

A Combined Differential Proteome and Transcriptome Profiling of Fast- and Slow-Twitch Skeletal Muscle in Pigs

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Samples Collection
2.2. iTRAQ Assays
2.2.1. Protein Extraction
2.2.2. Protein Digestion and iTRAQ Labelling
2.2.3. Strong Cation Exchange Fractionation
2.2.4. nLC-MS/MS Analysis
2.3. iTRAQ Data Processing and Analysis
2.3.1. Identification of Proteins and Analysis
2.3.2. Identification of Differentially Abundant Proteins (DAPs) and Analysis
2.4. Integrative Analysis of the Proteome and Transcriptome Data
3. Results
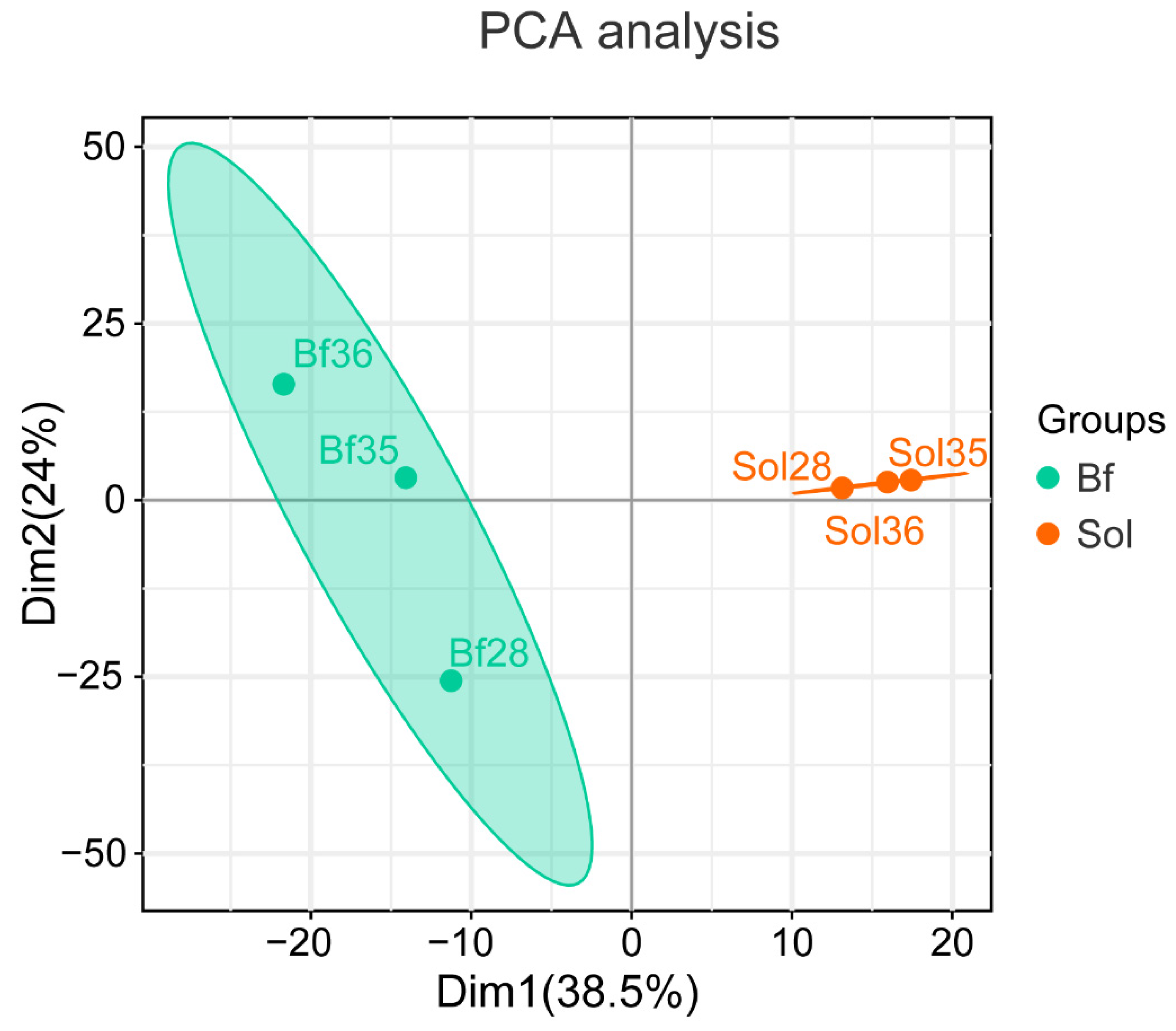
3.1. Summary of iTRAQ-Based Proteome Data
3.2. Functional Annotation and Classification of the Identified Proteins
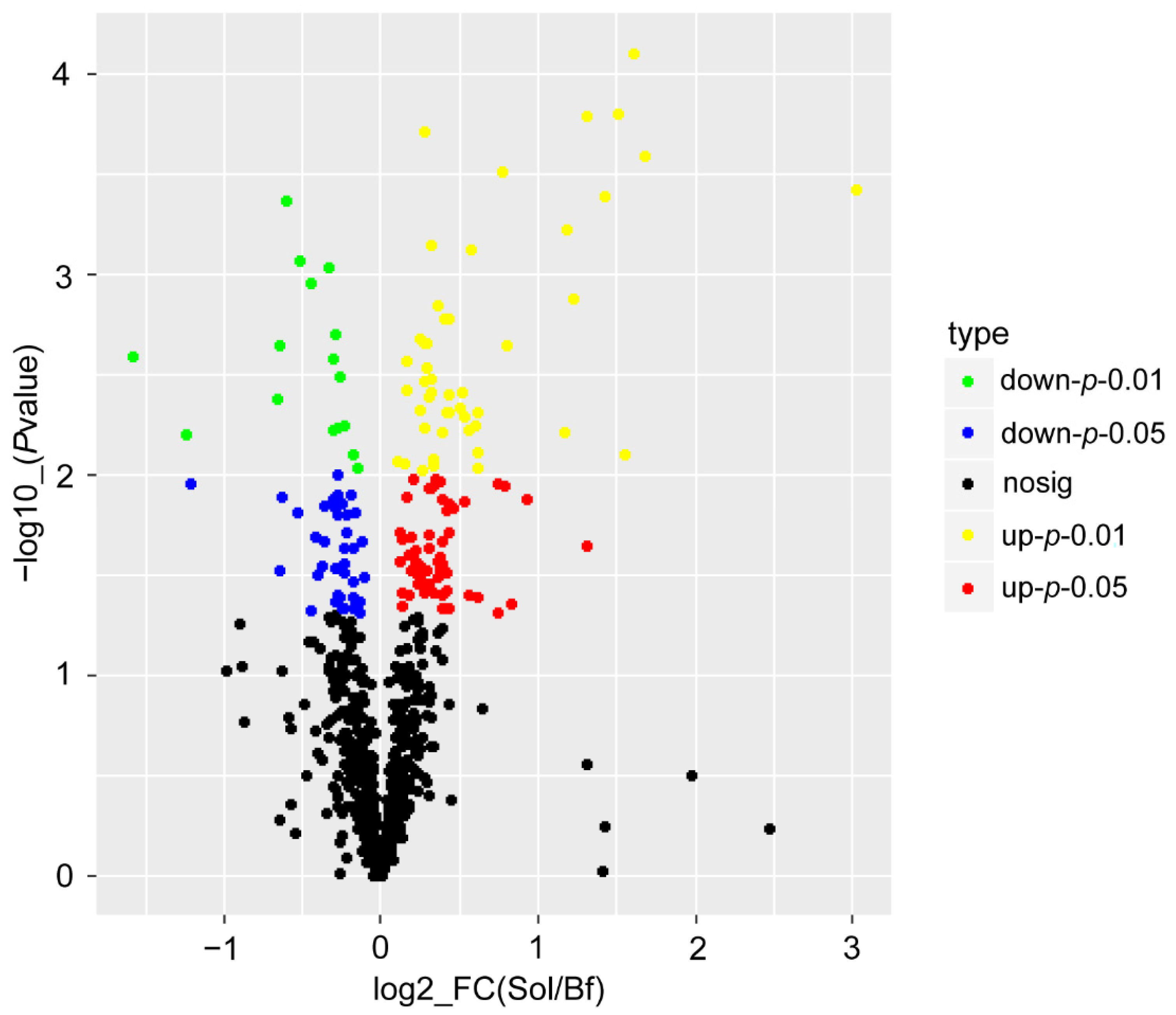
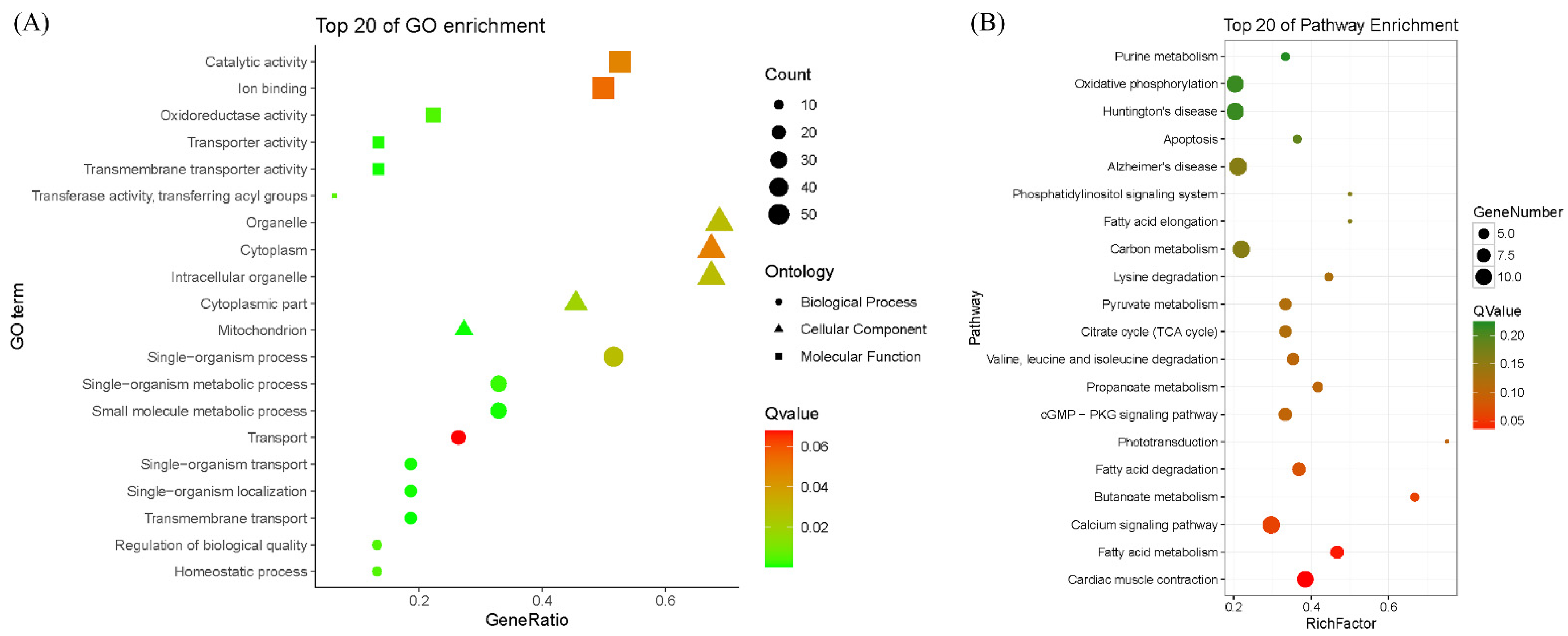
3.3. DAP Identification and GO Terms and KEGG Pathways Enrichment Analysis
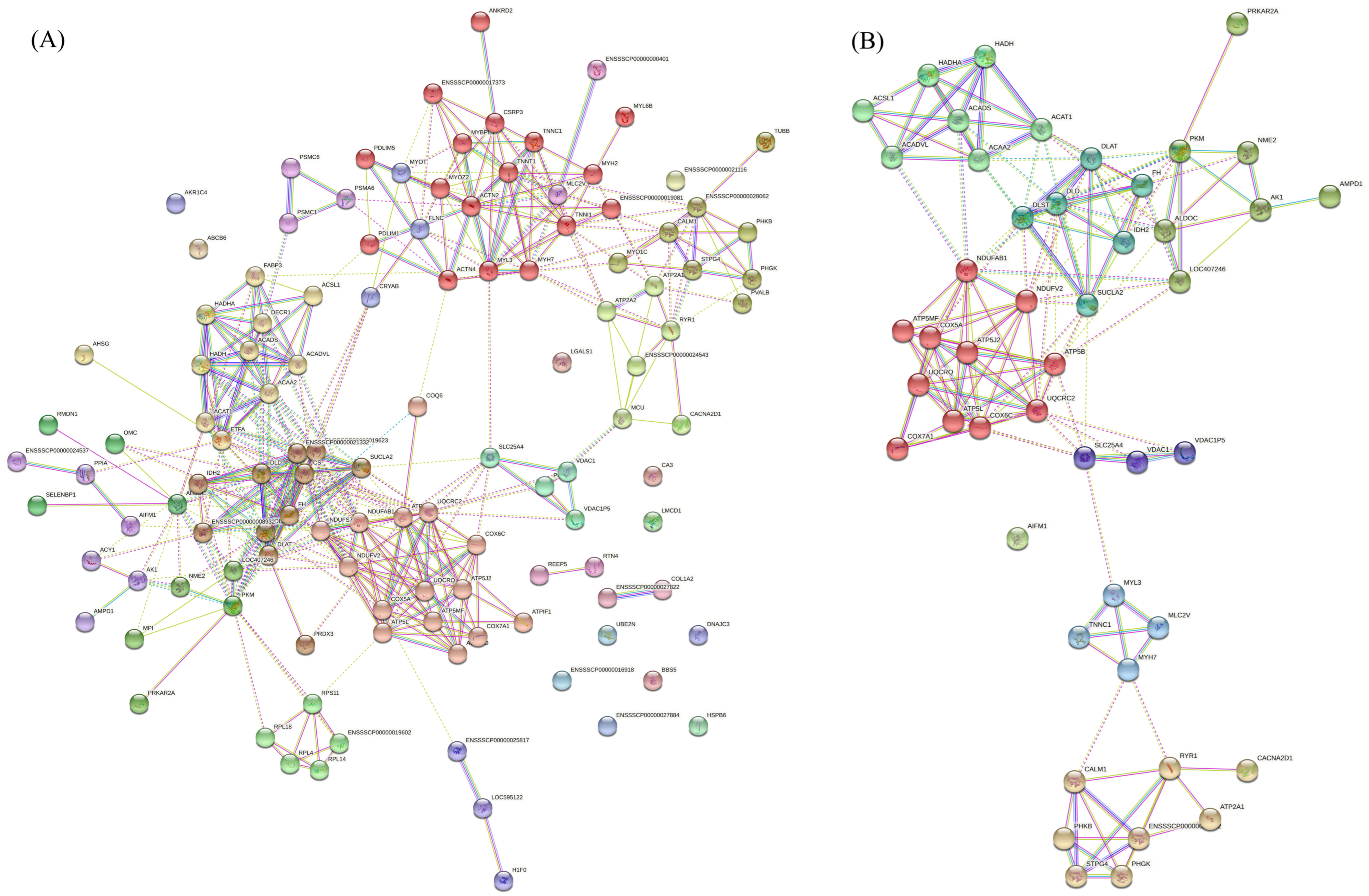
3.4. Interaction Network of DAPs
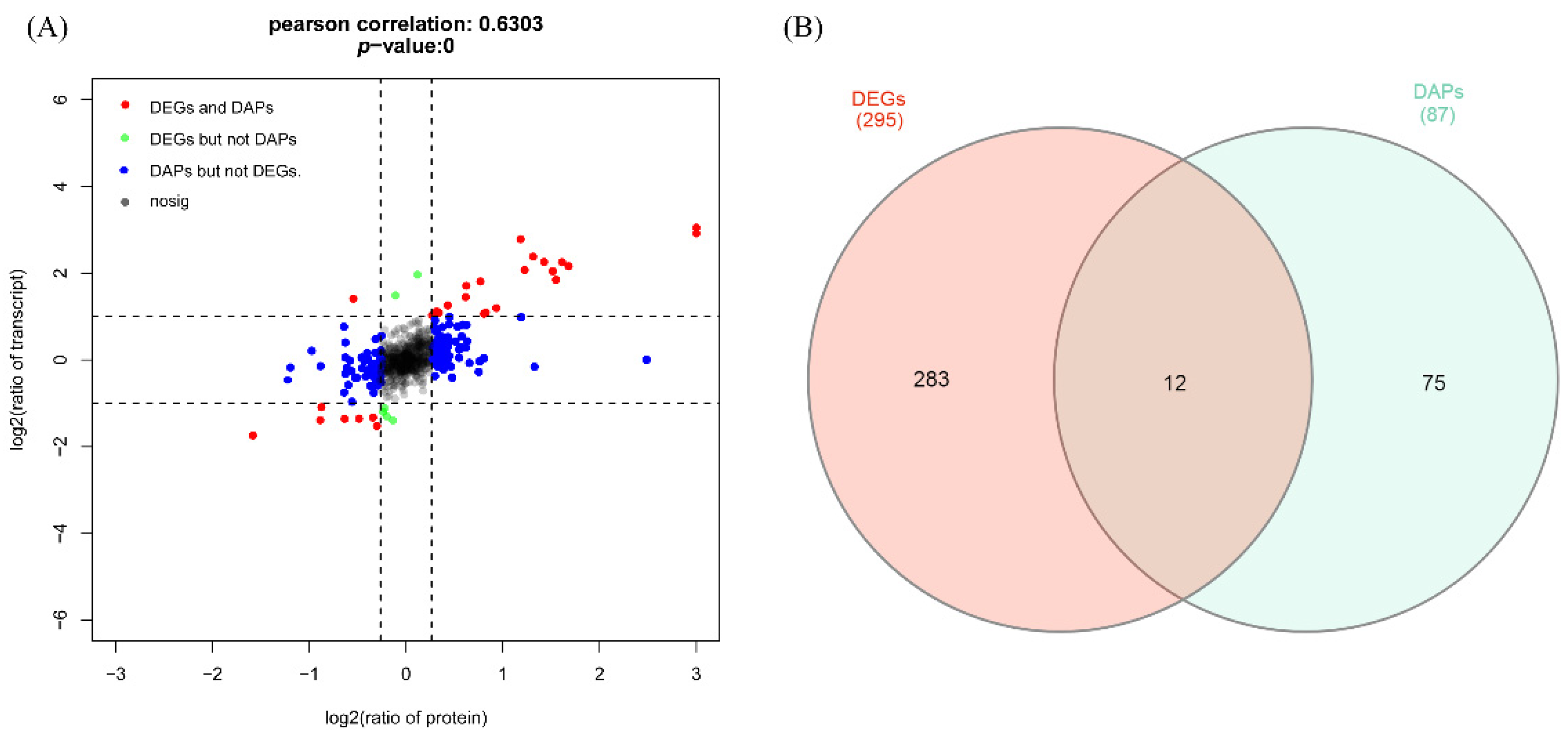
3.5. Association Analysis between Proteome and Transcriptome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gundersen, K. Excitation-transcription coupling in skeletal muscle: The molecular pathways of exercise. Biol. Rev. Camb. Philos. Soc. 2011, 86, 564–600. [Google Scholar] [CrossRef] [PubMed]
- Purslow, P.P.; Gagaoua, M.; Warner, R.D. Insights on meat quality from combining traditional studies and proteomics. Meat Sci. 2021, 174, 108423. [Google Scholar] [CrossRef] [PubMed]
- Listrat, A.; Lebret, B.; Louveau, I.; Astruc, T.; Bonnet, M.; Lefaucheur, L.; Picard, B.; Bugeon, J. How muscle structure and composition influence meat and flesh quality. Sci. World J. 2016, 2016, 3182746. [Google Scholar] [CrossRef] [PubMed]
