
A Water-Dispersible Carboxylated Carbon Nitride Nanoparticles-Based Electrochemical Platform for Direct Reporting of Hydroxyl Radical in Meat


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Synthesis of Carboxylated-g-C3N4 NPs
2.3. Apparatus and Instruments
2.4. Fabrication of MB/ssDNA/Carboxylated-g-C3N4/Chitosan/GCE
2.5. Electrochemical Measurements
2.6. Detection of Real Samples
3. Results and Discussion
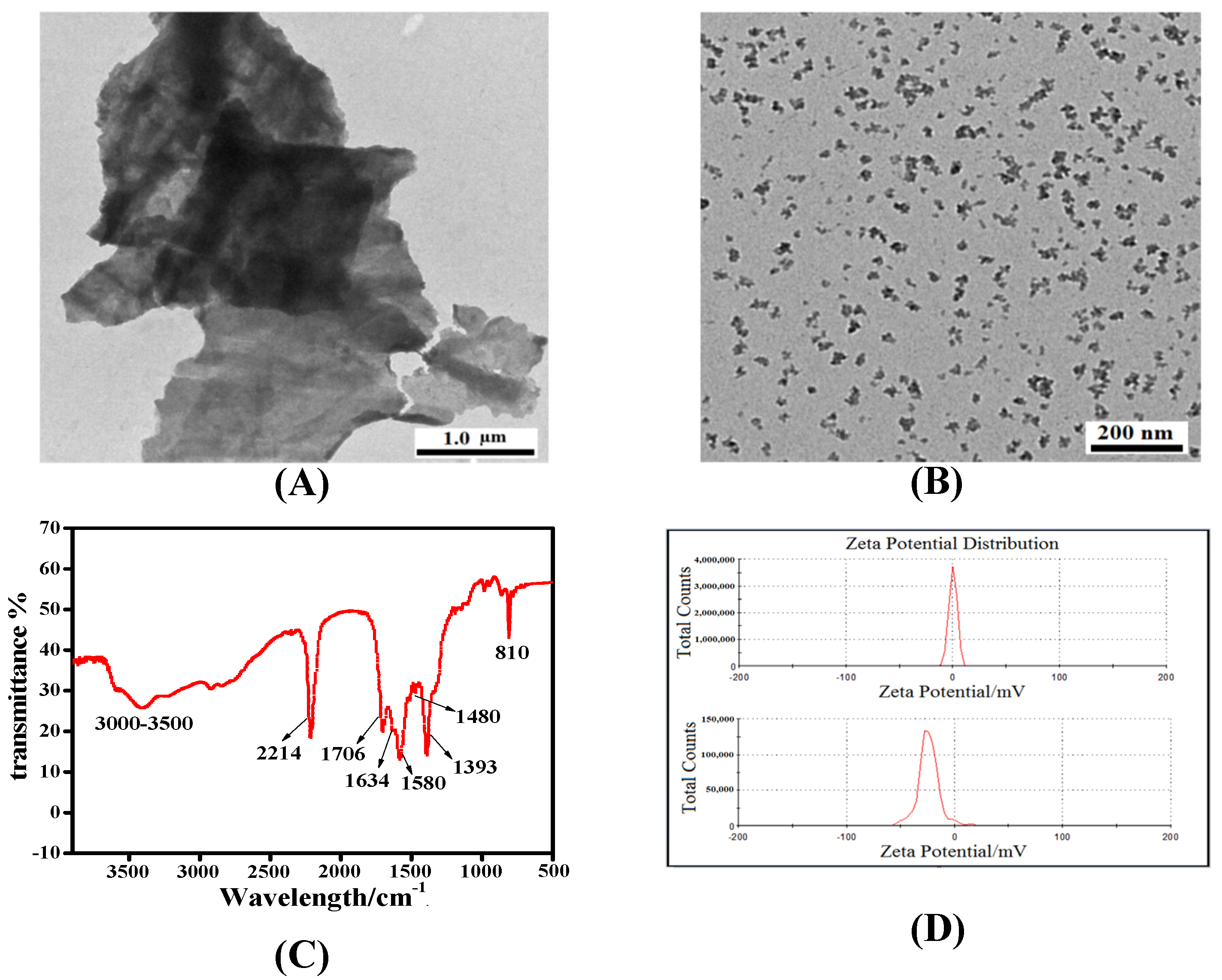
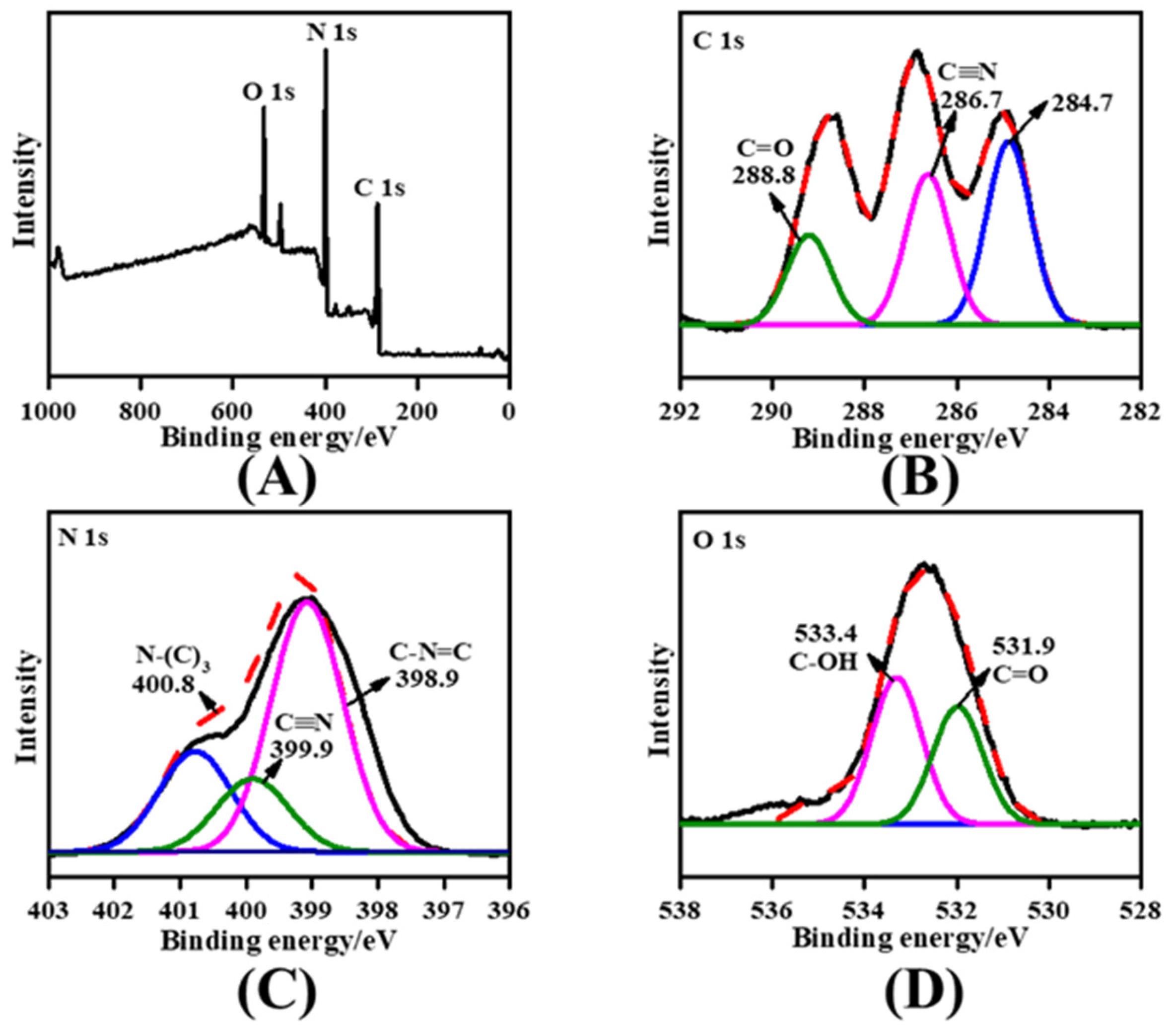
3.1. Characterization of the Carboxylated-g-C3N4 NPs
3.2. The Mechanism of Detection •OH
3.3. Optimization of Experimental Parameters
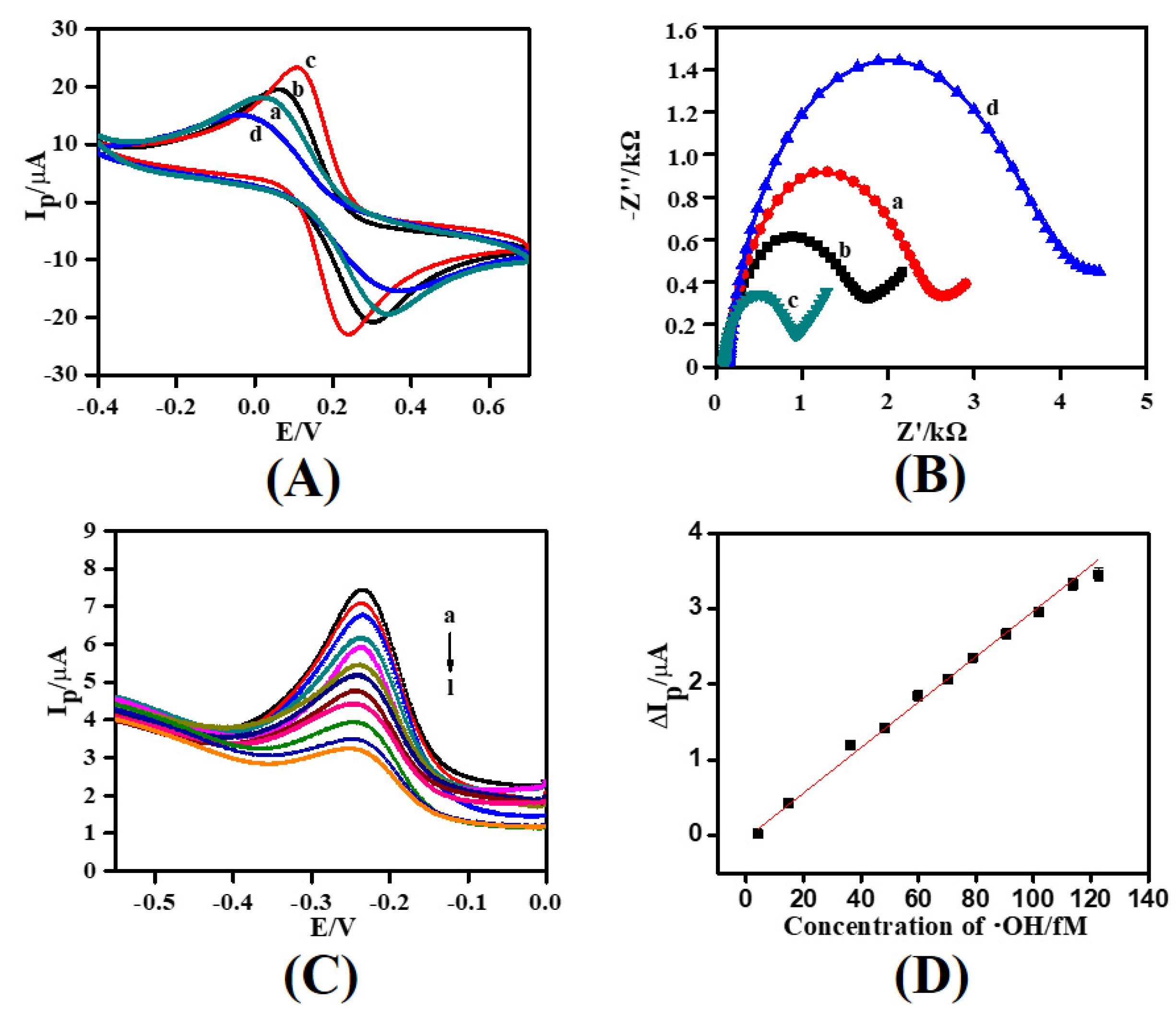
3.4. Electrochemical Characteristics of the Fabricated Electrode
3.5. Electrochemical Detection of •OH
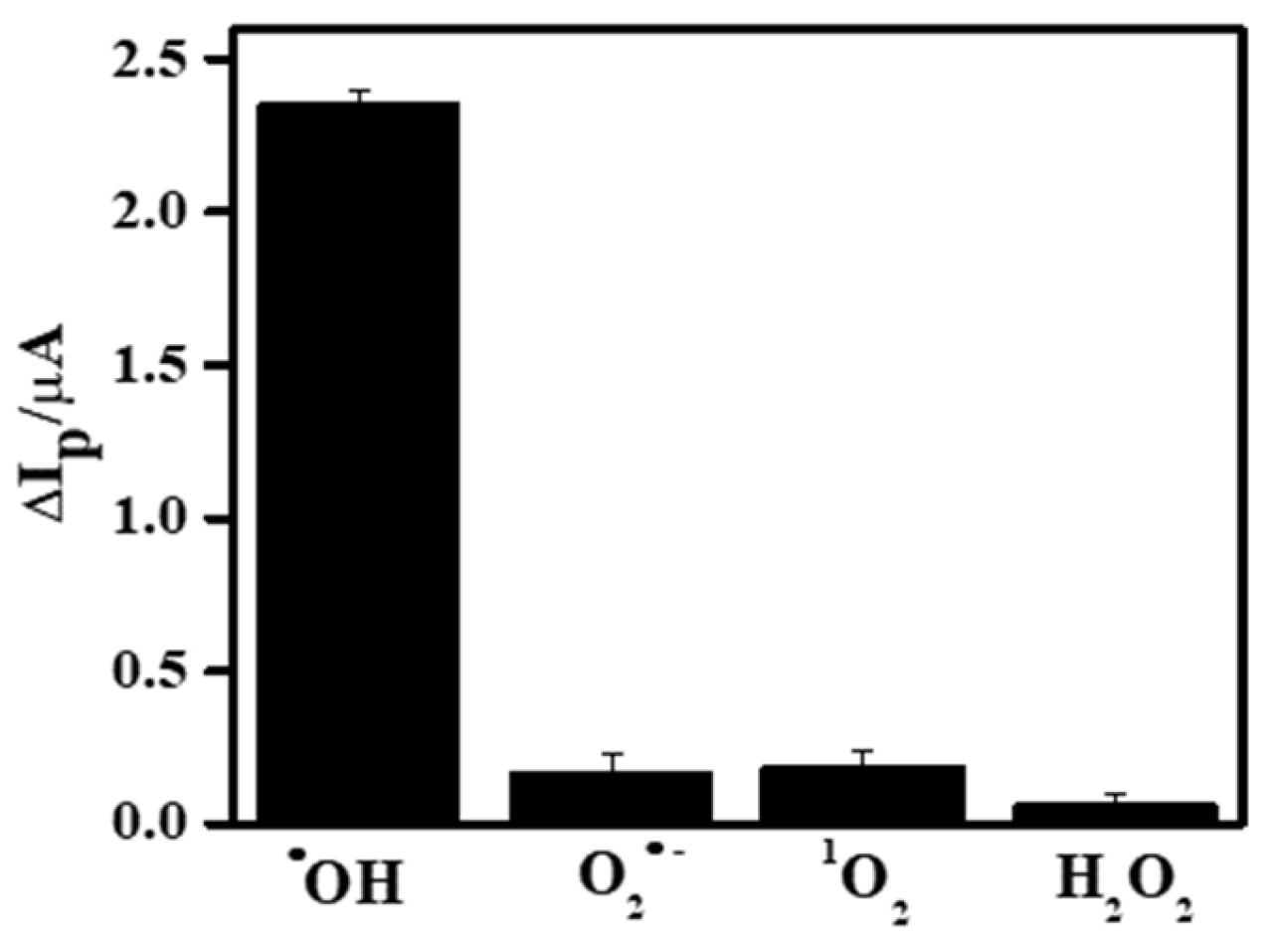
3.6. Specificity, Stability, and Reproducibility of the Fabricated Sensor
3.7. Real Sample Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pereira, P.F.M.; Picciani, P.H.D.; Calado, V.; Tonon, R.V. Electrical gas sensors for meat freshness assessment and quality monitoring: A review. Trends Food. Sci. Technol. 2021, 118, 36–44. [Google Scholar] [CrossRef]
- Balakrishna, M.; Ma, J.J.; Liu, T.; Geng, Z.M.; Li, P.P.; Wang, D.Y.; Zhang, M.H.; Xu, W.M. Hydrolysis of oxidized phosphatidylcholines by crude enzymes from chicken, pork and beef muscles. Food Chem. 2020, 313, 125956. [Google Scholar] [CrossRef]
- Falowo, A.B.; Fayemi, P.O.; Muchenje, V. Natural antioxidants against lipid–protein oxidative deterioration in meat and meat products: A review. Food Res. Int. 2014, 64, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Papuc, C.; Goran, G.V.; Predescu, C.N.; Nicorescu, V. Mechanisms of oxidative processes in meat and toxicity induced by postprandial degradation products: A Review. Compr. Rev. Food Sci. Food Saf. 2017, 16, 96–123. [Google Scholar] [CrossRef]
