
Microbial Community Affects Daqu Quality and the Production of Ethanol and Flavor Compounds in Baijiu Fermentation
 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Physicochemical Properties
2.3. Isolation and Enumeration of Different Culturable Microbes
2.4. Genomic DNA Extraction
2.5. DNA Amplification
2.6. Sequence Data Processing and Statistical Analysis
2.7. Laboratory-scale Fermentation
2.8. Flavor Compounds Analysis
3. Results
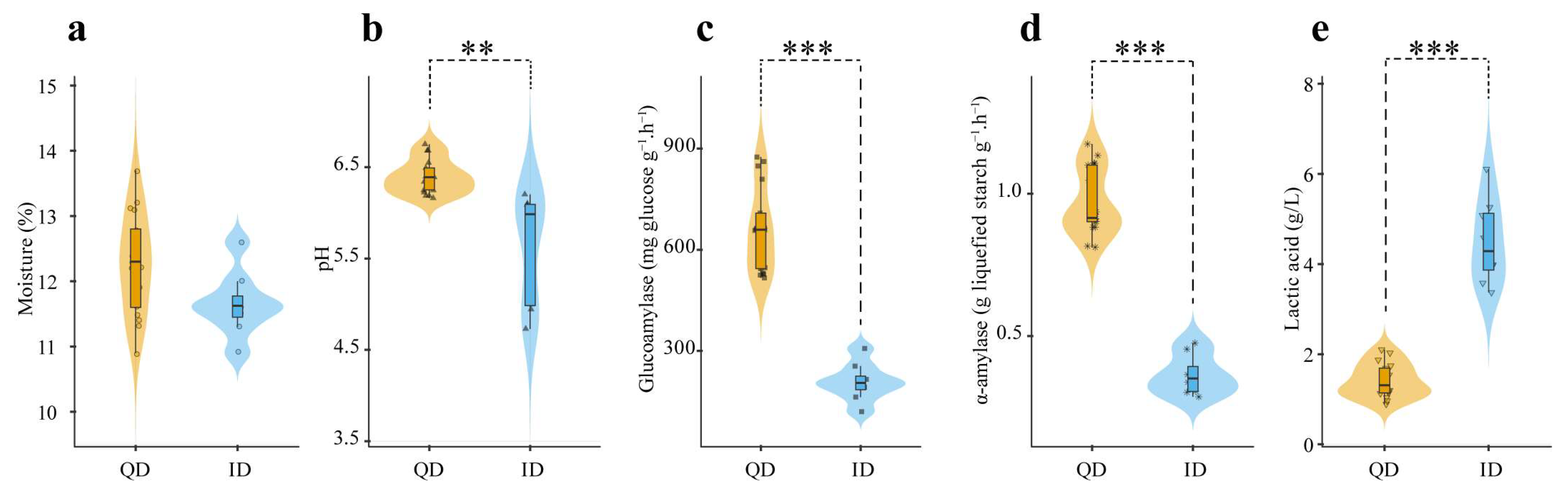
3.1. Physicochemical Characteristics of Qualified and Inferior Daqu
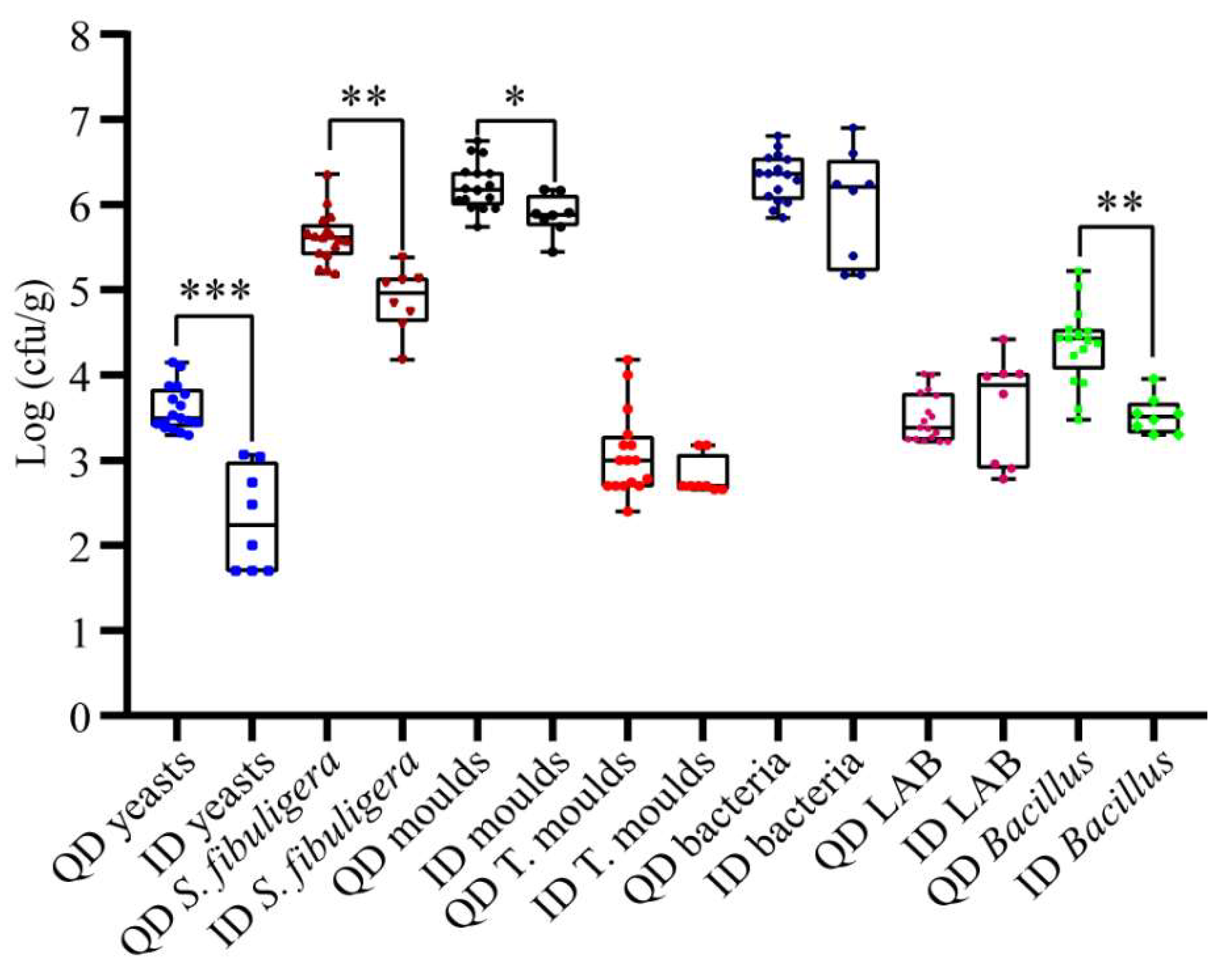
3.2. Enumeration of Microorganisms in Daqu
3.3. Microbial Landscape Revealed by PacBio SMRT Sequencing
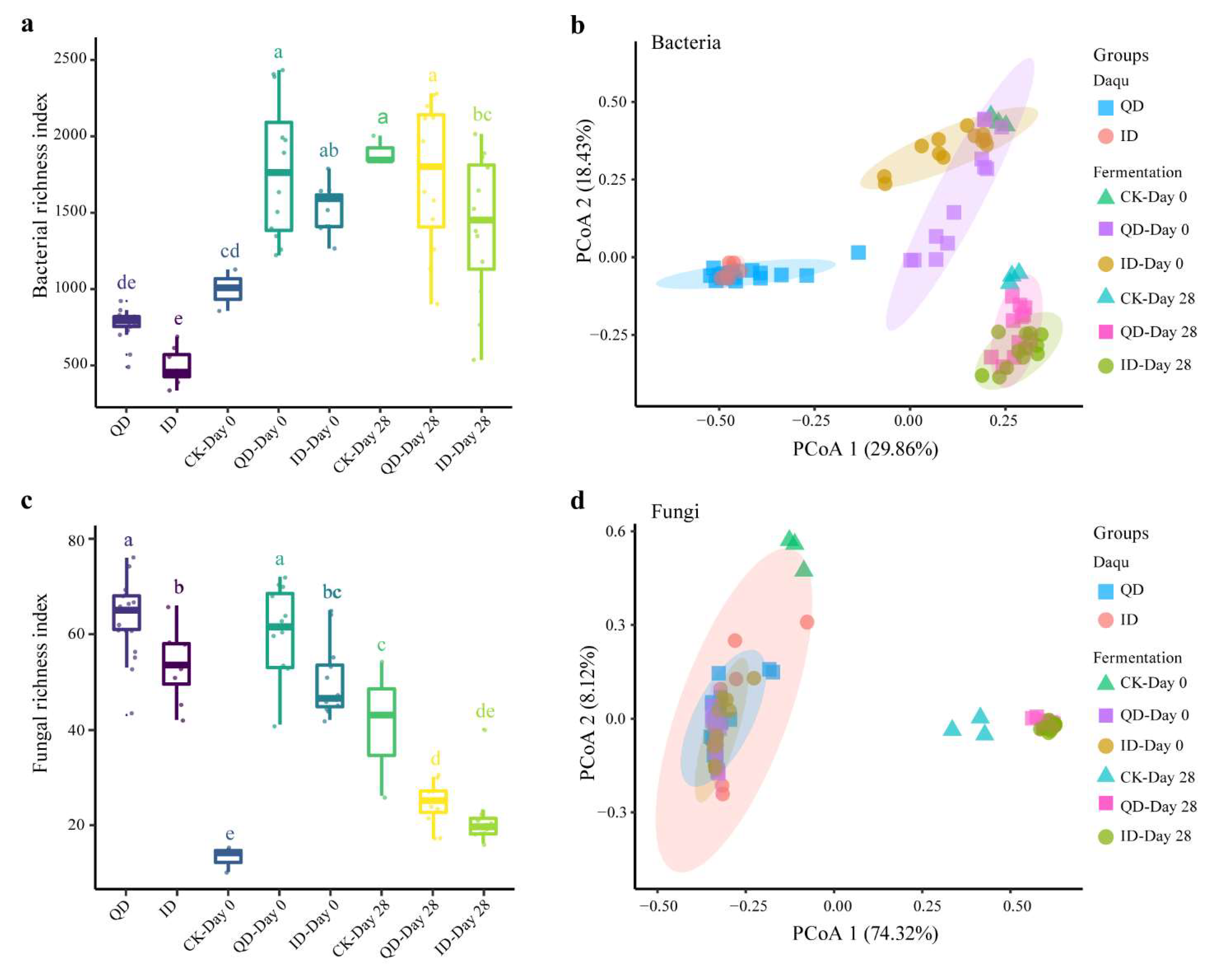
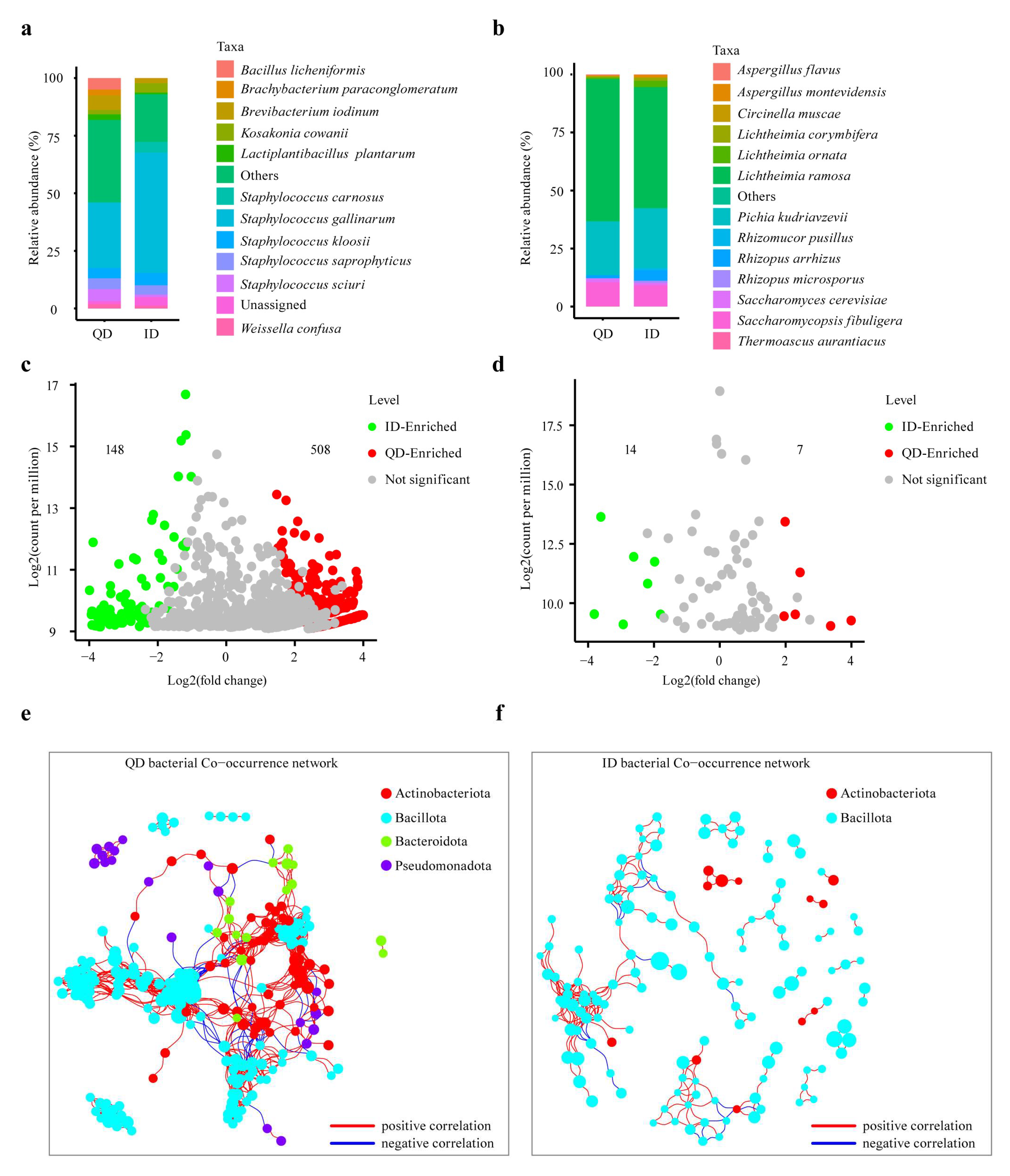
3.3.1. Microbial Community Composition and Co-occurrence Networks of QD and ID
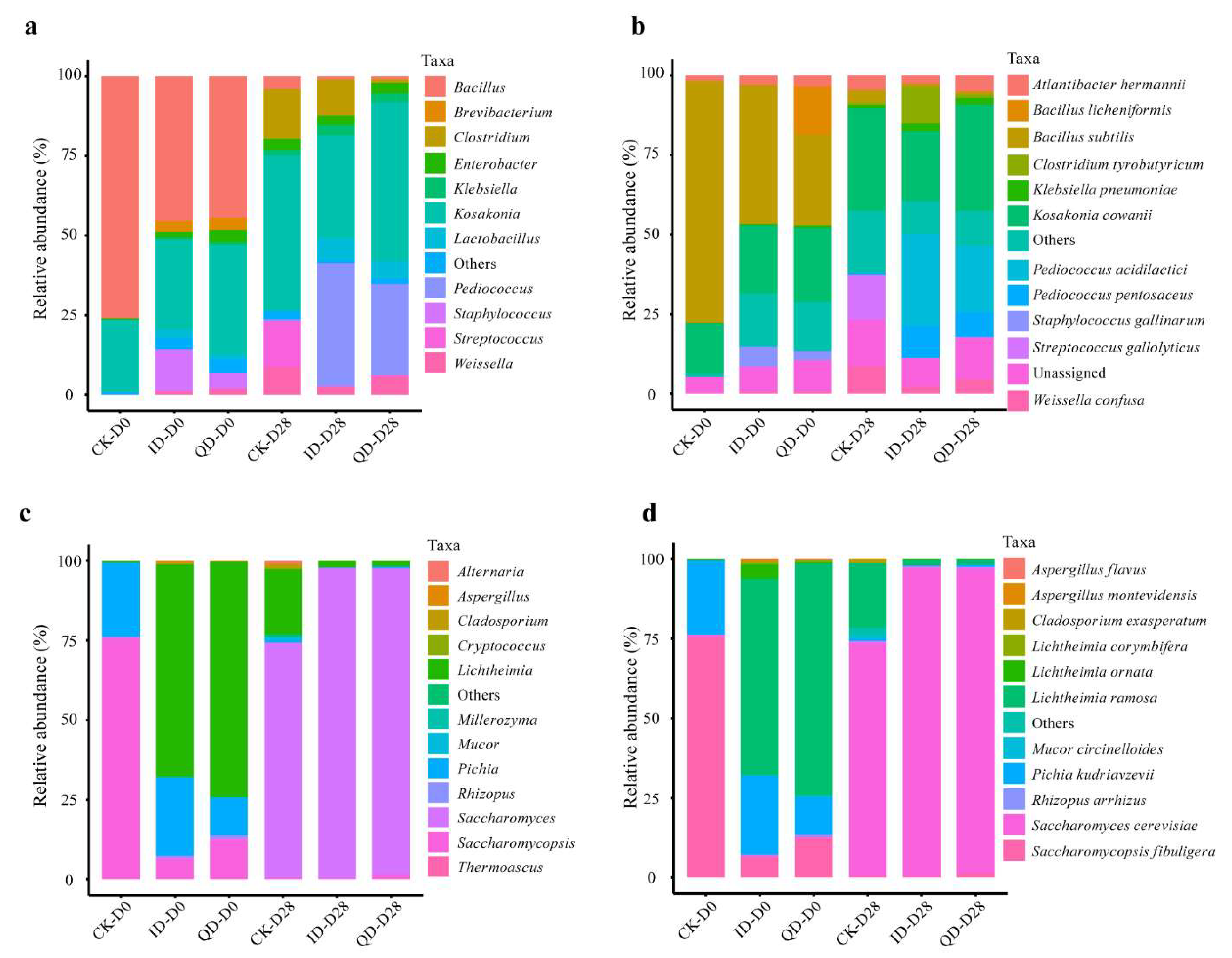
3.3.2. Microbial Composition during Fermentation
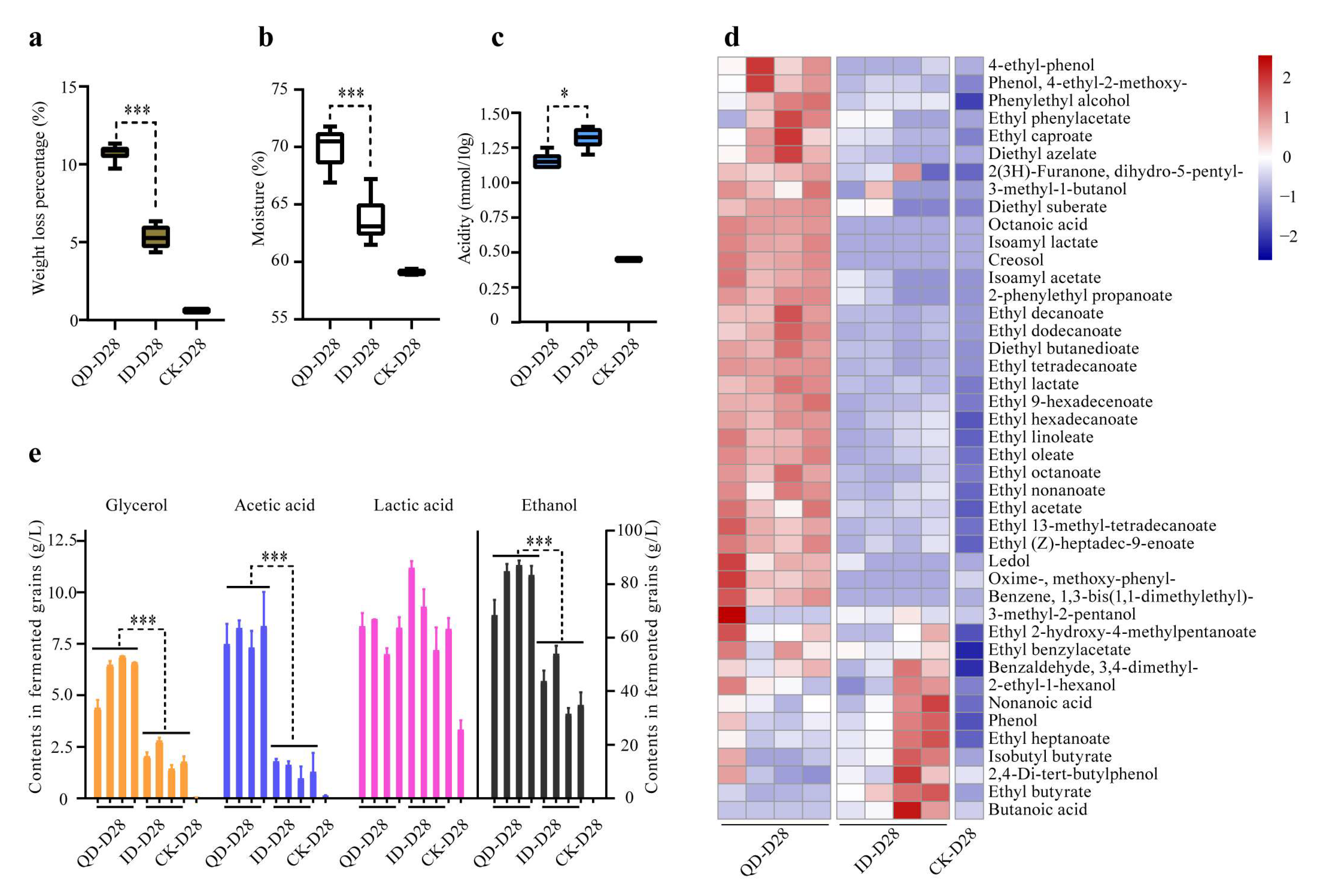
3.4. Physicochemical Characteristics and Metabolic Profiles of the Fermentation End Products
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jiang, X.; Xu, J.; Luo, J.; Zhao, F. Network Design towards Sustainability of Chinese Baijiu Industry from a Supply Chain Perspective. Discrete Dyn. Nat. Soc. 2018, 2018, 4391351. [Google Scholar] [CrossRef]
- Huang, Y.; Yi, Z.; Jin, Y.; Huang, M.; He, K.; Liu, D.; Luo, H.; Zhao, D.; He, H.; Fang, Y.; et al. Metatranscriptomics Reveals the Functions and Enzyme Profiles of the Microbial Community in Chinese Nong-Flavor Liquor Starter. Front. Microbiol. 2017, 8, 1747. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Jin, Y.; Xiao, Y.; Chen, L.; Tan, L.; Du, A.; He, K.; Liu, D.; Luo, H.; Fang, Y.; et al. Unraveling the Contribution of High Temperature Stage to Jiang-Flavor Daqu, a Liquor Starter for Production of Chinese Jiang-Flavor Baijiu, with Special Reference to Metatranscriptomics. Front. Microbiol. 2019, 10, 472. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.W.; Yan, Z.; Han, B.Z.; Zwietering, M.H.; Samson, R.A.; Boekhout, T.; Robert Nout, M.J. Complex Microbiota of a Chinese “Fen” Liquor Fermentation Starter (Fen-Daqu), Revealed by Culture-Dependent and Culture-Independent Methods. Food Microbiol. 2012, 31, 293–300. [Google Scholar] [CrossRef]
