
Multipronged Approach to Profiling Metabolites in Beta vulgaris L. Dried Pulp Extracts Using Chromatography, NMR and Other Spectroscopy Methods

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Sample Preparation
2.3. Extraction
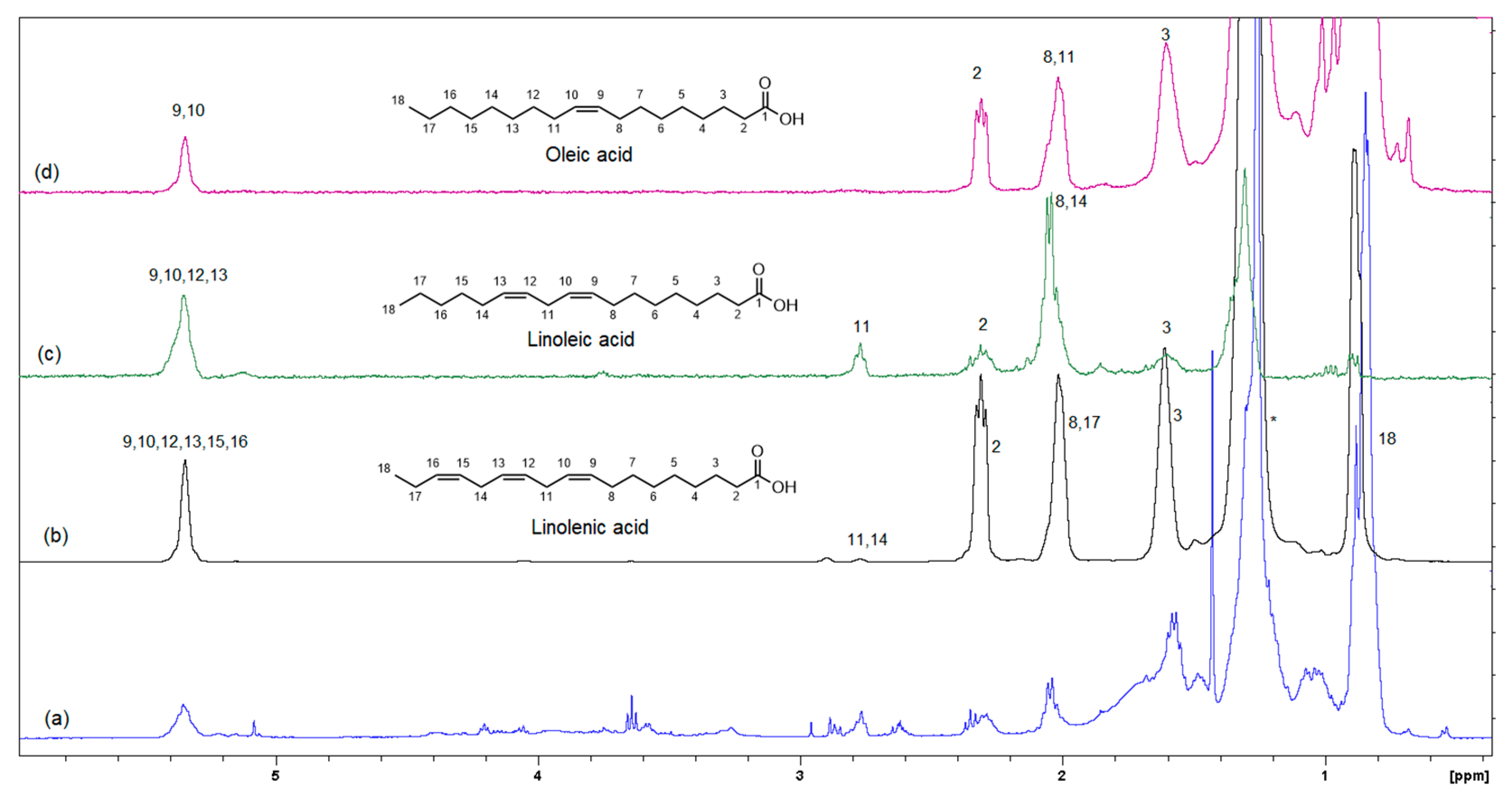
2.4. Instrumental Analysis
3. Results and Discussion
3.1. Extraction
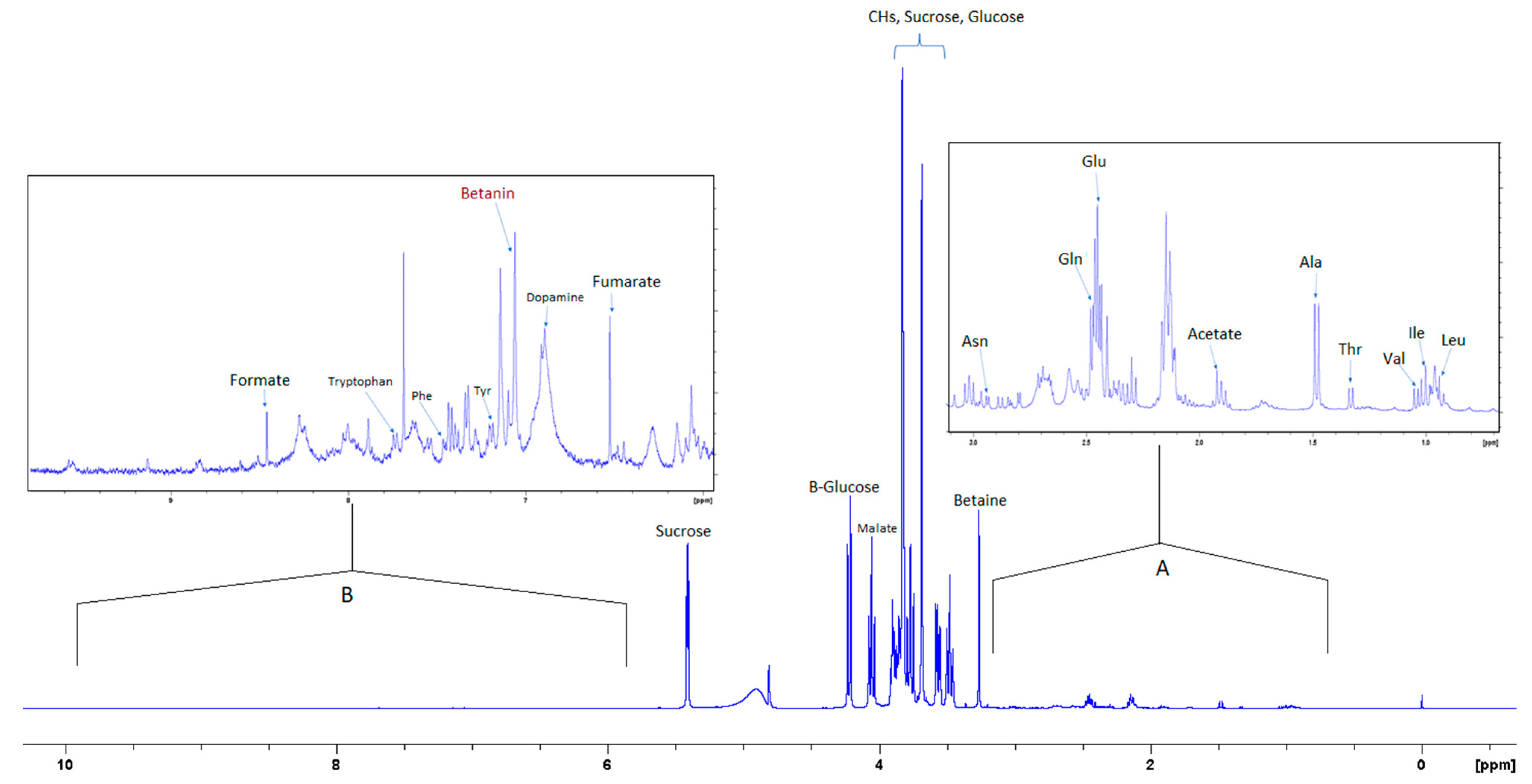
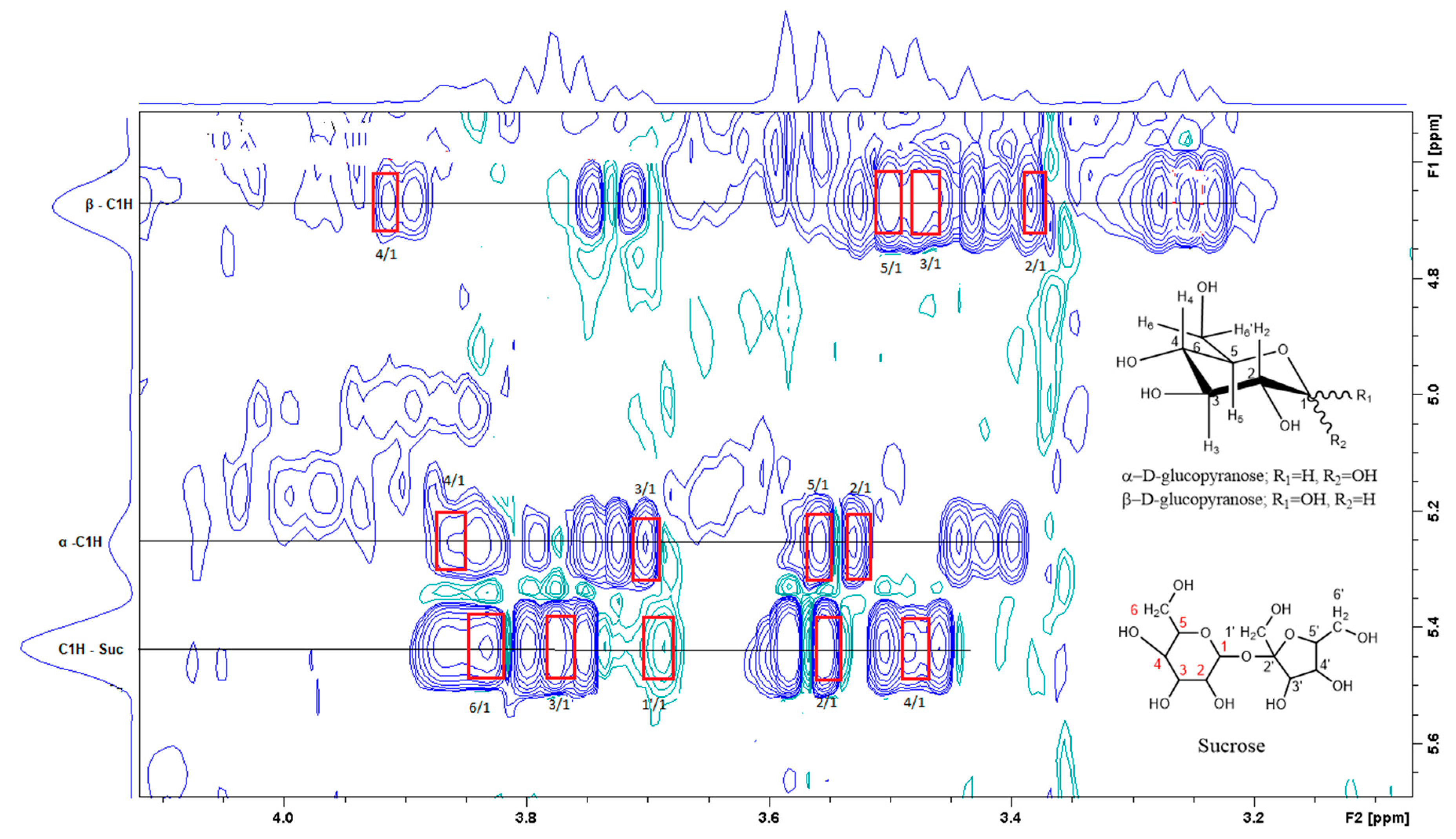
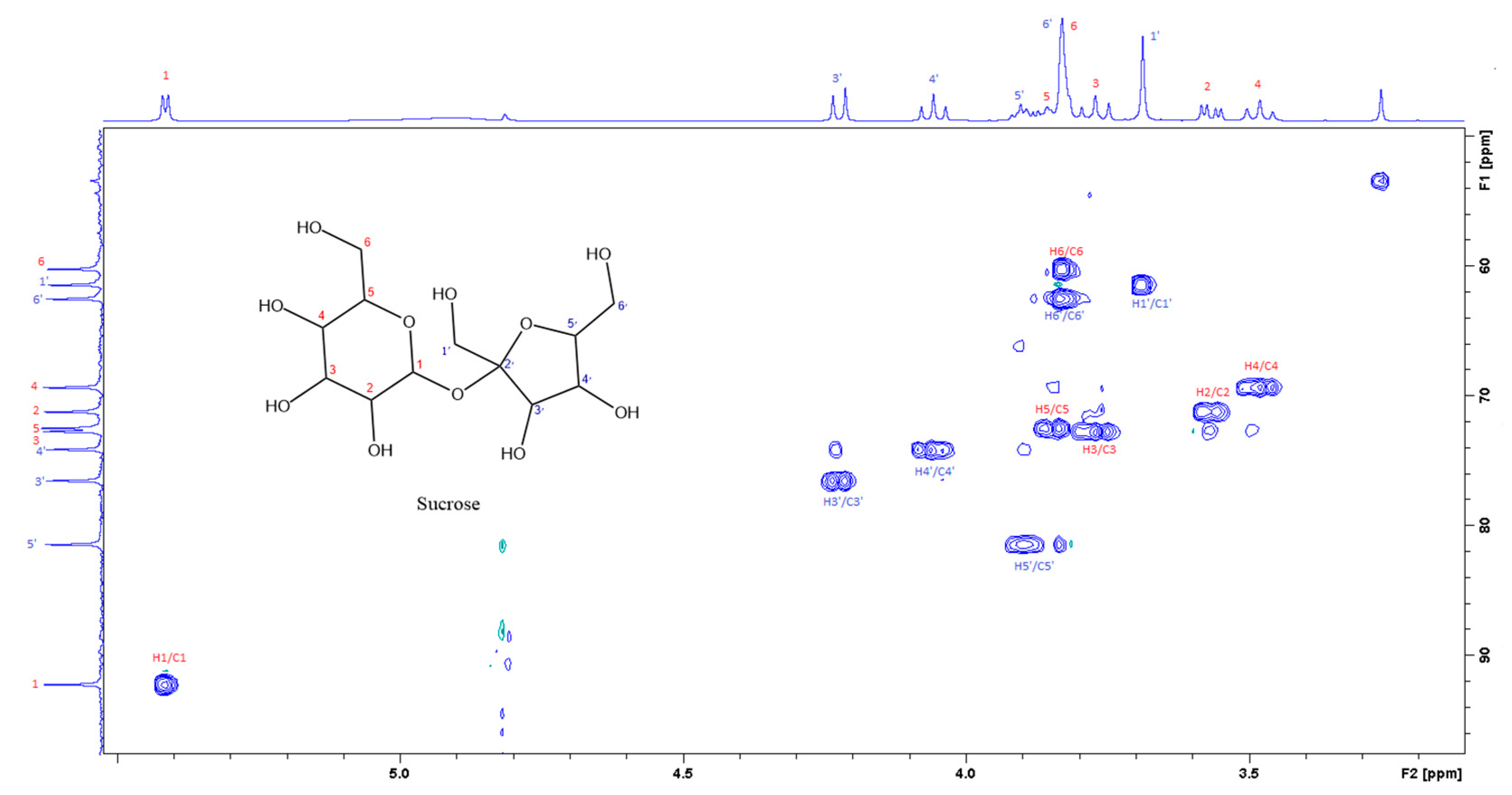
3.2. Aqueous (polar) Phase
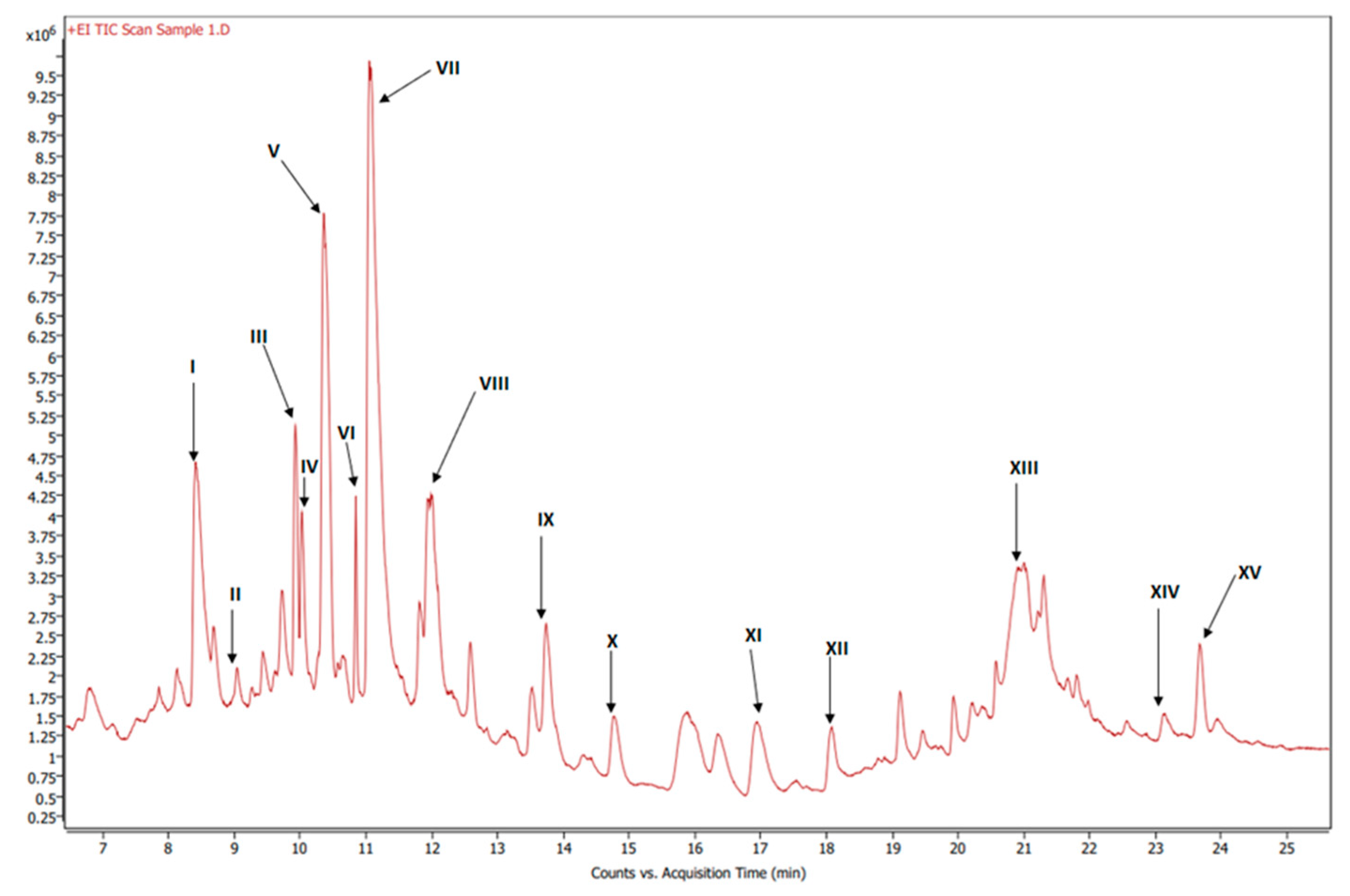
3.3. Organic Phase