- Mousavisani, S.Z.; Raoof, J.B.; Ojani, R.; Bagheryan, Z. An impedimetric biosensor for DNA damage detection and study of the protective effect of deferoxamine against DNA damage. Bioelectrochemistry 2018, 122, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liang, T.; Lv, S.W.; Zhuang, Q.G.; Liu, Z.H. A rationally designed upconversion nanoprobe for in vivo detection of hydroxyl radical. J. Am. Chem. Soc. 2015, 137, 11179–11185. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.J.; Zhu, Y.X.; Ren, X.P.; Zhang, Y.W.; Peng, Z.Q.; Zhou, G.H. Formation and inhibition of lipid alkyl radicals in roasted meat. Foods. 2020, 9, 572. [Google Scholar] [CrossRef]
- Hajkova, A.; Barek, J.; Vyskocil, V. Electrochemical DNA biosensor for detection of DNA damage induced by hydroxyl radicals. Bioelectrochemistry 2017, 116, 1–9. [Google Scholar] [CrossRef]
- Mason, R.P.; Hanna, P.M.; Burkitt, M.J.; Kadiiska, M.B. Detection of oxygen-derived radicals in biological systems using electron spin resonance. Environ. Health Perspect. 1994, 102, 33–36. [Google Scholar] [CrossRef]
- Wang, D.B.; Yu, W.S.; Pang, X.L.; Cao, J.M.; Qiu, J.; Kong, F.Y. A new method for hydroxyl radical detection by chemiluminescence of flue-cured tobacco extracts. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 204, 436–439. [Google Scholar] [CrossRef]
- Manzoor, K.; Mishra, S.K.; Podmore, I.D. Detection and identification of ethanal-derived spin-trapped free radicals using headspace thermal desorption gas chromatography-mass spectrometry (TD-GC-MS). Free Radic. Res. 2020, 54, 745–755. [Google Scholar] [CrossRef]
- Li, H.; Wang, Q.J. Evaluation of free hydroxyl radical scavenging activities of some Chinese herbs by capillary zone electrophoresis with amperometric detection. Anal. Bioanal. Chem. 2004, 378, 1801–1805. [Google Scholar] [CrossRef]