- Picard, B.; Gagaoua, M. Muscle Fiber Properties in Cattle and Their Relationships with Meat Qualities: An Overview. J. Agric. Food Chem. 2020, 68, 6021–6039. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Li, H.; Xin, X.; Duan, Y.; Hua, X.; Jin, Y. Muscle Fiber Types, Characteristics and Meat Quality. Adv. Mater. Res. 2013, 634–638, 1263–1267. [Google Scholar] [CrossRef]
- Li, B.; Dong, C.; Li, P.; Ren, Z.; Wang, H.; Yu, F.; Ning, C.; Liu, K.; Wei, W.; Huang, R.; et al. Identification of candidate genes associated with porcine meat color traits by genome-wide transcriptome analysis. Sci. Rep. 2016, 6, 35224. [Google Scholar] [CrossRef]
- Li, B.; Yin, D.; Li, P.; Zhang, Z.; Zhang, X.; Li, H.; Li, R.; Hou, L.; Liu, H.; Wu, W. Profiling and Functional Analysis of Circular RNAs in Porcine Fast and Slow Muscles. Front. Cell Dev. Biol. 2020, 8, 322. [Google Scholar] [CrossRef]
- Li, R.; Li, B.; Jiang, A.; Cao, Y.; Hou, L.; Zhang, Z.; Zhang, X.; Liu, H.; Kim, K.H.; Wu, W. Exploring the lncRNAs Related to Skeletal Muscle Fiber Types and Meat Quality Traits in Pigs. Genes 2020, 11, 883. [Google Scholar] [CrossRef]
- Yu, Q.; Tian, X.; Shao, L.; Xu, L.; Dai, R.; Li, X. Label-free proteomic strategy to compare the proteome differences between longissimus lumborum and psoas major muscles during early postmortem periods. Food Chem. 2018, 269, 427–435. [Google Scholar] [CrossRef]
