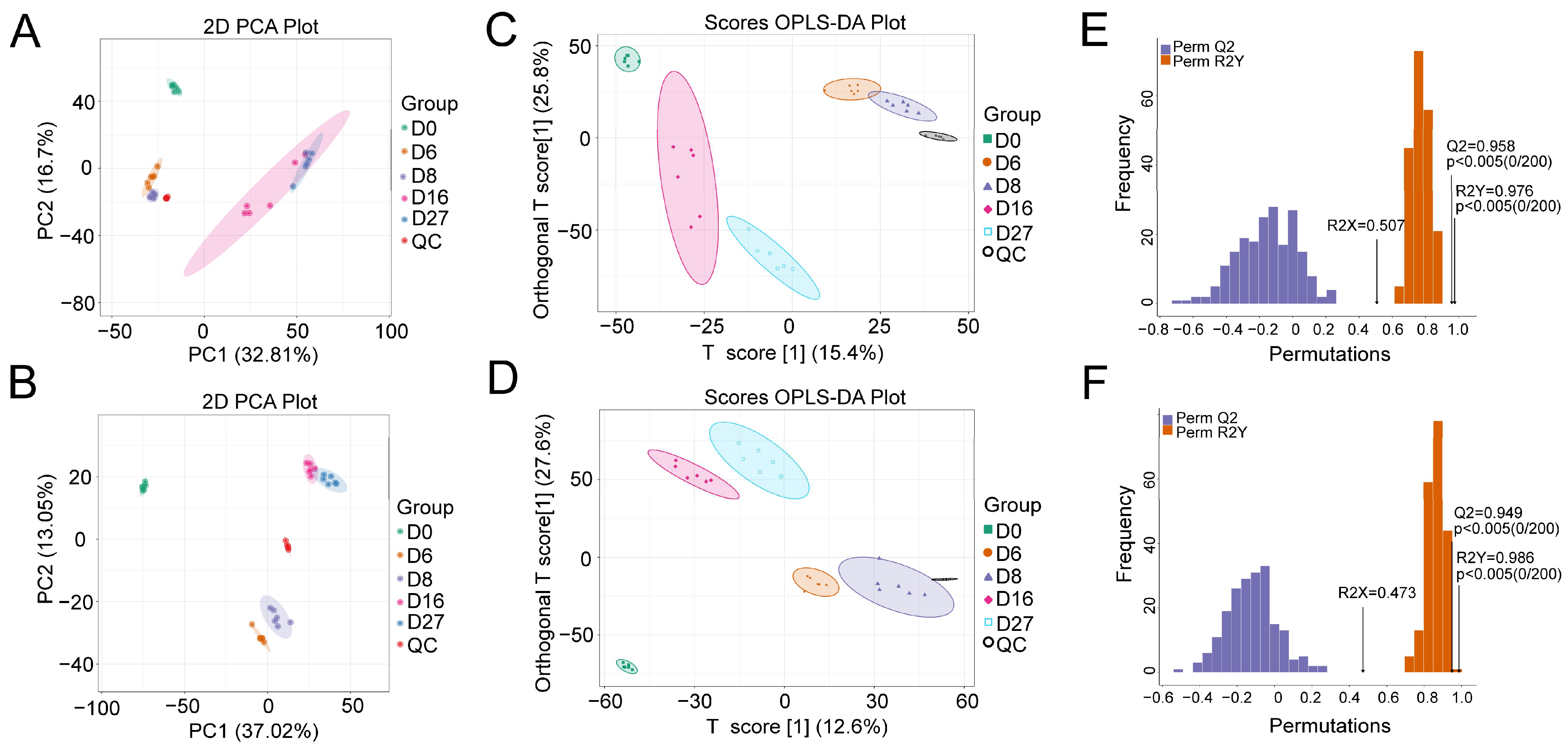
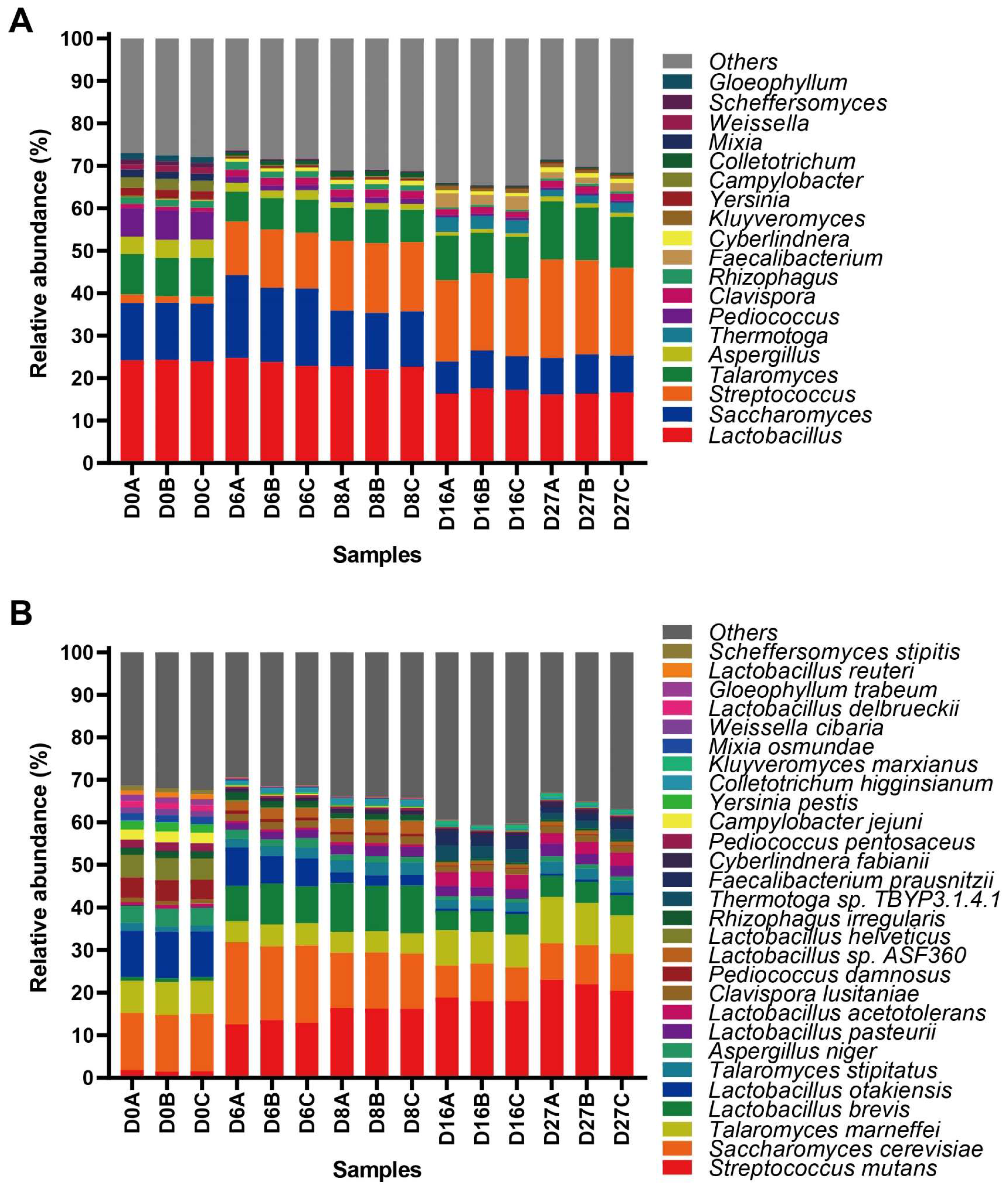
3.1. Active Microbial Community Diversity and Composition
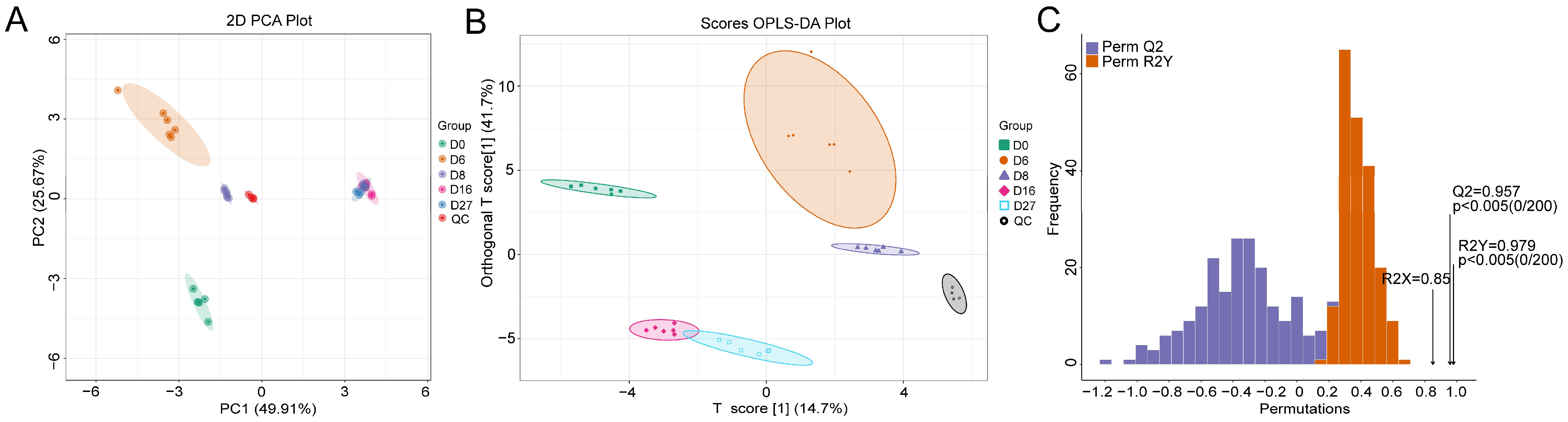
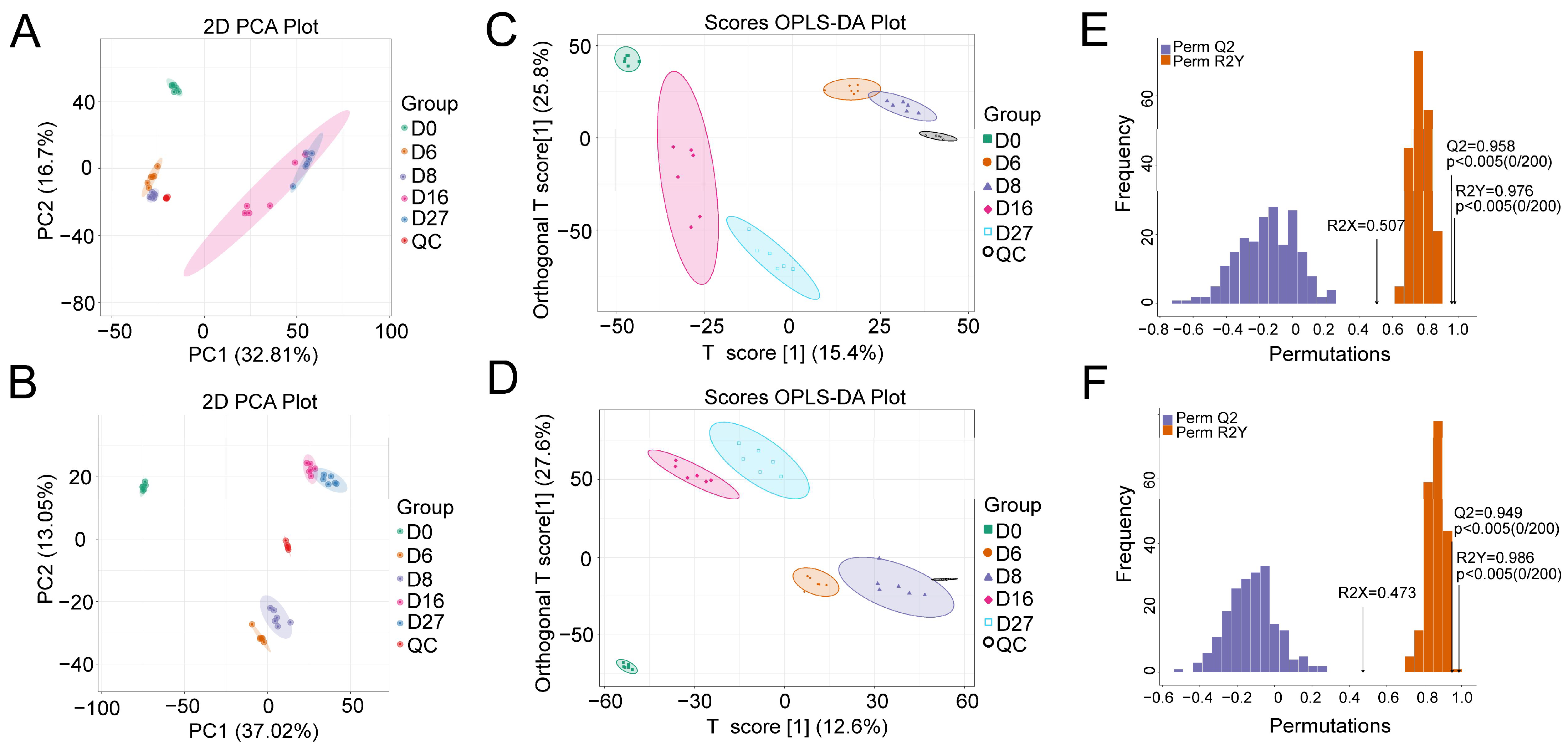
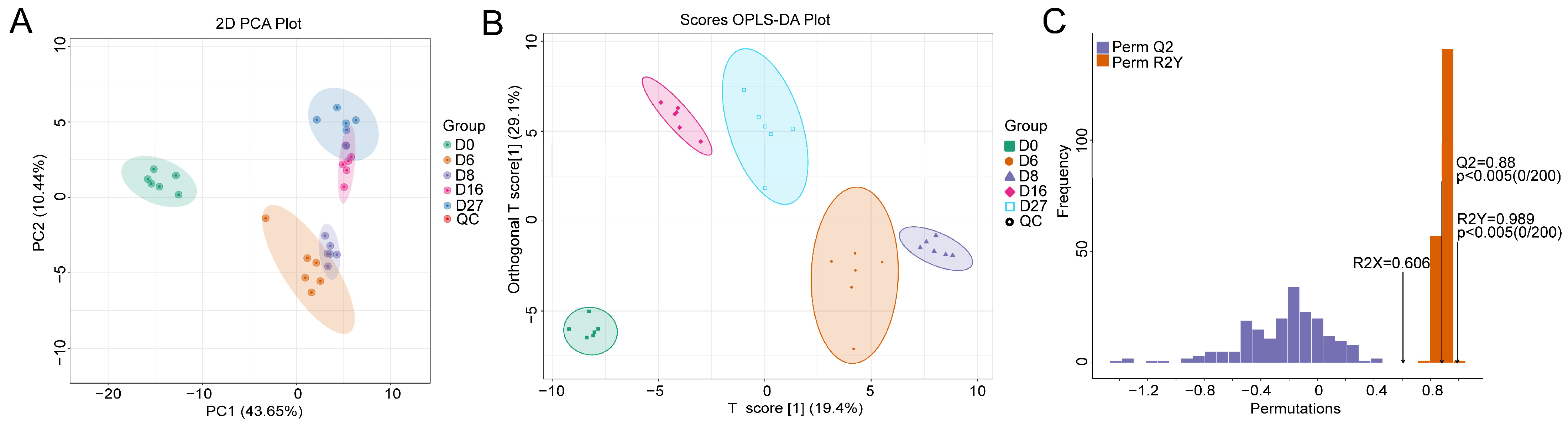
3.2. Characterization of the Polar Metabolites Based on LC-MS
3.3. Metabolite Characterization Based on the Derivatized GC-MS
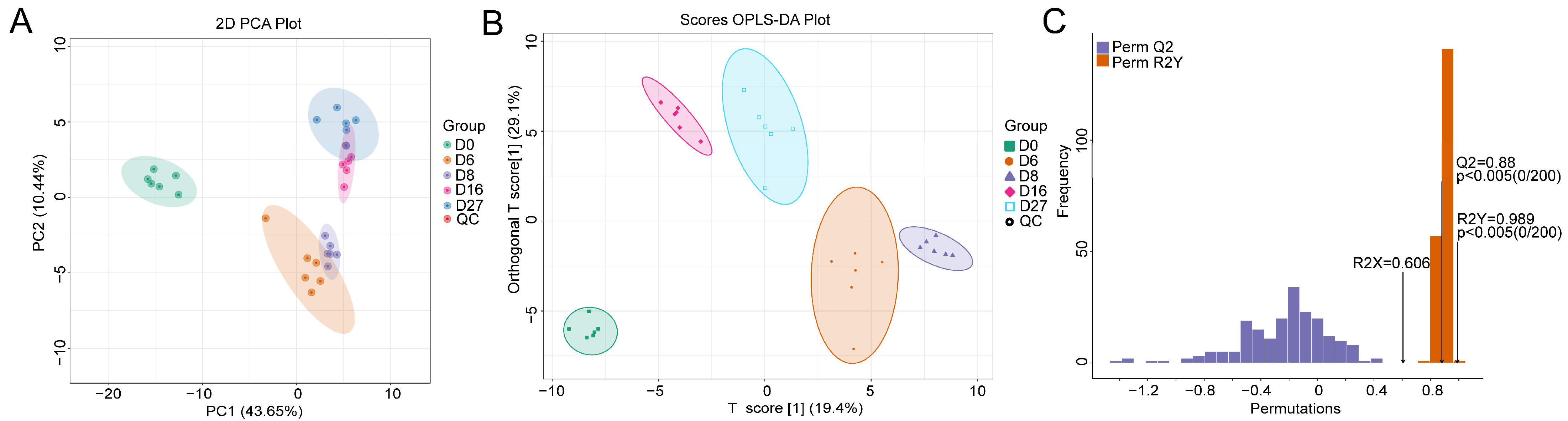
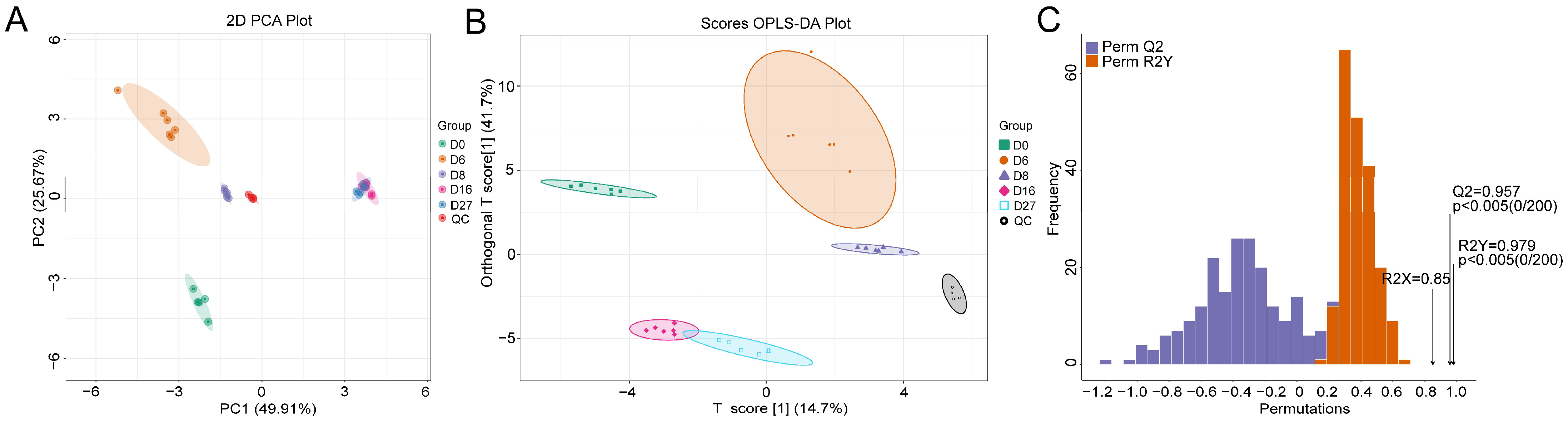
3.4. Metabolite Characterization Based on HS-SPME-GC-MS
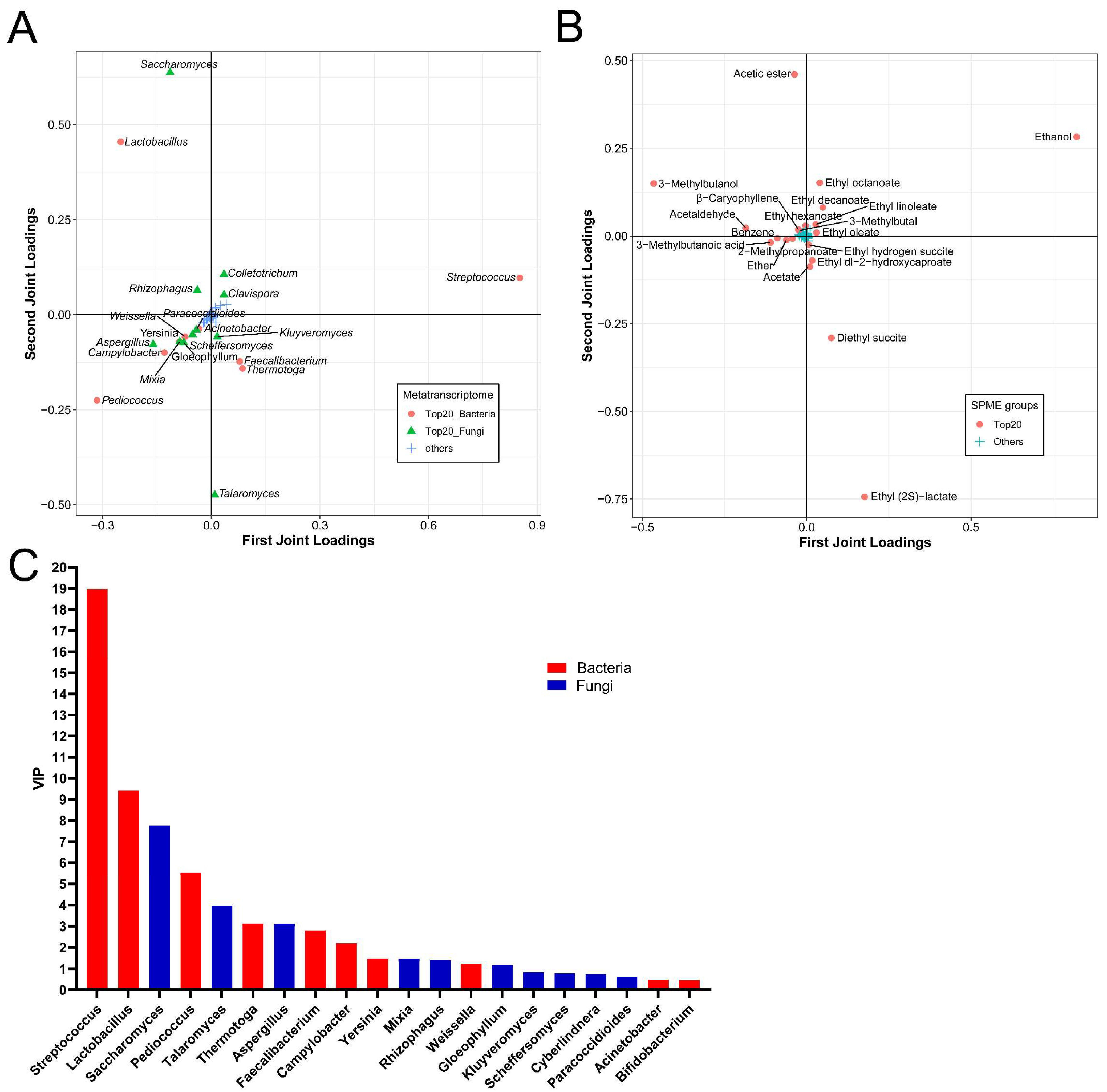
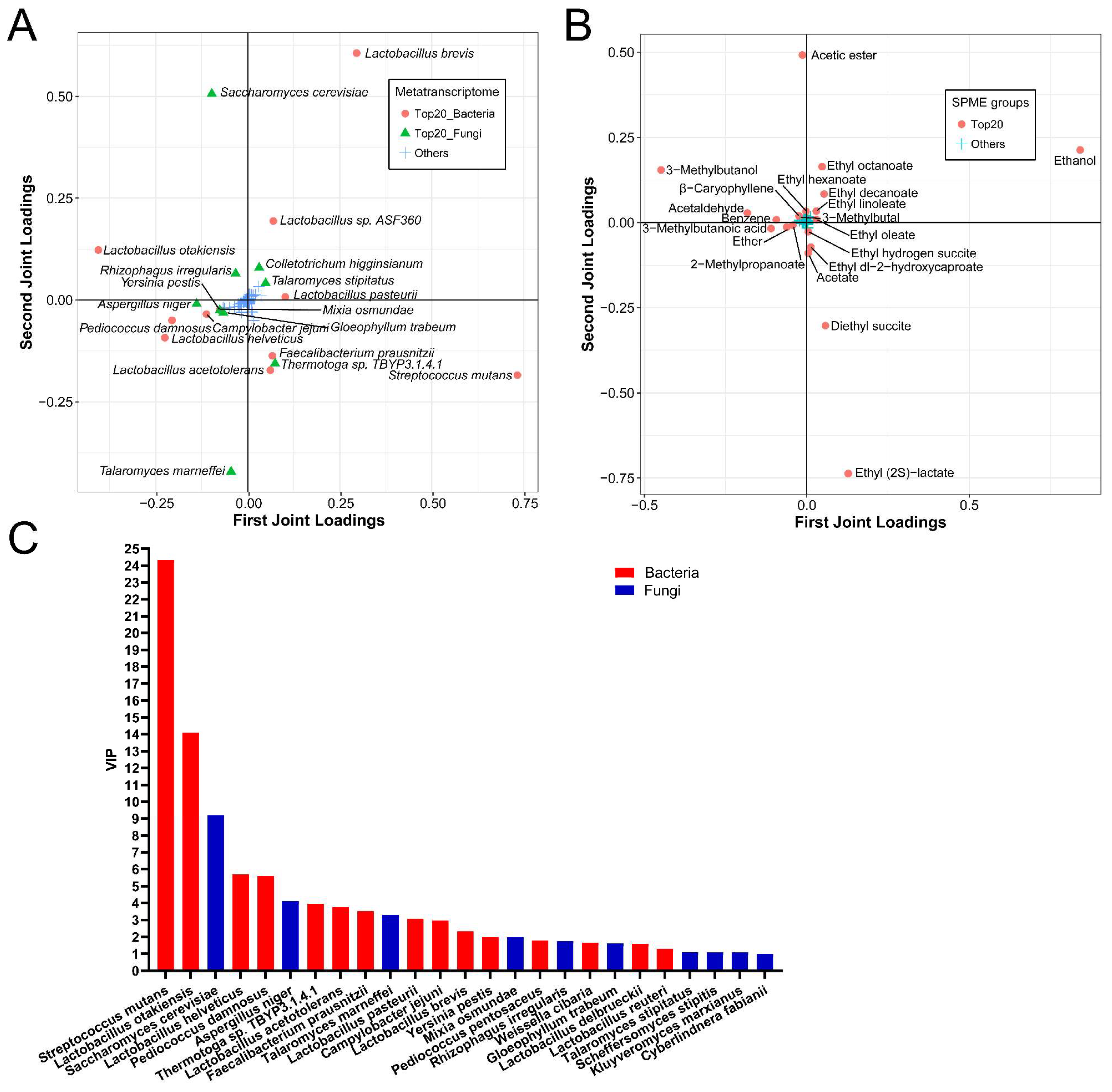
3.5. Functional Microorganisms and Volatile Compounds Identified Based on O2PLS-DA Model
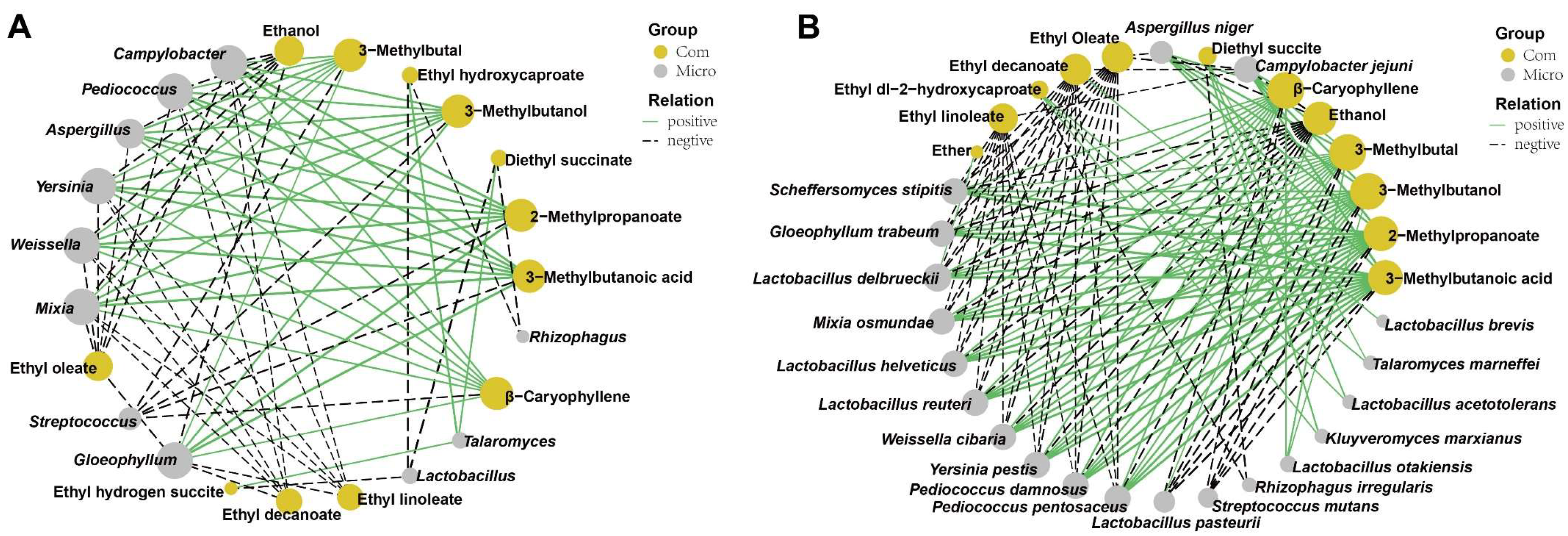
3.6. Correlations between the Active Microorganisms and the Volatile Flavors Based on O2PLS-DA Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L. Research trends in Jiang-flavor baijiu fermentation: From fermentation microecology to environmental ecology. J. Food Sci. 2022, 87, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, J.; Liu, X.; Zhang, C.; Zhao, Z.; Li, X.; Sun, B. Flavor mystery of Chinese traditional fermented baijiu: The great contribution of ester compounds. Food Chem. 2022, 369, 130920. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, J.; He, G. Effect of microbial interaction on flavor quality in Chinese baijiu fermentation. Front. Nutr. 2022, 9, 960712. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wang, J.; Shi, J.; Du, J.; Zhou, Y.; Huang, M.; Sun, B. Automatic and intelligent technologies of solid-state fermentation process of Baijiu production: Applications, challenges, and prospects. Foods 2021, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhu, Y.; Fang, C.; Wijffels, R.; Xu, Y. Can we control microbiota in spontaneous food fermentation?—Chinese liquor as a case example. Trends Food Sci. Technol. 2021, 110, 321–331. [Google Scholar] [CrossRef]
- Hong, J.; Tian, W.; Zhao, D. Research progress of trace components in sesame-aroma type of baijiu. Food Res. Int. 2020, 137, 109695. [Google Scholar] [CrossRef]
- Liu, H.; Sun, B. Effect of fermentation processing on the flavor of Baijiu. J. Agric. Food Chem. 2018, 66, 5425–5432. [Google Scholar] [CrossRef]
- Huang, X.; Fan, Y.; Lu, T.; Kang, J.; Pang, X.; Han, B.; Chen, J. Composition and metabolic functions of the microbiome in fermented grain during light-flavor baijiu fermentation. Microorganisms 2020, 8, 1281. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Zhu, T.; Wang, J.; Huang, M.; Wei, J.; Ye, H.; Wu, J.; Zhang, J.; Meng, N. Characterization of the key odorants and their content variation in Niulanshan Baijiu with different storage years using flavor sensory omics analysis. Food Chem. 2021, 376, 131851. [Google Scholar] [CrossRef]
- Ling, Y.; Li, W.; Tong, T.; Li, Z.; Li, Q.; Bai, Z.; Wang, G.; Chen, J.; Wang, Y. Assessing the microbial communities in four different Daqus by using PCR-DGGE, PLFA, and biolog analyses. Pol. J. Microbiol. 2020, 69, 1–11. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, Y.; Hao, W.; Duan, C.; Wang, S.; Wei, J.; Liu, G. Metatranscriptomics unravel composition, drivers, and functions of the active microorganisms in light-flavor liquor fermentation. Microbiol. Spectr. 2022, 10, e0215121. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Han, P.J.; Han, D.Y.; Zhou, S.; Li, K.; He, P.Y.; Zhen, P.; Yu, H.X.; Liang, Z.R.; Wang, X.W.; et al. Genetic diversity and population structure of the amylolytic yeast Saccharomycopsis fibuligera associated with Baijiu fermentation in China. J. Microbiol. 2021, 59, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.N.; Huang, X.N.; Chen, J.Y.; Yu, H.X.; Wang, X.Y.; Han, B.Z. Exploring the diversity and role of microbiota during material pretreatment of light-flavor Baijiu. Food Microbiol. 2020, 91, 103514. [Google Scholar] [CrossRef]
- Tang, J.; Liu, Y.; Lin, B.; Zhu, H.; Jiang, W.; Yang, Q.; Chen, S. Effects of ultra-long fermentation time on the microbial community and flavor components of light-flavor Xiaoqu Baijiu based on fermentation tanks. World J. Microbiol. Biotechnol. 2021, 38, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dong, K.; Wu, Y.; An, F.; Luo, Z.; Huang, Q.; Wei, S. Exploring the formation mechanism of off-flavor of irradiated yak meat based on metabolomics. Food Chem. X 2022, 16, 100494. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef]
- Jia, S.; Wang, Y.; Hu, J.; Ding, Z.; Liang, Q.; Zhang, Y.; Wang, H. Mineral and metabolic profiles in tea leaves and flowers during flower development. Plant Physiol. Biochem. 2016, 106, 316–326. [Google Scholar] [CrossRef]