- Xia, Y.; Luo, H.; Wu, Z.; Zhang, W. Microbial Diversity in Jiuqu and Its Fermentation Features: Saccharification, Alcohol Fermentation and Flavors Generation. Appl. Microbiol. Biotechnol. 2023, 107, 25–41. [Google Scholar] [CrossRef]
- Li, P.; Lin, W.; Liu, X.; Wang, X.; Luo, L. Environmental Factors Affecting Microbiota Dynamics during Traditional Solid-State Fermentation of Chinese Daqu Starter. Front. Microbiol. 2016, 7, 1237. [Google Scholar] [CrossRef]
- Zou, W.; Zhao, C.; Luo, H. Diversity and Function of Microbial Community in Chinese Strong-Flavor Baijiu Ecosystem: A Review. Front. Microbiol. 2018, 9, 671. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.; Sun, B.; Fu, Z.; Xia, Y.; Huang, M.; Xu, C.; Li, X. Analysis of Physicochemical Indices, Volatile Flavor Components, and Microbial Community of a Light-Flavor Daqu. J. Am. Soc. Brew. Chem. 2018, 76, 209–218. [Google Scholar] [CrossRef]
- Liu, J.; Chen, J.; Fan, Y.; Huang, X.; Han, B. Biochemical Characterisation and Dominance of Different Hydrolases in Different Types of Daqu—A Chinese Industrial Fermentation Starter. J. Sci. Food Agric. 2018, 98, 113–121. [Google Scholar] [CrossRef]
- Wang, B.; Wu, Q.; Xu, Y.; Sun, B. Synergistic Effect of Multiple Saccharifying Enzymes on Alcoholic Fermentation for Chinese Baijiu Production. Appl. Environ. Microbiol. 2020, 86, e00013-20. [Google Scholar] [CrossRef]
- Wang, H.Y.; Xu, Y. Effect of Temperature on Microbial Composition of Starter Culture for Chinese Light Aroma Style Liquor Fermentation. Lett. Appl. Microbiol. 2015, 60, 85–91. [Google Scholar] [CrossRef]
- Cai, W.; Wang, Y.; Ni, H.; Liu, Z.; Liu, J.; Zhong, J.; Hou, Q.; Shan, C.; Yang, X.; Guo, Z. Diversity of Microbiota, Microbial Functions, and Flavor in Different Types of Low-Temperature Daqu. Food Res. Int. 2021, 150, 110734. [Google Scholar] [CrossRef]
- Dai, Y.J.; Li, Z.J.; Tian, Z.Q. Analysis of Maotai-Flavor Daqu and Fungal Diversity of Fermented Grains. Mod. Food Sci. Technol. 2018, 34, 97–104. [Google Scholar] [CrossRef]