- Nair, M.N.; Li, S.; Beach, C.M.; Rentfrow, G.; Suman, S.P. Changes in the Sarcoplasmic Proteome of Beef Muscles with Differential Color Stability during Postmortem Aging. Meat Muscle Biol. 2018, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wu, S.; Hopkins, D.L.; Liang, R.; Zhu, L.; Zhang, Y.; Luo, X. Proteomic analysis to investigate color changes of chilled beef longissimus steaks held under carbon monoxide and high oxygen packaging. Meat Sci. 2018, 142, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pedrouso, M.; Lorenzo, J.M.; Di Stasio, L.; Brugiapaglia, A.; Franco, D. Quantitative proteomic analysis of beef tenderness of Piemontese young bulls by SWATH-MS. Food Chem. 2021, 356, 129711. [Google Scholar] [CrossRef] [PubMed]
- Malheiros, J.M.; Enriquez-Valencia, C.E.; Braga, C.P.; Vieira, J.C.S.; Vieira, D.S.; Pereira, G.L.; Curi, R.A.; Neto, O.R.M.; Oliveira, H.N.; Padilha, P.M.; et al. Application of proteomic to investigate the different degrees of meat tenderness in Nellore breed. J. Proteom. 2021, 248, 104331. [Google Scholar] [CrossRef]
- Gao, X.; Zhao, D.; Wang, L.; Cui, Y.; Wang, S.; Lv, M.; Zang, F.; Dai, R. Proteomic Changes in Sarcoplasmic and Myofibrillar Proteins Associated with Color Stability of Ovine Muscle during Post-Mortem Storage. Foods 2021, 10, 2989. [Google Scholar] [CrossRef] [PubMed]
- Della Malva, A.; De Palo, P.; Lorenzo, J.M.; Maggiolino, A.; Albenzio, M.; Marino, R. Application of proteomic to investigate the post-mortem tenderization rate of different horse muscles. Meat Sci. 2019, 157, 107885. [Google Scholar] [CrossRef]
- Chu, W.; Zhang, F.; Song, R.; Li, Y.; Wu, P.; Chen, L.; Cheng, J.; Du, S.; Zhang, J. Proteomic and microRNA Transcriptome Analysis revealed the microRNA-SmyD1 network regulation in Skeletal Muscle Fibers performance of Chinese perch. Sci. Rep. 2017, 7, 16498. [Google Scholar] [CrossRef]