- Tomita, S.; Nakamura, T.; Okada, S. NMR- and GC/MS-based metabolomic characterization of sunki, an unsalted fermented pickle of turnip leaves. Food Chem. 2018, 258, 25–34. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Ferrocino, I.; Bellio, A.; Giordano, M.; Macori, G.; Romano, A.; Rantsiou, K.; Decastelli, L.; Cocolin, L. Shotgun metagenomics and volatilome profile of the microbiota of fermented sausages. Appl. Environ. Microbiol. 2018, 84, e02120-17. [Google Scholar] [CrossRef]
- Luo, L.; Song, L.; Han, Y.; Zhen, P.; Han, D.; Zhao, X.; Zhou, X.; Wei, Y.; Yu, H.; Han, P.; et al. Microbial communities and their correlation with flavor compound formation during the mechanized production of light-flavor Baijiu. Food Res. Int. 2023, 172, 113139. [Google Scholar] [CrossRef]
- Hu, X.; Wang, K.; Chen, M.; Fan, J.; Han, S.; Hou, J.; Chi, L.; Liu, Y.; Wei, T. Profiling the composition and metabolic activities of microbial community in fermented grain for the Chinese strong-flavor Baijiu production by using the metatranscriptome, high-throughput 16S rRNA and ITS gene sequencings. Food Res. Int. 2020, 138, 109765. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, Q.; Chen, D.; Fu, B.; Zhang, Y.; Zhang, Y.; Xia, X.; Peng, N.; Liang, Y.; Zhao, S. Study on microbial communities and higher alcohol formations in the fermentation of Chinese Xiaoqu Baijiu produced by traditional and new mechanical technologies. Food Res. Int. 2021, 140, 109876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tan, Y.; Wei, J.; Du, H.; Xu, Y. Fungal interactions strengthen the diversity-functioning relationship of solid-state fermentation systems. mSystems 2022, 7, e0040122. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Yang, N.; Yang, J.; Hao, J.; Zhao, J.; Shi, S.; Hu, B. Effects of microbial interspecies relationships and physicochemical parameters on volatile flavors in sorghum-based fermented grains during the fermentation of Shanxi light-flavored liquor. Food Sci. Nutr. 2023, 11, 1452–1462. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Zheng, L.; Meng, L.; Liu, G.; Yang, T.; Lu, Z.; Chai, L.; Wang, S.; Shi, J.; et al. Machine learning based age-authentication assisted by chemo-kinetics: Case study of strong-flavor Chinese Baijiu. Food Res. Int. 2023, 167, 112594. [Google Scholar] [CrossRef]
- Wang, Z.; Ji, X.; Wang, S.; Wu, Q.; Xu, Y. Sugar profile regulates the microbial metabolic diversity in Chinese Baijiu fermentation. Int. J. Food Microbiol. 2021, 359, 109426. [Google Scholar] [CrossRef]
- Qian, W.; Lu, Z.M.; Chai, L.J.; Zhang, X.J.; Li, Q.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Cooperation within the microbial consortia of fermented grains and pit mud drives organic acid synthesis in strong-flavor Baijiu production. Food Res. Int. 2021, 147, 110449. [Google Scholar] [CrossRef]
- Cong, S.; Tian, K.; Zhang, X.; Lu, F.; Singh, S.; Prior, B.; Wang, Z.X. Synthesis of flavor esters by a novel lipase from Aspergillus niger in a soybean-solvent system. 3 Biotech 2019, 9, 244. [Google Scholar] [CrossRef]
- Ding, X.; Wu, C.; Huang, J.; Zhou, R. Changes in volatile compounds of Chinese Luzhou-flavor liquor during the fermentation and distillation process. J. Food Sci. 2015, 80, C2373–C2381. [Google Scholar] [CrossRef]