- Deng, L.; Mao, X.; Liu, D.; Ning, X.Q.; Shen, Y.; Chen, B.; Nie, H.F.; Huang, D.; Luo, H.B. Comparative Analysis of Physicochemical Properties and Microbial Composition in High-Temperature Daqu With Different Colors. Front. Microbiol. 2020, 11, 588117. [Google Scholar] [CrossRef]
- Gan, S.H.; Yang, F.; Sahu, S.K.; Luo, R.Y.; Liao, S.L.; Wang, H.Y.; Jin, T.; Wang, L.; Zhang, P.F.; Liu, X.; et al. Deciphering the Composition and Functional Profile of the Microbial Communities in Chinese Moutai Liquor Starters. Front. Microbiol. 2019, 10, 1540. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.H.; Xiao, Y.P.; Li, X.R.; Ma, E.B.; Du, X.W.; Quan, Z.X. Analyses of Microbial Consortia in the Starter of Fen Liquor. Lett. Appl. Microbiol. 2009, 48, 478–485. [Google Scholar] [CrossRef]
- Wang, H.Y.; Gao, Y.B.; Fan, Q.W.; Xu, Y. Characterization and Comparison of Microbial Community of Different Typical Chinese Liquor Daqus by PCR-DGGE. Lett. Appl. Microbiol. 2011, 53, 134–140. [Google Scholar] [CrossRef]
- Zheng, X.W.; Yan, Z.; Nout, M.J.R.; Boekhout, T.; Han, B.Z.; Zwietering, M.H.; Smid, E.J. Characterization of the Microbial Community in Different Types of Daqu Samples as Revealed by 16S RRNA and 26S RRNA Gene Clone Libraries. World J. Microbiol. Biotechnol. 2015, 31, 199–208. [Google Scholar] [CrossRef]
- Jiang, Q.; Wu, X.; Xu, Y.; Zhang, Y.; Wang, Z.; Shen, L.; Yang, W.; Sun, J.; Liu, Y. Microbial Composition and Dynamic Succession during the Daqu Production Process of Northern Jiang-Flavored Liquor in China. 3 Biotech 2021, 11, 224. [Google Scholar] [CrossRef]
- Li, X.R.; Ma, E.B.; Yan, L.Z.; Meng, H.; Du, X.W.; Quan, Z.X. Bacterial and Fungal Diversity in the Starter Production Process of Fen Liquor, a Traditional Chinese Liquor. J. Microbiol. 2013, 51, 430–438. [Google Scholar] [CrossRef]
- Mao, J.; Liu, X.; Gao, T.; Gu, S.; Wu, Y.; Zhao, L.; Ma, J.; Li, X.; Zhang, J. Unraveling the Correlations between Bacterial Diversity, Physicochemical Properties and Bacterial Community Succession during the Fermentation of Traditional Chinese Strong-Flavor Daqu. LWT 2022, 154, 112764. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, Y.; Cheng, W.; Wang, X.; Xue, Y.; Chen, X.; Han, B.Z. Understanding the Shifts of Microbial Community and Metabolite Profile From Wheat to Mature Daqu. Front. Microbiol. 2021, 12, 714726. [Google Scholar] [CrossRef]