- Ohira, T.; Ino, Y.; Nakai, Y.; Morita, H.; Kimura, A.; Kurata, Y.; Kagawa, H.; Kimura, M.; Egashira, K.; Moriya, S.; et al. Proteomic analysis revealed different responses to hypergravity of soleus and extensor digitorum longus muscles in mice. J. Proteom. 2020, 217, 103686. [Google Scholar] [CrossRef]
- Mach, N.; Keuning, E.; Kruijt, L.; Hortos, M.; Arnau, J.; Te Pas, M.F. Comparative proteomic profiling of 2 muscles from 5 different pure pig breeds using surface-enhanced laser desorption/ionization time-of-flight proteomics technology. J. Anim. Sci. 2010, 88, 1522–1534. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Pan, J.; Liu, X.; Chen, H.; Zhou, X.; Yuan, Z.; Wang, X.; Mo, D. iTRAQ-based quantitative proteomic analysis reveals the distinct early embryo myofiber type characteristics involved in landrace and miniature pig. BMC Genom. 2016, 17, 137. [Google Scholar] [CrossRef]
- Wang, Z.; Shang, P.; Li, Q.; Wang, L.; Chamba, Y.; Zhang, B.; Zhang, H.; Wu, C. iTRAQ-based proteomic analysis reveals key proteins affecting muscle growth and lipid deposition in pigs. Sci. Rep. 2017, 7, 46717. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, X.; Zhang, Y.; Zhang, H.; Zhang, X.; Zhang, H. Comparative transcriptomic and proteomic analyses provide insights into functional genes for hypoxic adaptation in embryos of Tibetan chickens. Sci. Rep. 2020, 10, 11213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, D.; Xu, X.; Xu, W.; Zhou, G. iTRAQ-based proteomic analysis of duck muscle related to lipid oxidation. Poult. Sci. 2021, 100, 101029. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.D.; Ryu, Y.C.; Jeong, J.Y.; Yang, H.S.; Joo, S.T. Relationship between pork quality and characteristics of muscle fibers classified by the distribution of myosin heavy chain isoforms. J. Anim. Sci. 2013, 91, 5525–5534. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, J.M.; Ryu, Y.C.; Ko, K.S. Effects of Morphological Characteristics of Muscle Fibers on Porcine Growth Performance and Pork Quality. Korean J. Food Sci. Anim. Resour. 2016, 36, 583–593. [Google Scholar] [CrossRef]
- Weng, K.Q.; Huo, W.R.; Li, Y.; Zhang, Y.; Zhang, Y.; Chen, G.H.; Xu, Q. Fiber characteristics and meat quality of different muscular tissues from slow- and fast-growing broilers. Poultry Sci. 2022, 101, 101537. [Google Scholar] [CrossRef]
- Huo, W.R.; Weng, K.Q.; Gu, T.T.; Zhang, Y.; Zhang, Y.; Chen, G.H.; Xu, Q. Effect of muscle fiber characteristics on meat quality in fast- and slow-growing ducks. Poultry Sci. 2021, 100, 101264. [Google Scholar] [CrossRef]
- Jiang, A.; Yin, D.; Zhang, L.; Li, B.; Li, R.; Zhang, X.; Zhang, Z.; Liu, H.; Kim, K.; Wu, W. Parsing the microRNA genetics basis regulating skeletal muscle fiber types and meat quality traits in pigs. Anim. Genet. 2021, 52, 292–303. [Google Scholar] [CrossRef]
- Unwin, R.D. Quantification of proteins by iTRAQ. Methods Mol. Biol. 2010, 658, 205–215. [Google Scholar] [CrossRef]