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitfield, F.B.; Last, J.H. Vegetables. In Volatile Compounds in Foods and Beverages; Routledge: Oxfordshire, UK, 2017; pp. 203–281. [Google Scholar]
- Kale, R.; Sawate, A.; Kshirsagar, R.; Patil, B.; Mane, R. Studies on evaluation of physical and chemical composition of beetroot (Beta vulgaris L.) Kale RG, Sawate AR, Kshirsagar RB, Patil BM and Mane RP. Int. J. Chem. Stud. 2018, 6, 2977–2979. [Google Scholar]
- Bunkar, D.S.; Anand, A.; Kumar, K.; Meena, M.; Goyal, S.K.; Paswan, V.K. Development of production technology for preparation of beetroot powder using different drying methods. Ann. Phytomed. Int. J. 2020, 9, 293–301. [Google Scholar] [CrossRef]
- Brzezińska-rojek, J.; Rutkowska, M.; Brzezicha, J.; Konieczka, P.; Prokopowicz, M.; Grembecka, M. Mineral composition of dietary supplements-analytical and chemometric approach. Nutrients 2022, 14, 106. [Google Scholar] [CrossRef] [PubMed]
- Chhikara, N.; Kushwaha, K.; Sharma, P.; Gat, Y.; Panghal, A. Bioactive compounds of beetroot and utilization in food processing industry: A critical review. Food Chem. 2019, 272, 192–200. [Google Scholar] [CrossRef]
- Liliana, C.; Oana-Viorela, N. Red Beetroot: Composition and Health Effects—A Review. J. Nutr. Med. Diet Care 2020, 6, 043. [Google Scholar] [CrossRef]