- Zha, M.; Sun, B.; Wu, Y.; Yin, S.; Wang, C. Improving flavor metabolism of Saccharomyces cerevisiae by mixed culture with Wickerhamomyces anomalus for Chinese Baijiu making. J. Biosci. Bioeng. 2018, 126, 189–195. [Google Scholar] [CrossRef]
- Wang, X.; Wang, B.; Sun, Z.; Tan, W.; Zheng, J.; Zhu, W. Effects of modernized fermentation on the microbial community succession and ethyl lactate metabolism in Chinese baijiu fermentation. Food Res. Int. 2022, 159, 111566. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, H.; Lu, H.; Wu, M.; Lin, M.; Zhang, C.; Zhao, Z.; Li, W.; Zhang, C.; Li, X.; et al. Characterization of an Aspergillus niger for efficient fatty acid ethyl ester synthesis in aqueous phase and the molecular mechanism. Front. Microbiol. 2021, 12, 820380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, F.; Guo, J.; Liu, G.; Guo, X.; Xiao, D. Enhanced ethyl caproate production of Chinese liquor yeast by overexpressing EHT1 with deleted FAA1. J. Ind. Microbiol. Biotechnol. 2014, 41, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, W.; Gong, R.; Xue, X.; Guan, X.; Song, L.; Guo, X.; Xiao, D. Improved ethyl caproate production of Chinese liquor yeast by overexpressing fatty acid synthesis genes with OPI1 deletion. J. Ind. Microbiol. Biotechnol. 2016, 43, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.T.; Ruan, S.L.; Zhao, Y.P.; Li, J.M.; Yang, C.; Cao, H. Multi-objective evaluation of freshly distilled brandy: Characterisation and distribution patterns of key odour-active compounds. Food Chem. X 2022, 14, 100276. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Fan, W.; Xu, Y. Characterization of the key odorants in light aroma type chinese liquor by gas chromatography-olfactometry, quantitative measurements, aroma recombination, and omission studies. J. Agric. Food Chem. 2014, 62, 5796–5804. [Google Scholar] [CrossRef]
- Niu, Y.; Yao, Z.; Xiao, Q.; Xiao, Z.; Ma, N.; Zhu, J. Characterization of the key aroma compounds in different light aroma type Chinese liquors by GC-olfactometry, GC-FPD, quantitative measurements, and aroma recombination. Food Chem. 2017, 233, 204–215. [Google Scholar] [CrossRef]
- Qian, Y.L.; An, Y.; Chen, S.; Qian, M.C. Characterization of Qingke Liquor Aroma from Tibet. J. Agric. Food Chem. 2019, 67, 13870–13881. [Google Scholar] [CrossRef]
- Fan, S.; Tang, K.; Xu, Y.; Chen, S. Characterization of the potent odorants in Tibetan Qingke Jiu by sensory analysis, aroma extract dilution analysis, quantitative analysis and odor activity values. Food Res. Int. 2020, 137, 109349. [Google Scholar] [CrossRef]
- Chen, Y.; Li, P.; Liao, L.; Qin, Y.; Jiang, L.; Liu, Y. Characteristic fingerprints and volatile flavor compound variations in Liuyang Douchi during fermentation via HS-GC-IMS and HS-SPME-GC-MS. Food Chem. 2021, 361, 130055. [Google Scholar] [CrossRef]
- Zhao, Z.; Sugimachi, M.; Yoshizaki, Y.; Yin, X.; Han, X.L.; Okutsu, K.; Futagami, T.; Tamaki, H.; Takamine, K. Impact of solid-state saccharification on the flavor of rice-flavor baijiu. J. Food Sci. 2021, 86, 4958–4968. [Google Scholar] [CrossRef] [PubMed]