- Chen, S.; Huang, J.; Qin, H.; He, G.; Zhou, R.; Yang, Y.; Qiu, C.; Zhang, S. Evolving the Core Microbial Community in Pit Mud Based on Bioturbation of Fortified Daqu. Can. J. Microbiol. 2021, 67, 396–405. [Google Scholar] [CrossRef]
- He, G.; Huang, J.; Wu, C.; Jin, Y.; Zhou, R. Bioturbation Effect of Fortified Daqu on Microbial Community and Flavor Metabolite in Chinese Strong-Flavor Liquor Brewing Microecosystem. Food Res. Int. 2020, 129, 108851. [Google Scholar] [CrossRef]
- He, G.; Dong, Y.; Huang, J.; Wang, X.; Zhang, S.; Wu, C.; Jin, Y.; Zhou, R. Alteration of Microbial Community for Improving Flavor Character of Daqu by Inoculation with Bacillus Velezensis and Bacillus Subtilis. LWT 2019, 111, 1–8. [Google Scholar] [CrossRef]
- Li, P.; Lin, W.; Liu, X.; Wang, X.; Gan, X.; Luo, L.; Lin, W.T. Effect of Bioaugmented Inoculation on Microbiota Dynamics during Solid-State Fermentation of Daqu Starter Using Autochthonous of Bacillus, Pediococcus, Wickerhamomyces and Saccharomycopsis. Food Microbiol. 2017, 61, 83–92. [Google Scholar] [CrossRef]
- Li, W.; Fan, G.; Fu, Z.; Wang, W.; Xu, Y.; Teng, C.; Zhang, C.; Yang, R.; Sun, B.; Li, X. Effects of Fortification of Daqu with Various Yeasts on Microbial Community Structure and Flavor Metabolism. Food Res. Int. 2020, 129, 108837. [Google Scholar] [CrossRef]
- Xiao, C.; Lu, Z.M.; Zhang, X.J.; Wang, S.T.; Ao, L.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Bio-Heat Is a Key Environmental Driver Shaping the Microbial Community of Medium-Temperature Daqu. Appl. Environ. Microbiol. 2017, 83, e01550-17. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.; Fu, Z.; Teng, C.; Wu, Q.; Liu, P.; Yang, R.; Minhazul, K.A.H.M.; Li, X. Comprehensive Analysis of Different Grades of Roasted-Sesame-like Flavored Daqu. Int. J. Food Prop. 2019, 22, 1205–1222. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhao, Q.; Yang, J.; Shao, G.; Dong, D.; Ma, W. Construction of Quality Evaluation System of Medium-High-Temperature Daqu. Liquor.-Mak. Sci. Technol. 2022, 2, 74–78. [Google Scholar] [CrossRef]
