
Development of an RNA Extraction Protocol for Norovirus from Raw Oysters and Detection by qRT-PCR and Droplet-Digital RT-PCR

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Oyster Samples
2.2. Preparation of Murine Norovirus (MNV) Stock
2.3. RNA Extractions
2.3.1. Lysis Followed by Extraction with Direct-Zol Columns
2.3.2. Lysis Followed by Extraction with QIAamp Columns
2.3.3. The ISO 15216-1:2017 Method
2.3.4. The Method E
2.4. Further RNA Clean-Up
2.4.1. Lithium Chloride Precipitation
2.4.2. Cetyltrimethylammonium Bromide (CTAB) and Lithium Chloride Precipitation
2.4.3. Precipitation in the Presence of Glycogen
2.5. Detection by qRT-PCR
2.6. Detection by Droplet Digital PCR
2.7. Recovery Rates
2.8. PCR Inhibition
2.9. Statistical Analyses
3. Results
3.1. Comparing RNA Extraction Efficiency Using Tri-Reagent
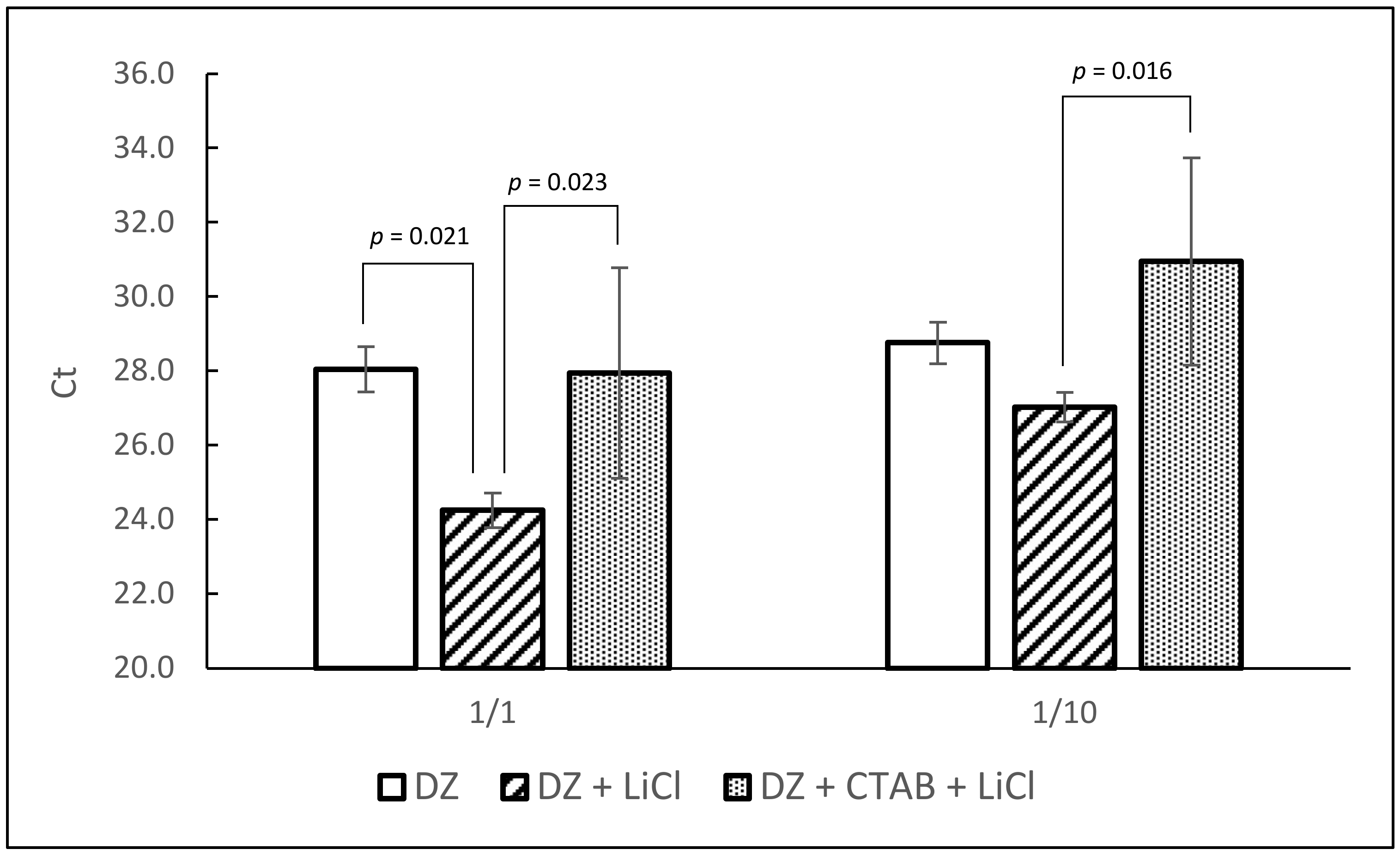
3.2. Removal of Potential RT-PCR Inhibitors
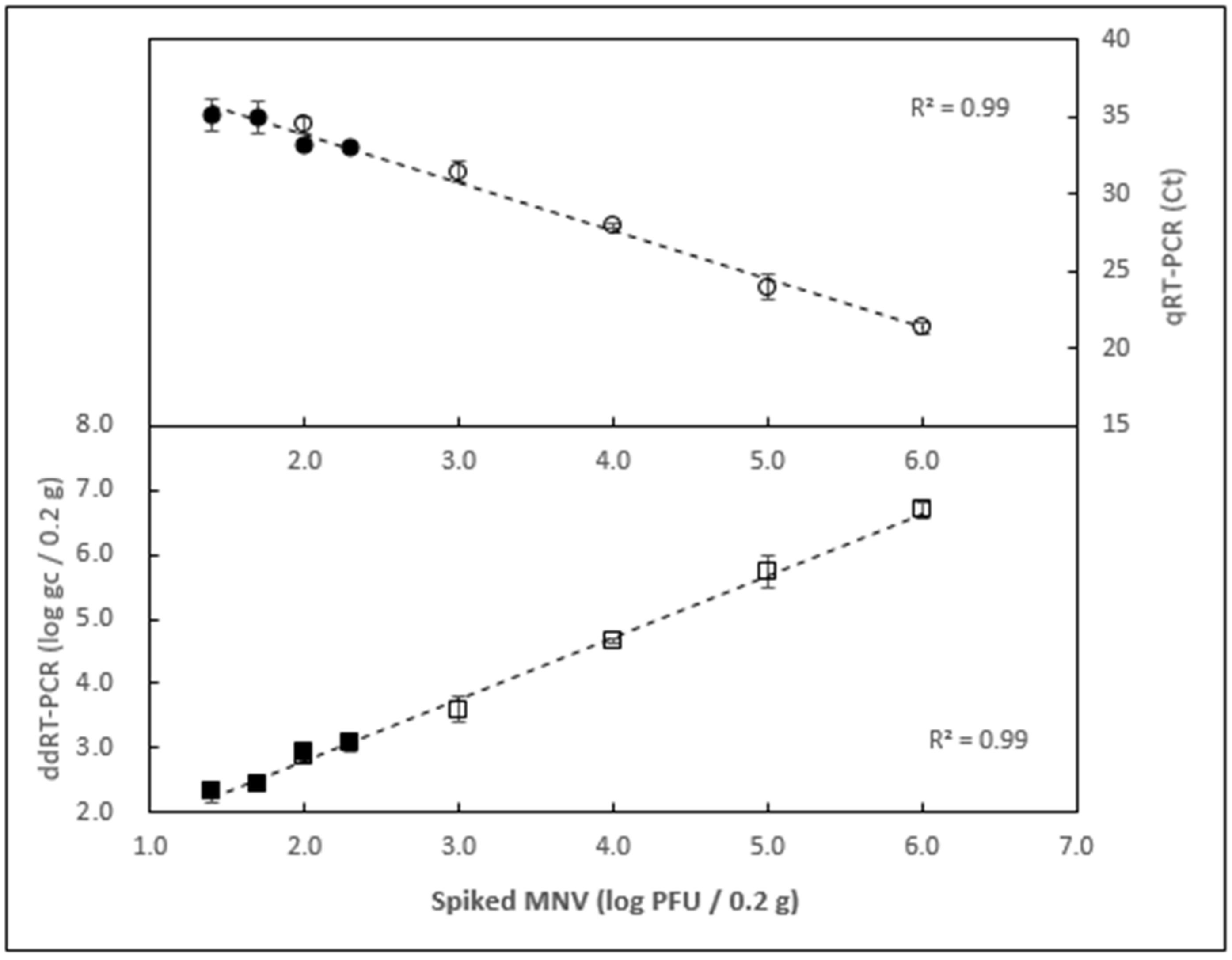
3.3. Examining the Limit of Detection
3.4. Determining the Extraction Efficiency
3.5. Calculation of RT-PCR Inhibition
3.6. Comparison with Other Extraction Methods
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopman, B.A.; Steele, D.; Kirkwood, C.D.; Parashar, U.D. The Vast and Varied Global Burden of Norovirus: Prospects for Prevention and Control. PLoS Med. 2016, 13, e1001999. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Surveillance for Foodborne Disease Outbreaks, United States, 2016, Annual Report; Department of Health and Human Services, CDC: Atlanta, GA, USA, 2018.