- Liu, L.Z.; Dong, F.Q.; Sun, S.Y.; He, X.C.; Wang, M.C. Quantitative detection of hydroxyl radical generated in quartz powder/phosphate buffer solution system by fluorescence spectrophotometry. Spectrosc. Spect. Anal. 2014, 34, 1886–1889. [Google Scholar] [CrossRef]
- Sui, Y.K.; Zorman, C.A. Inkjet printing of metal structures for electrochemical sensor applications: A review. J. Electrochem. Soc. 2020, 167, 037571. [Google Scholar] [CrossRef]
- Xu, Y.W.; Wang, D.Y.; Zhang, Y.; Zhang, J.J.; Jiang, S.Y.; Liang, W.C.; Zhu, T.T.; Ye, B.C. A novel electrochemical sensor for determination of hydroxyl radicals in living cells by coupling nanoporous gold layer with self-assembled 6-(Ferrocenyl) hexanethiol. Anal. Chim. Acta 2020, 1096, 69–75. [Google Scholar] [CrossRef]
- Duanghathaipornsuk, S.; Alateeq, F.A.O.; Kim, S.S.; Kim, D.S.; Alba-Rubio, A.C. The effects of size and content of cerium oxide nanoparticles on a composite sensor for hydroxyl radicals detection. Sens. Actuators B Chem. 2020, 321, 128467. [Google Scholar] [CrossRef]
- Shoda, S.; Hyodo, F.; Tachibana, Y.; Kiniwa, M.; Naganuma, T.; Eto, H.; Koyasu, N.; Murata, M.; Matsuo, M. Imaging of hydroxyl-radical generation using dynamic nuclear polarization-magnetic resonance imaging and a spin-trapping agent. Anal. Chem. 2020, 92, 14408–14414. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Ansari, M.S.; Devnani, H.; Satsangee, S.P.; Jain, R. CeO2/g-C3N4 nanocomposite: A perspective for electrochemical sensing of anti-depressant drug. Sens. Actuators B Chem. 2018, 273, 1226–1236. [Google Scholar] [CrossRef]
- Xiang, M.H.; Li, N.; Liu, J.W.; Yu, R.Q.; Jiang, J.H. A tumour mRNA-triggered nanoassembly for enhanced fluorescence imaging-guided photodynamic therapy. Nanoscale 2020, 12, 8727–8731. [Google Scholar] [CrossRef]