- Giampaoli, O.; Sciubba, F.; Conta, G.; Capuani, G.; Tomassini, A.; Giorgi, G.; Brasili, E.; Aureli, W.; Miccheli, A. Red Beetroot’s NMR-Based Metabolomics: Phytochemical Profile Related to Development Time and Production Year. Foods 2021, 10, 1887. [Google Scholar] [CrossRef]
- Pedreño, M.A.; Escribano, J. Correlation between antiradical activity and stability of betanine from Beta vulgaris L roots under different pH, temperature and light conditions. J. Sci. Food Agric. 2001, 81, 627–631. [Google Scholar] [CrossRef]
- Kujala, T.S.; Loponen, J.M.; Klika, K.D.; Pihlaja, K. Phenolics and betacyanins in red beetroot (Beta vulgaris) root: Distribution and effect of cold storage on the content of total phenolics and three individual compounds. J. Agric. Food Chem. 2000, 48, 5338–5342. [Google Scholar] [CrossRef]
- Guenther, B.D.; Christensen, C.R.; Upatnieks, J. Coherent Optical Processing: Another Approach. IEEE J. Quantum Electron. 1979, 15, 1348–1362. [Google Scholar] [CrossRef]
- Szalay, J. What Are Flavonoids? 20 October 2015. Available online: https://www.livescience.com/52524-flavonoids.html (accessed on 2 February 2023).