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global Economic Burden of Norovirus Gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef]
- Green, K.Y. Caliciviridae: The Noroviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; p. 948. [Google Scholar]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.W.; Calci, K.R.; Marchant-Tambone, J.G.; Burkhardt, W., 3rd. Detection and molecular characterization of norovirus from oysters implicated in outbreaks in the US. Food Microbiol. 2016, 59, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Meghnath, K.; Hasselback, P.; McCormick, R.; Prystajecky, N.; Taylor, M.; McIntyre, L.; Man, S.; Whitfield, Y.; Warshawsky, B.; McKinley, M.; et al. Outbreak Team Outbreaks of Norovirus and Acute Gastroenteritis Associated with British Columbia Oysters, 2016–2017. Food Environ. Virol. 2019, 11, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Hardstaff, J.L.; Clough, H.E.; Lutje, V.; McIntyre, K.M.; Harris, J.P.; Garner, P.; O’Brien, S.J. Foodborne and Food-Handler Norovirus Outbreaks: A Systematic Review. Foodborne Pathog. Dis. 2018, 15, 589–597. [Google Scholar] [CrossRef]
- Pouillot, R.; Smith, M.; Van Doren, J.M.; Catford, A.; Holtzman, J.; Calci, K.R.; Edwards, R.; Goblick, G.; Roberts, C.; Stobo, J.; et al. Risk Assessment of Norovirus Illness from Consumption of Raw Oysters in the United States and in Canada. Risk Anal. 2021. [Google Scholar] [CrossRef]
- Le Guyader, F.S.; Atmar, R.L.; Le Pendu, J. Transmission of viruses through shellfish: When specific ligands come into play. Curr. Opin. Virol. 2012, 2, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Strubbia, S.; Schaeffer, J.; Besnard, A.; Wacrenier, C.; Le Mennec, C.; Garry, P.; Desdouits, M.; Le Guyader, F.S. Metagenomic to evaluate norovirus genomic diversity in oysters: Impact on hexamer selection and targeted capture-based enrichment. Int. J. Food Microbiol. 2020, 323, 108588. [Google Scholar] [CrossRef]
- Anonymous Microbiology of Food and Animal Feed—Horizontal Method for Determination of Hepatitis A Virus and Norovirus in Food Using Real-time RT-PCR—Part 1: Method for Quantification. ISO/TS 15216-1:2013; International Organization for Standardization: Geneva, Switzerland, 2013. [Google Scholar]
- Anonymous Microbiology of Food and Animal Feed—Horizontal Method for Determination of Hepatitis A Virus and Norovirus in Food Using Real-Time RT-PCR—Part 2: Method for Qualitative Detection; ISO/TS 15216-2:2013; International Organization for Standardization: Geneva, Switzerland, 2013.
- Anonymous Microbiology of the Food Chain—Horizontal Method for Determination of Hepatitis A Virus and Norovirus Using Real-Time RT-PCR—Part 1: Method for Quantification; ISO 15216-1:2017; International Organization for Standardization: Geneva, Switzerland, 2017.
- Lowther, J.A.; Bosch, A.; Butot, S.; Ollivier, J.; Made, D.; Rutjes, S.A.; Hardouin, G.; Lombard, B.; In’t Veld, P.; Leclercq, A. Validation of EN ISO method 15216—Part 1—Quantification of hepatitis A virus and norovirus in food matrices. Int. J. Food Microbiol 2019, 288, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Eriksson, R.; Lowther, J.; Ellstrom, P.; Simonsson, M. Comparison between RT droplet digital PCR and RT real-time PCR for quantification of noroviruses in oysters. Int. J. Food Microbiol. 2018, 284, 73–83. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA) Analysis of the European baseline survey of norovirus in oysters. EFSA J. 2019, 17, e05762. [CrossRef]
- Suffredini, E.; Magnabosco, C.; Civettini, M.; Rossetti, E.; Arcangeli, G.; Croci, L. Norovirus contamination in different shellfish species harvested in the same production areas. J. Appl. Microbiol. 2012, 113, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, P.; Kc, S.; Beale, D.J.; Hewitt, J. Current and Emerging Technologies for the Detection of Norovirus from Shellfish. Foods 2019, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Baek, C.; Cong, V.T.; Min, J. The microfluidic chip module for the detection of murine norovirus in oysters using charge switchable micro-bead beating. Biosens. Bioelectron. 2015, 67, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, D.H. High Pressure Processing of Bivalve Shellfish and HPP’s Use as a Virus Intervention. Foods 2014, 3, 336–350. [Google Scholar] [CrossRef]