- Mohanraj, J.; Durgalakshmi, D.; Saravanan, R. Water-soluble graphitic carbon nitride for clean environmental applications. Environ. Pollut. 2021, 269, 116172. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wu, S.L.; Liu, Y.; Sun, Y.J.; Cao, Y.; Hsu, J.P.; Wee, A.T.S.; Jiang, J.Z. An ultra-sensitive electrochemical sensor based on 2D g-C3N4/CuO nanocomposites for dopamine detection. Carbon 2018, 130, 652–663. [Google Scholar] [CrossRef]
- Zhou, P.; Lai, J.P.; Tang, Y.H.; Chao, Y.G.; Lin, F.; Guo, S.J. Amorphous FeCoPOx nanowires coupled to g-C3N4 nanosheets with enhanced interfacial electronic transfer for boosting photocatalytic hydrogen production. Appl. Catal. B 2018, 238, 161–167. [Google Scholar] [CrossRef]
- Shao, M.M.; Shao, Y.F.; Ding, S.J.; Wang, J.W.; Xu, J.C.; Qu, Y.J.; Zhong, X.W.; Chen, X.M.; Ip, W.F.; Wang, N.; et al. Vanadium disulfide decorated graphitic carbon nitride for super-efficient solar-driven hydrogen evolution. Appl. Catal. B 2018, 237, 295–301. [Google Scholar] [CrossRef]
- Zhou, X.B.; Li, Y.F.; Xing, Y.; Li, J.S.; Jiang, X. Effects of the preparation method of Pt/g-C3N4 photocatalysts on their efficiency for visible-light hydrogen production. Dalton. Trans. 2019, 48, 15068–15073. [Google Scholar] [CrossRef] [PubMed]
- Henle, E.S.; Linn, S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J. Biol. Chem. 1997, 272, 19095–19098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.P.; Wang, J.S.; Ma, P.P.; Yao, H.C.; Zhang, L.; Li, Z.J. Synthesis of fluorescent polymeric carbon nitride quantum dots in molten salts for security inks. New J. Chem. 2017, 41, 14918–14923. [Google Scholar] [CrossRef]
- Bolumar, T.; Andersen, M.L.; Orlien, V. Mechanisms of radical formation in beef and chicken meat during high pressure processing evaluated by electron spin resonance detection and the addition of antioxidants. Food Chem. 2014, 150, 422–428. [Google Scholar] [CrossRef]