- Zheng, X.W.; Tabrizi, M.R.; Robert Nout, M.J.; Han, B.Z. Daqu-a Traditional Chinese Liquor Fermentation Starter. J. Inst. Brew. 2011, 117, 82–90. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, F.; Shen, Y.; Cheng, W.; Xue, Y.; Han, B.-Z.; Chen, X. Characteristics of the Microbiota and Metabolic Profile of High-Temperature Daqu with Different Grades. World J. Microbiol. Biotechnol. 2022, 38, 137. [Google Scholar] [CrossRef]
- Fan, G.; Fu, Z.; Sun, B.; Zhang, Y.; Wang, X.; Xia, Y.; Huang, M.; Li, X. Roles of Aging in the Production of Light-Flavored Daqu. J. Biosci. Bioeng. 2019, 127, 309–317. [Google Scholar] [CrossRef]
- Guan, K.-L.; Han, P.J.; Zhou, S.; Ji, F.; Bai, F.Y. Development of Mold-Inhibiting Methods for Selective Isolation and Counting of Yeasts from Low-Temperature Liquor Starter “Daqu”. Mycosystema 2019, 38, 1191–1201. [Google Scholar] [CrossRef]
- Pitt, J.I. An Appraisal of Identification Methods for Penicillium Species: Novel Taxonomic Criteria Based on Temperature and Water Relations. Mycologia 1973, 65, 1135–1157. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zhang, S.; Liu, X.Z.; Wen, H.A.; Wang, M. A Simple Method of Genomic DNA Extraction Suitable for Analysis of Bulk Fungal Strains. Lett. Appl. Microbiol. 2010, 51, 114–118. [Google Scholar] [CrossRef]
- Zain Hasan, S.M.; Shafie, M.S.B.; Shah, R.M. Efficient Method for the Extraction of Genomic DNA from Wormwood (Artemisia Capillaris). Afr. J. Biotechnol. 2008, 7, 3211–3216. [Google Scholar]
- Luo, L.-J.; Song, L.; Han, Y.; Zhen, P.; Han, D.-Y.; Zhao, X.; Zhou, X.; Wei, Y.-H.; Yu, H.-X.; Han, P.-J.; et al. Microbial Communities and Their Correlation with Flavor Compound Formation during the Mechanized Production of Light-Flavor Baijiu. Food Res. Int. 2023, 172, 113139. [Google Scholar] [CrossRef]