- Chang, H.; Jiang, S.F.; Ma, X.F.; Peng, X.; Zhang, J.; Wang, Z.; Xu, S.H.; Wang, H.P.; Gao, Y.F. Proteomic analysis reveals the distinct energy and protein metabolism characteristics involved in myofiber type conversion and resistance of atrophy in the extensor digitorum longus muscle of hibernating Daurian ground squirrels. Comp. Biochem. Phys. D 2018, 26, 20–31. [Google Scholar] [CrossRef]
- Ge, C.M.; Lu, W.Q.; Chen, A. Quantitative proteomic reveals the dynamic of protein profile during final oocyte maturation in zebrafish. Biochem. Biophys. Res. Commun. 2017, 490, 657–663. [Google Scholar] [CrossRef]
- Song, X.; Chen, Z.Z.; Jia, R.Y.; Cao, M.; Zou, Y.F.; Li, L.X.; Liang, X.X.; Yin, L.Z.; He, C.L.; Yue, G.Z.; et al. Transcriptomics and proteomic studies reveal acaricidal mechanism of octadecanoic acid-3, 4-tetrahydrofuran diester against Sarcoptes scabiei var. cuniculi. Sci. Rep. 2017, 7, 45479. [Google Scholar] [CrossRef] [PubMed]
- Hunt, L.C.; Graca, F.A.; Pagala, V.; Wang, Y.D.; Li, Y.; Yuan, Z.F.; Fan, Y.; Labelle, M.; Peng, J.; Demontis, F. Integrated genomic and proteomic analyses identify stimulus-dependent molecular changes associated with distinct modes of skeletal muscle atrophy. Cell Rep. 2021, 37, 109971. [Google Scholar] [CrossRef] [PubMed]
- Williams, Z.J.; Velez-Irizarry, D.; Gardner, K.; Valberg, S.J. Integrated proteomic and transcriptomic profiling identifies aberrant gene and protein expression in the sarcomere, mitochondrial complex I, and the extracellular matrix in Warmblood horses with myofibrillar myopathy. BMC Genom. 2021, 22, 438. [Google Scholar] [CrossRef]
- Mohammadabadi, M.; Bordbar, F.; Jensen, J.; Du, M.; Guo, W. Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals. Animals 2021, 11, 835. [Google Scholar] [CrossRef]