- Dahham, S.S.; Tabana, Y.M.; Iqbal, M.A.; Ahamed, M.B.; Ezzat, M.O.; Majid, A.S.; Majid, A.M. The anticancer, antioxidant and antimicrobial properties of the sesquiterpene β-caryophyllene from the essential oil of Aquilaria crassna. Molecules 2015, 20, 11808–11829. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Wang, Y.; Hao, W.; Zhou, S.; Duan, C.; Li, Q.; Wei, J.; Liu, G. Exploring the Role of Active Functional Microbiota in Flavor Generation by Integrated Metatranscriptomics and Metabolomics during Niulanshan Baijiu Fermentation. Foods 2023, 12, 4140. https://doi.org/10.3390/foods12224140
Pan Y, Wang Y, Hao W, Zhou S, Duan C, Li Q, Wei J, Liu G. Exploring the Role of Active Functional Microbiota in Flavor Generation by Integrated Metatranscriptomics and Metabolomics during Niulanshan Baijiu Fermentation. Foods. 2023; 12(22):4140. https://doi.org/10.3390/foods12224140
Chicago/Turabian StylePan, Yuanyuan, Ying Wang, Wenjun Hao, Sen Zhou, Chengbao Duan, Qiushi Li, Jinwang Wei, and Gang Liu. 2023. "Exploring the Role of Active Functional Microbiota in Flavor Generation by Integrated Metatranscriptomics and Metabolomics during Niulanshan Baijiu Fermentation" Foods 12, no. 22: 4140. https://doi.org/10.3390/foods12224140
APA StylePan, Y., Wang, Y., Hao, W., Zhou, S., Duan, C., Li, Q., Wei, J., & Liu, G. (2023). Exploring the Role of Active Functional Microbiota in Flavor Generation by Integrated Metatranscriptomics and Metabolomics during Niulanshan Baijiu Fermentation. Foods, 12(22), 4140. https://doi.org/10.3390/foods12224140