- Banerjee, S.; Walder, F.; Büchi, L.; Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; van der Heijden, M.G.A. Agricultural Intensification Reduces Microbial Network Complexity and the Abundance of Keystone Taxa in Roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S RRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE Database for Molecular Identification of Fungi: Handling Dark Taxa and Parallel Taxonomic Classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Tedersoo, L.; Nilsson, R.H.; Vellak, K.; Saar, I.; Veldre, V.; Parmasto, E.; Prous, M.; Aan, A.; Ots, M.; et al. Plutof-a Web Based Workbench for Ecological and Taxonomic Research, with an Online Implementation for Fungal Its Sequences. Evol. Bioinform. 2010, 6, 189–196. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package Vegan: Community Ecology Package, 2013. In R Package. Software. Available online: http://CRAN.R-project.org/package=vegan (accessed on 13 May 2023).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wu, Q.; Nie, Y.; Wu, J.; Xu, Y. Construction of Synthetic Microbiota for Reproducible Flavor Compound Metabolism in Chinese Light-Aroma-Type Liquor Produced by Solid-State Fermentation. Appl. Environ. Microbiol. 2019, 85, e03090-18. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Sui, L.; Zhang, J.; Hu, J.; Liu, P. Polyphasic Characterization of Yeasts and Lactic Acid Bacteria Metabolic Contribution in Semi-Solid Fermentation of Chinese Baijiu (Traditional Fermented Alcoholic Drink): Towards the Design of a Tailored Starter Culture. Microorganisms 2019, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Han, P.J.; Han, D.Y.; Zhou, S.; Li, K.; He, P.Y.; Zhen, P.; Yu, H.X.; Liang, Z.R.; Wang, X.W.; et al. Genetic Diversity and Population Structure of the Amylolytic Yeast Saccharomycopsis fibuligera Associated with Baijiu Fermentation in China. J. Microbiol. 2021, 59, 753–762. [Google Scholar] [CrossRef]
- Hui, W.; Hou, Q.; Cao, C.; Xu, H.; Zhen, Y.; Kwok, L.Y.; Sun, T.; Zhang, H.; Zhang, W. Identification of Microbial Profile of Koji Using Single Molecule, Real-Time Sequencing Technology. J. Food Sci. 2017, 82, 1193–1199. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Zhang, S.; Liu, T.; Qin, H.; Shen, C.; Liu, H.; Yang, F.; Yang, C.; Yin, Q.; et al. Spatiotemporal Distribution of Environmental Microbiota in Spontaneous Fermentation Workshop: The Case of Chinese Baijiu. Food Res. Int. 2022, 156, 111126. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, X.; Yang, B.; Zhang, X.; Han, Y.; Chen, X.X.; Han, B.Z. Contrasting the Microbial Community and Metabolic Profile of Three Types of Light-Flavor Daqu. Food Biosci. 2021, 44, 101395. [Google Scholar] [CrossRef]
- Mabrouk, S.S.; Hashem, A.M.; El-Shayeb, N.M.A.; Ismail, A.M.S.; Abdel-Fattah, A.F. Optimization of Alkaline Protease Productivity by Bacillus Licheniformis ATCC 21415. Bioresour. Technol. 1999, 69, 155–159. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y.; Chen, L.; Li, J.; Wang, L.; Du, G. Genome Sequencing and Flavor Compound Biosynthesis Pathway Analyses of Bacillus Licheniformis Isolated from Chinese Maotai-Flavor Liquor-Brewing Microbiome. Food Biotechnol. 2020, 34, 193–211. [Google Scholar] [CrossRef]
- Pang, X.N.; Han, B.Z.; Huang, X.N.; Zhang, X.; Hou, L.F.; Cao, M.; Gao, L.J.; Hu, G.H.; Chen, J.Y. Effect of the Environment Microbiota on the Flavour of Light-Flavour Baijiu during Spontaneous Fermentation. Sci. Rep. 2018, 8, 3396. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.N.; Chen, C.; Huang, X.N.; Yan, Y.Z.; Chen, J.Y.; Han, B.Z. Influence of Indigenous Lactic Acid Bacteria on the Volatile Flavor Profile of Light-Flavor Baijiu. LWT 2021, 147, 111540. [Google Scholar] [CrossRef]
- Collins, N.E.; Kirschner, L.A.M.; von Holy, A. Characterization of Bacillus Isolates from Ropey Bread, Bakery Equipment and Raw Materials. S. Afr. J. Sci. 1991, 87, 62–66. [Google Scholar]
- Ito, T.; Konno, M.; Shimura, Y.; Watanabe, S.; Takahashi, H.; Hashizume, K. Formation of Guaiacol by Spoilage Bacteria from Vanillic Acid, a Product of Rice Koji Cultivation, in Japanese Sake Brewing. J. Agric. Food Chem. 2016, 64, 4599–4605. [Google Scholar] [CrossRef]
- Klein, M.; Swinnen, S.; Thevelein, J.M.; Nevoigt, E. Glycerol Metabolism and Transport in Yeast and Fungi: Established Knowledge and Ambiguities. Environ. Microbiol. 2017, 19, 878–893. [Google Scholar] [CrossRef] [Green Version]