- Boo, Y.C. p-coumaric acid as an active ingredient in cosmetics: A review focusing on its antimelanogenic effects. Antioxidants 2019, 8, 275. [Google Scholar] [CrossRef]
- Cai, Y.Z.; Sun, M.; Corke, H. Characterization and application of betalain pigments from plants of the Amaranthaceae. Trends Food Sci. Technol. 2005, 16, 370–376. [Google Scholar] [CrossRef]
- Anson, N.M.; van den Berg, R.; Havenaar, R.; Bast, A.; Haenen, G.R.M.M. Bioavailability of ferulic acid is determined by its bioaccessibility. J. Cereal Sci. 2009, 49, 296–300. [Google Scholar] [CrossRef]
- Dongowski, G. Enzymatic degradation studies of pectin and cellulose from red beets. Nahr.-Food 2001, 45, 324–331. [Google Scholar] [CrossRef]
- Lovegrove, A.; Edwards, C.H.; De Noni, I.; Patel, H.; El, S.N.; Grassby, T.; Zielke, C.; Ulmius, M.; Nilsson, L.; Butterworth, P.J.; et al. Role of polysaccharides in food, digestion, and health. Crit. Rev. Food Sci. Nutr. 2017, 57, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Pérez, F.; Steigerwald, H.; Schülke, S.; Vieths, S.; Toda, M.; Scheurer, S. The Dietary Fiber Pectin: Health Benefits and Potential for the Treatment of Allergies by Modulation of Gut Microbiota. Curr. Allergy Asthma Rep. 2021, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Shi, J.; Xie, S.Y.; Zhang, T.Y.; Soladoye, O.P.; Aluko, R.E. Red Beetroot Betalains: Perspectives on Extraction, Processing, and Potential Health Benefits. J. Agric. Food Chem. 2020, 68, 11595–11611. [Google Scholar] [CrossRef]
- Ibrahim, K.O.O.; Adepoju, G.F.; Owoeye, J.F.A.; Abdulmajeed, A.A.; Folaranmi, O.O. Orbital Mesenchymal Chondrosarcoma: Report of a Rare Tumor in a Nigerian Girl. Ann. Trop. Pathol. 2020, 11, 20–23. [Google Scholar]
- Ivrea, M.A.A.L. Distribution of Betalain Pigments in Red Blood Cells after Consumption of Cactus Pear Fruits and Increased Resistance of the Cells to ex Vivo Induced Oxidative Hemolysis in Humans. J. Agric. Food Chem. 2005, 53, 1266–1270. [Google Scholar]
- Sadowska-Bartosz, I.; Bartosz, G. Biological properties and applications of betalains. Molecules 2021, 26, 2520. [Google Scholar] [CrossRef]
- Khan, M.I. Plant Betalains: Safety, Antioxidant Activity, Clinical Efficacy, and Bioavailability. Compr. Rev. Food Sci. Food Saf. 2016, 15, 316–330. [Google Scholar] [CrossRef]
- Farag, M.A.; Sharaf El-Din, M.G.; Selim, M.A.; Owis, A.I.; Abouzid, S.F.; Porzel, A.; Wessjohann, L.A.; Otify, A. Nuclear magnetic resonance metabolomics approach for the analysis of major legume sprouts coupled to chemometrics. Molecules 2021, 26, 761. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Xu, J.; Wen, H.; Du, J.; Zhang, S.; Lv, M.; Xu, H. Recent Advances on Biological Activities and Structural Modifications of Dehydroabietic Acid. Toxins 2022, 14, 632. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Huang, R.; Pang, F.; Li, A.; Huang, G.; Zhou, X.; Li, Q.; Li, F.; Ma, X. Synthesis and biological evaluation of dehydroabietic acid-pyrimidine hybrids as antitumor agents. RSC Adv. 2020, 10, 18008–18015. [Google Scholar] [CrossRef] [PubMed]