- Zeng, H.M.; Liu, Y.L.; Xu, Z.G.; Wang, Y.; Chai, Y.Q.; Yuan, R.; Liu, H.Y. Construction of a Z-scheme g-C3N4/Ag/AgI heterojunction for highly selective photoelectrochemical detection of hydrogen sulfide. Chem. Commun. 2019, 55, 11940–11943. [Google Scholar] [CrossRef]
- Wang, Y.P.; Li, Y.K.; Ju, W.; Wang, J.C.; Yao, H.C.; Zhang, L.; Wang, J.S.; Li, Z.J. Molten salt synthesis of water-dispersible polymeric carbon nitride nanoseaweeds and their application as luminescent probes. Carbon 2016, 102, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Tang, L.; Zeng, G.M.; Fang, S.Y.; Ouyang, X.L.; Long, B.Q.; Zhou, Y.Y.; Deng, Y.C.; Liu, Y.N.; Wang, J.J. Self-powered photoelectrochemical aptasensor based on phosphorus doped porous ultrathin g-C3N4 nanosheets enhanced by surface plasmon resonance effect. Biosens. Bioelectron. 2018, 121, 19–26. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Su, C.S.; Ren, H.; Li, M.L.; Zhu, L.F.; Ge, S.; Wang, M.; Zhang, Z.L.; Li, L.; Cao, X.B. In-situ fabrication of g-C3N4/ZnO nanocomposites for photocatalytic degradation of methylene blue: Synthesis procedure does matter. Nanomaterials 2019, 9, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.B.; Gong, Y.J.; Zhang, J.S.; Zhan, L.; Ma, L.L.; Fang, Z.Y.; Vajtai, R.; Wang, X.C.; Ajayan, P.M. Exfoliated graphitic carbon nitride nanosheets as efficient catalysts for hydrogen evolution under visible light. Adv. Mater. 2013, 25, 2452–2456. [Google Scholar] [CrossRef]
- Xu, J.H.; Li, D.N.; Chen, Y.; Tan, L.H.; Kou, B.; Wan, F.S.; Jiang, W.; Li, F.S. Constructing sheet-on-sheet structured graphitic carbon nitride/rReduced graphene oxide/layered MnO2 ternary nanocomposite with outstanding catalytic properties on thermal decomposition of ammonium perchlorate. Nanomaterials 2017, 7, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiravand, G.; Badiei, A.; Ziarani, G.M. Carboxyl-rich g-C3N4 nanoparticles: Synthesis, characterization and their application for selective fluorescence sensing of Hg2+ and Fe3+ in aqueous media. Sens. Actuators B Chem. 2017, 242, 244–252. [Google Scholar] [CrossRef]