- Cespi, D.; Passarini, F.; Mastragostino, G.; Vassura, I.; Larocca, S.; Iaconi, A.; Chieregato, A.; Dubois, J.L.; Cavani, F. Glycerol as Feedstock in the Synthesis of Chemicals: A Life Cycle Analysis for Acrolein Production. Green Chemistry 2015, 17, 343–355. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, J.; Liu, X.; Zhang, C.; Zhao, Z.; Li, X.; Sun, B. Flavor Mystery of Chinese Traditional Fermented Baijiu: The Great Contribution of Ester Compounds. Food Chem. 2022, 369, 130920. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.J.; Xu, P.X.; Qian, W.; Zhang, X.J.; Ma, J.; Lu, Z.M.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Profiling the Clostridia with Butyrate-Producing Potential in the Mud of Chinese Liquor Fermentation Cellar. Int. J. Food Microbiol. 2019, 297, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Bao, Y.; Zhang, Z.; Zhao, W.; Xiao, J.; He, S.; Zhang, G.; Li, Y.; Zhao, G.; Chen, R. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, P.-J.; Luo, L.-J.; Han, Y.; Song, L.; Zhen, P.; Han, D.-Y.; Wei, Y.-H.; Zhou, X.; Wen, Z.; Qiu, J.-Z.; et al. Microbial Community Affects Daqu Quality and the Production of Ethanol and Flavor Compounds in Baijiu Fermentation. Foods 2023, 12, 2936. https://doi.org/10.3390/foods12152936
Han P-J, Luo L-J, Han Y, Song L, Zhen P, Han D-Y, Wei Y-H, Zhou X, Wen Z, Qiu J-Z, et al. Microbial Community Affects Daqu Quality and the Production of Ethanol and Flavor Compounds in Baijiu Fermentation. Foods. 2023; 12(15):2936. https://doi.org/10.3390/foods12152936
Chicago/Turabian StyleHan, Pei-Jie, Lu-Jun Luo, Ying Han, Liang Song, Pan Zhen, Da-Yong Han, Yu-Hua Wei, Xin Zhou, Zhang Wen, Jun-Zhi Qiu, and et al. 2023. "Microbial Community Affects Daqu Quality and the Production of Ethanol and Flavor Compounds in Baijiu Fermentation" Foods 12, no. 15: 2936. https://doi.org/10.3390/foods12152936
APA StyleHan, P.-J., Luo, L.-J., Han, Y., Song, L., Zhen, P., Han, D.-Y., Wei, Y.-H., Zhou, X., Wen, Z., Qiu, J.-Z., & Bai, F.-Y. (2023). Microbial Community Affects Daqu Quality and the Production of Ethanol and Flavor Compounds in Baijiu Fermentation. Foods, 12(15), 2936. https://doi.org/10.3390/foods12152936