- Popova, L.; Ivanchenko, O.; Pochkaeva, E.; Klotchenko, S.; Plotnikova, M.; Tsyrulnikova, A.; Aronova, E. Rosin derivatives as a platform for the antiviral drug design. Molecules 2021, 26, 3836. [Google Scholar] [CrossRef]
- Gokhale, S.V.; Lele, S.S. Dehydration of Red Beet Root (Beta vulgaris) by Hot Air Drying: Process Optimization and Mathematical Modeling. Food Sci. Biotechnol. 2011, 20, 955–964. [Google Scholar] [CrossRef]
- Hadaruga, D.I.; Hadaruga, N.G.; Hermenean, A.; Rivis, A.; Paslaru, V.; Codina, G. Bionanomaterials: Thermal stability of the oleic acid/α- and β-cyclodextrin complexes. Rev. Chim. 2008, 59, 994–998. [Google Scholar] [CrossRef]
- Thrippleton, M.J.; Keeler, J. Elimination of zero-quantum interference in two-dimensional NMR spectra. Angew. Chem. Int. Ed. 2003, 42, 3938–3941. [Google Scholar] [CrossRef]
- Aue, W.P.; Karhan, J.; Ernst, R.R. Homonuclear broad band decoupling and two-dimensional J-resolved NMR spectroscopy. J. Chem. Phys. 1976, 64, 4226–4227. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1969, 65, 355–360. [Google Scholar] [CrossRef]
- Palmer, A.G.; Cavanagh, J.; Wright, P.E.; Rance, M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 1969, 93, 151–170. [Google Scholar] [CrossRef]
- Pomin, V.H. Unravelling Glycobiology by NMR Spectroscopy. In Glycosylation; IntechOpen: London, UK, 2012; pp. 63–98. [Google Scholar]
- Ritota, M.; Marini, F.; Sequi, P.; Valentini, M. Metabolomic characterization of italian sweet pepper (Capsicum annum L.) by means of HRMAS-NMR spectroscopy and multivariate analysis. J. Agric. Food Chem. 2010, 58, 9675–9684. [Google Scholar] [CrossRef] [PubMed]
- Magritek. Characterizing Fatty Acids with advanced multinuclear NMR methods. In Magritek-Spin Solve; Magritek: Malvern, PA, USA, 2018; Volume 6. [Google Scholar]
- Cade-Menun, B.J. Improved peak identification in 31P-NMR spectra of environmental samples with a standardized method and peak library. Geoderma 2015, 257–258, 102–114. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Formula | Measured [M + H]+ (m/z) | Measured Rt (min) | Standard [M + H]+ (m/z) | Rt (min) of Standard | Confirmed by NMR |
|---|---|---|---|---|---|---|
| Lysine | C6H14N2O2 | 147.1131 | 0.58 | 147.1126 | NA | No |
| Histidine | C6H9N3O2 | 156.0771 | 0.61 | 156.0765 | 0.61 | No |
| Arginine | C6H14N4O2 | 175.1193 | 0.64 | N/A | NA | No |
| Threonine | C4H9NO3 | 120.0659 | 0.68 | N/A | NA | No |
| Glutamic acid | C5H9NO4 | 148.0606 | 0.68 | N/A | NA | No |
| Valine | C5H11NO2 | 118.0867 | 0.71 | N/A | NA | Yes |
| Proline | C5H9NO2 | 116.0710 | 0.77 | N/A | NA | No |
| Sucrose | C12H22O11 | 343.1239 | 0.98 | 343.1229 | 0.98 | Yes |
| Glucose | C6H12O6 | 181.0710 | 0.77 | N/A | NA | Yes |
| Methionine | C5H11NO2S | 150.0587 | 1.25 | N/A | NA | No |
| Leucine | C6H13NO2 | 132.1022 | 2.53 | 132.1018 | 2.49 | Yes |
| Isoleucine | C6H13NO2 | 132.1023 | 2.68 | N/A | NA | Yes |
| Tyrosine | C9H11NO3 | 182.0816 | 2.68 | N/A | NA | No |