- Li, C.; Yang, X.G.; Yang, B.J.; Yan, Y.; Qian, Y.T. Synthesis and characterization of nitrogen-rich graphitic carbon nitride. Mater. Chem. Phys. 2007, 103, 427–432. [Google Scholar] [CrossRef]
- Jiang, J.A.; Zhu, L.H.; Zou, J.; Lei, O.Y.; Zheng, A.M.; Tang, H.Q. Micro/nano-structured graphitic carbon nitride-Ag nanoparticle hybrids as surface-enhanced Raman scattering substrates with much improved long-term stability. Carbon 2015, 87, 193–205. [Google Scholar] [CrossRef]
- Zhang, X.D.; Xie, X.; Wang, H.; Zhang, J.J.; Pan, B.C.; Xie, Y. Enhanced photoresponsive ultrathin graphitic-phase C3N4 nanosheets for bioimaging. J. Am. Chem. Soc. 2013, 135, 18–21. [Google Scholar] [CrossRef]
- Yoon, S.R.; Dang, Y.M.; Kim, S.Y.; You, S.Y.; Kim, M.K.; Ha, J.H. Correlating capsaicinoid levels and physicochemical properties of kimchi and its perceived spiciness. Foods 2021, 10, 86. [Google Scholar] [CrossRef]
- Xu, Y.J.; Lei, W.; Su, J.; Hu, J.J.; Yu, X.D.; Zhou, T.; Yang, Y.; Mandler, D.; Hao, Q.L. A high-performance electrochemical sensor based on g-C3N4-E-PEDOT for the determination of acetaminophen. Electrochim. Acta 2018, 259, 994–1003. [Google Scholar] [CrossRef]
- Huang, J.Z.; Zeng, Q.; Bai, S.L.; Wang, L.S. Application of coal in electrochemical sensing. Anal. Chem. 2017, 89, 8358–8365. [Google Scholar] [CrossRef]
- Teengam, P.; Siangproh, W.; Tuantranont, A.; Vilaivan, T.; Chailapakul, O.; Henry, C.S. Electrochemical impedance-based DNA sensor using pyrrolidinyl peptide nucleic acids for tuberculosis detection. Anal. Chim. Acta 2018, 1044, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Hai, X.; Guo, Z.Y.; Lin, X.; Chen, X.W.; Wang, J. Fluorescent TPA@GQDs probe for sensitive assay and quantitative imaging of hydroxyl radicals in living cells. ACS Appl. Mater. Interfaces 2018, 10, 5853–5861. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Hwang, J.H.; Li, R.; Rodriguez, K.L. Flexible copper-biopolymer nanocomposite sensors for trace level lead detection in water. Sens. Actuators B Chem. 2021, 344, 130263. [Google Scholar] [CrossRef]
- Ganea, G.M.; Kolic, P.E.; El-Zahab, B.; Warner, I.M. Ratiometric coumarin-neutral red (CONER) nanoprobe for detection of hydroxyl radicals. Anal. Chem. 2011, 83, 2576–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, N.; Guo, L.H.; Zhu, B.Z. An electrochemical biosensor for the detection of tyrosine oxidation induced by Fenton reaction. Biosens. Bioelectron. 2011, 26, 2292–2296. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, J.W.; Zhang, Y.S.; Zou, Q.; Zhang, X.H.; Chen, J.H. Sensitive electrochemical detection of hydroxyl radical based on MBs-DNA-AgNPs nanocomposite. Sens. Actuators B Chem. 2013, 182, 504–509. [Google Scholar] [CrossRef]
- Jia, S.P.; Liang, M.M.; Guo, L.H. Photoelectrochemical detection of oxidative DNA damage induced by Fenton Reaction with low concentration and DNA-associated Fe2+. J. Phys. Chem. B 2008, 112, 4461–4464. [Google Scholar] [CrossRef]
- Huang, W.T.; Xie, W.Y.; Shi, Y.; Luo, H.Q.; Li, N.B. A simple and facile strategy based on Fenton-induced DNA cleavage for fluorescent turn-on detection of hydroxyl radicals and Fe2+. J. Mater. Chem. 2012, 22, 1477–1481. [Google Scholar] [CrossRef]
- Wu, L.; Yang, Y.; Zhang, H.J.; Zhu, G.B.; Zhang, X.H.; Chen, J.H. Sensitive electrochemical detection of hydroxyl radical with biobarcode amplification. Anal. Chim. Acta 2012, 756, 1–6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detection Methods | R2 | Linear Range | Detection Limit | Ref. |
|---|---|---|---|---|
| Fluorescent TPA@GQDs probe | 0.9972 | 0.018–6 μM | 12 nM | [42] |
| Ratiometric fluorescence | - | 10–1000 μM | 730 nM | [44] |