- Bourdeau Julien, I.; Sephton, C.F.; Dutchak, P.A. Metabolic Networks Influencing Skeletal Muscle Fiber Composition. Front. Cell Dev. Biol. 2018, 6, 125. [Google Scholar] [CrossRef]
- Mendes, T.B.; Nozima, B.H.; Budu, A.; de Souza, R.B.; Braga Catroxo, M.H.; Delcelo, R.; Gazarini, M.L.; Cerutti, J.M. PVALB diminishes [Ca2+] and alters mitochondrial features in follicular thyroid carcinoma cells through AKT/GSK3beta pathway. Endocr.-Relat. Cancer 2016, 23, 769–782. [Google Scholar] [CrossRef]
- Hou, X.; Liu, Q.; Meng, Q.; Wang, L.; Yan, H.; Zhang, L.; Wang, L. TMT-based quantitative proteomic analysis of porcine muscle associated with postmortem meat quality. Food Chem. 2020, 328, 127133. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.W.; Yang, J.; Zhou, L.S.; Ren, J.; Liu, X.X.; Zhang, H.; Yang, B.; Zhang, Z.Y.; Ma, H.B.; Xie, X.H.; et al. A Splice Mutation in the PHKG1 Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle. PLoS Genet. 2014, 10, e1004710. [Google Scholar] [CrossRef]
- Fujii, J.; Otsu, K.; Zorzato, F.; Deleon, S.; Khanna, V.K.; Weiler, J.E.; Obrien, P.J.; Maclennan, D.H. Identification of a Mutation in Porcine Ryanodine Receptor Associated with Malignant Hyperthermia. Science 1991, 253, 448–451. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of carbonic anhydrase CA3 in skeletal muscle. Expert Rev. Proteom. 2021, 18, 1073–1086. [Google Scholar] [CrossRef]
- Wu, W.J.; Zhang, Z.K.; Chao, Z.; Li, B.J.; Li, R.Y.; Jiang, A.W.; Kim, K.H.; Liu, H.L. Transcriptome analysis reveals the genetic basis of skeletal muscle glycolytic potential based on a pig model. Gene 2021, 766, 145157. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, G.; Liao, J.; Chen, X. A functional mutation in the AMPD1 promoter region affects promoter activity and breast meat freshness in chicken. Anim. Genet. 2021, 52, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Sieczkowska, H.; Zybert, A.; Krzecio, E.; Antosik, K.; Kocwin-Podsiadla, M.; Pierzchala, M.; Urbanski, P. The expression of genes PKM2 and CAST in the muscle tissue of pigs differentiated by glycolytic potential and drip loss, with reference to the genetic group. Meat Sci. 2010, 84, 137–142. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, W.; Zha, C.; Jiang, A.; Chao, Z.; Hou, L.; Liu, H.; Huang, R.; Wu, W. A Combined Differential Proteome and Transcriptome Profiling of Fast- and Slow-Twitch Skeletal Muscle in Pigs. Foods 2022, 11, 2842. https://doi.org/10.3390/foods11182842
Wei W, Zha C, Jiang A, Chao Z, Hou L, Liu H, Huang R, Wu W. A Combined Differential Proteome and Transcriptome Profiling of Fast- and Slow-Twitch Skeletal Muscle in Pigs. Foods. 2022; 11(18):2842. https://doi.org/10.3390/foods11182842
Chicago/Turabian StyleWei, Wei, Chengwan Zha, Aiwen Jiang, Zhe Chao, Liming Hou, Honglin Liu, Ruihua Huang, and Wangjun Wu. 2022. "A Combined Differential Proteome and Transcriptome Profiling of Fast- and Slow-Twitch Skeletal Muscle in Pigs" Foods 11, no. 18: 2842. https://doi.org/10.3390/foods11182842
APA StyleWei, W., Zha, C., Jiang, A., Chao, Z., Hou, L., Liu, H., Huang, R., & Wu, W. (2022). A Combined Differential Proteome and Transcriptome Profiling of Fast- and Slow-Twitch Skeletal Muscle in Pigs. Foods, 11(18), 2842. https://doi.org/10.3390/foods11182842