| Betacyanin | C24H26N2O13 | 551.1520 | 2.96 | N/A | NA | Yes |
| Phenylalanine | C9H11NO2 | 166.0867 | 3.02 | 166.0860 | 3.02 | No |
| Tryptophan | C11H12N2O2 | 205.0975 | 3.48 | N/A | NA | No |
| Riboflavin | C17H20N4O6 | 377.1442 | 3.85 | N/A | NA | No |
| Betaxanthin | C18H18N2O6 | 359.1247 | 4.11 | N/A | NA | Yes |
| Theanine | C7H14N2O3 | 175.1078 | 13.82 | N/A | NA | No |
| Label | RT (min) | Base Peak S/N Ratio | Base Peak Area | Compound Name | Formula | Match/Similarity Score (%) |
|---|---|---|---|---|---|---|
| I | 8.41 | 9.73 × 102 | 6.15 × 106 | Hexadecanoic acid, methyl ester | C17H34O2 | 93.9 |
| II | 9.33 | 3.88 × 101 | 2.69 × 105 | 3-Methylbenzoic acid, 2,5-dichlorophenyl ester | C14H10Cl2O2 | 88.7 |
| III | 9.93 | 1.0 × 103 | 2.69 × 106 | Methyl stearate | C19H38O2 | 96.5 |
| IV | 10.03 | 2.35 × 102 | 7.04 × 105 | 9-Octadecenoic acid, methyl ester, (E)- | C19H36O2 | 90.7 |
| V | 10.36 | 3.42 × 102 | 3.70 × 106 | 9,12-Octadecadienoic acid (Z,Z)-, methyl ester | C19H34O2 | 91.9 |
| VI | 10.85 | 3.08 × 102 | 6.45 × 105 | 9,12,15-Octadecatrienoic acid, methyl ester, (Z,Z,Z)- | C19H32O2 | 96.4 |
| VII | 11.99 | 3.58 × 103 | 2.42 × 107 | Dibutyl phthalate | C16H22O4 | 91.4 |
| VIII | 12.59 | 6.44 × 101 | 1.04 × 106 | Pentacosane | C25H52 | 91.7 |
| IX | 13.74 | 1.66 × 102 | 1.57 × 106 | n-Hexadecanoic acid | C16H32O2 | 92.7 |
| X | 14.77 | 4.59 × 101 | 1.07 × 106 | Octacosane | C28H58 | 90.1 |
| XI | 16.95 | 8.14 × 101 | 1.31 × 106 | Octadecanoic acid | C18H36O2 | 92.6 |
| XII | 19.46 | 2.31 × 102 | 4.92 × 105 | 3,5-di-tert-Butyl-4-hydroxyphenylpropionic acid | C17H26O3 | 90.0 |
| XIII | 21.21 | 1.50 × 102 | 1.02 × 106 | Oxybis(propane-1,2-diyl) dibenzoate | C20H22O5 | 90.7 |
| XIV | 23.14 | 2.38 × 102 | 1.20 × 106 | Diethylene glycol dibenzoate | C18H18O5 | 97.2 |
| XV | 23.94 | 1.87 × 102 | 3.80 × 105 | Dehydroabietic acid | C20H28O2 | 88.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiadorwu, J.; Subedi, K.; Todd, D.; Basti, M.M. Multipronged Approach to Profiling Metabolites in Beta vulgaris L. Dried Pulp Extracts Using Chromatography, NMR and Other Spectroscopy Methods. Foods 2023, 12, 3510. https://doi.org/10.3390/foods12183510
Fiadorwu J, Subedi K, Todd D, Basti MM. Multipronged Approach to Profiling Metabolites in Beta vulgaris L. Dried Pulp Extracts Using Chromatography, NMR and Other Spectroscopy Methods. Foods. 2023; 12(18):3510. https://doi.org/10.3390/foods12183510
Chicago/Turabian StyleFiadorwu, Joshua, Kiran Subedi, Daniel Todd, and Mufeed M. Basti. 2023. "Multipronged Approach to Profiling Metabolites in Beta vulgaris L. Dried Pulp Extracts Using Chromatography, NMR and Other Spectroscopy Methods" Foods 12, no. 18: 3510. https://doi.org/10.3390/foods12183510
APA StyleFiadorwu, J., Subedi, K., Todd, D., & Basti, M. M. (2023). Multipronged Approach to Profiling Metabolites in Beta vulgaris L. Dried Pulp Extracts Using Chromatography, NMR and Other Spectroscopy Methods. Foods, 12(18), 3510. https://doi.org/10.3390/foods12183510