| Electrochemical peptide biosensor | - | - | 1.0 × 104 nM | [45] |
| MBs-DNA-AgNPs/GCE | 0.9987 | 0.05–4 μM | 10 nM | [46] |
| Photochemical sensor | - | - | 1.0 × 104 nM | [47] |
| ssDNA/carboxylated-g-C3N4/chitosan/GCE | 0.9933 | 4.06–122.79 fM | 1.35 fM | This work |
| Detected by sensor (fM·g−1) | Detected by ESR (fM·g−1) | ||||||
|---|---|---|---|---|---|---|---|
| Samples | Added | Initial | Found | Recovery (n = 3) | Initial | Found | Recovery (n = 3) |
| Shrimp | 0.61 | 0.0032 | 0.60 | 97.84% ± 3.69% | ND | 0.63 | 103.27% ± 4.84% |
| Chicken | 3.66 | 0.0079 | 3.61 | 98.42% ± 4.25% | ND | 3.58 | 97.81% ± 3.53% |
| Pork | 12.20 | 0.0095 | 12.13 | 99.34% ± 4.87% | ND | 12.15 | 99.59% ± 4.66% |
| Sausage | 48.80 | 0.0156 | 48.49 | 99.33% ± 3.76% | ND | 48.91 | 100.23% ± 3.27% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, T.; Huang, Y.; Sun, C.; Wang, D.; Xu, W. A Water-Dispersible Carboxylated Carbon Nitride Nanoparticles-Based Electrochemical Platform for Direct Reporting of Hydroxyl Radical in Meat. Foods 2022, 11, 40. https://doi.org/10.3390/foods11010040
Han T, Huang Y, Sun C, Wang D, Xu W. A Water-Dispersible Carboxylated Carbon Nitride Nanoparticles-Based Electrochemical Platform for Direct Reporting of Hydroxyl Radical in Meat. Foods. 2022; 11(1):40. https://doi.org/10.3390/foods11010040
Chicago/Turabian StyleHan, Tingting, Yang Huang, Chong Sun, Daoying Wang, and Weimin Xu. 2022. "A Water-Dispersible Carboxylated Carbon Nitride Nanoparticles-Based Electrochemical Platform for Direct Reporting of Hydroxyl Radical in Meat" Foods 11, no. 1: 40. https://doi.org/10.3390/foods11010040
APA StyleHan, T., Huang, Y., Sun, C., Wang, D., & Xu, W. (2022). A Water-Dispersible Carboxylated Carbon Nitride Nanoparticles-Based Electrochemical Platform for Direct Reporting of Hydroxyl Radical in Meat. Foods, 11(1), 40. https://doi.org/10.3390/foods11010040