- Quang Le, H.; Suffredini, E.; Tien Pham, D.; Kim To, A.; De Medici, D. Development of a method for direct extraction of viral RNA from bivalve molluscs. Lett. Appl. Microbiol. 2018, 67, 426–434. [Google Scholar] [CrossRef]
- Nasheri, N.; Harlow, J.; Chen, A.; Corneau, N.; Bidawid, S. Survival and Inactivation by Advanced Oxidative Process of Foodborne Viruses in Model Low-Moisture Foods. Food Environ. Virol. 2021, 13, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, S.; Mattison, K. Evaluation of murine norovirus persistence in environments relevant to food production and processing. J. Food Prot. 2011, 74, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Schwab, K.J. Evaluation of murine norovirus, feline calicivirus, poliovirus, and MS2 as surrogates for human norovirus in a model of viral persistence in surface water and groundwater. Appl. Environ. Microbiol. 2008, 74, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Nasheri, N.; Harlow, J.; Chen, A.; Corneau, N.; Bidawid, S. Evaluation of Bead-Based Assays in the Isolation of Foodborne Viruses from Low-Moisture Foods. J. Food Prot. 2020, 83, 388–396. [Google Scholar] [CrossRef]
- Meijerink, J.; Mandigers, C.; van de Locht, L.; Tonnissen, E.; Goodsaid, F.; Raemaekers, J. A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J. Mol. Diagn. 2001, 3, 55–61. [Google Scholar] [CrossRef][Green Version]
- Rutledge, R.G.; Cote, C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res. 2003, 31, e93. [Google Scholar] [CrossRef]
- Kuypers, J.; Jerome, K.R. Applications of Digital PCR for Clinical Microbiology. J. Clin. Microbiol. 2017, 55, 1621–1628. [Google Scholar] [CrossRef]
- Bustin, S.A.; Nolan, T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 2004, 15, 155–166. [Google Scholar]
- Smith, C.J.; Osborn, A.M. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 2009, 67, 6–20. [Google Scholar] [CrossRef]
- Kralik, P.; Ricchi, M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Dingle, T.C.; Sedlak, R.H.; Cook, L.; Jerome, K.R. Tolerance of droplet-digital PCR vs real-time quantitative PCR to inhibitory substances. Clin. Chem. 2013, 59, 1670–1672. [Google Scholar] [CrossRef] [PubMed]
- Hall Sedlak, R.; Jerome, K.R. The potential advantages of digital PCR for clinical virology diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Cheong, S.; Lee, H.J.; Kwon, M.; Kang, I.; Oh, E.G.; Yu, H.S.; Shin, S.B.; Kim, S.J. Evaluation of the sensitivity and specificity of primer pairs and the efficiency of RNA extraction procedures to improve noroviral detection from oysters by nested reverse transcription-polymerase chain reaction. J. Microbiol. 2010, 48, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Benabbes, L.; Ollivier, J.; Schaeffer, J.; Parnaudeau, S.; Rhaissi, H.; Nourlil, J.; Le Guyader, F.S. Norovirus and other human enteric viruses in moroccan shellfish. Food Environ. Virol. 2013, 5, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Nybogard, L.; Simonsson, M.; Eriksson, R. Optimisation and evaluation of an automated system for extraction of viral RNA from oysters. Int. J. Food Microbiol. 2020, 315, 108386. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inoculum (PFU) | Extraction from Inoculum | Extraction from Oysters | ||
|---|---|---|---|---|
| gc | gc/PFU | gc | Extraction Efficiency | |
| 100,000 | 2,850,900 ± 23,284 | 29 ± 0.2 | 627,900 ± 374,279 | 22% |
| 10,000 | 285,967 ± 5560 | 29 ± 0.6 | 44,773 ± 3878 | 16% |
| 1000 | 20,715 ± 552 | 21 ± 0.6 | 4232 ± 2103 | 20% |
| 200 | 4102 ± 219 | 21 ± 1.1 | 1234 ± 386 | 30% |
| 100 | 3235 ± 1502 | 32 ± 15 | 820 ± 154 | 25% |
| 50 | 337 ± 93 | 7 ± 1.2 | 268 ± 35 | 80% |
| 25 | 345 ± 46 | 14 ± 1.2 | 220 ± 91 | 64% |
| Weighted Average | 28 ± 1.0 | 21 ± 3% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plante, D.; Bran Barrera, J.A.; Lord, M.; Iugovaz, I.; Nasheri, N. Development of an RNA Extraction Protocol for Norovirus from Raw Oysters and Detection by qRT-PCR and Droplet-Digital RT-PCR. Foods 2021, 10, 1804. https://doi.org/10.3390/foods10081804
Plante D, Bran Barrera JA, Lord M, Iugovaz I, Nasheri N. Development of an RNA Extraction Protocol for Norovirus from Raw Oysters and Detection by qRT-PCR and Droplet-Digital RT-PCR. Foods. 2021; 10(8):1804. https://doi.org/10.3390/foods10081804
Chicago/Turabian StylePlante, Daniel, Julio Alexander Bran Barrera, Maude Lord, Irène Iugovaz, and Neda Nasheri. 2021. "Development of an RNA Extraction Protocol for Norovirus from Raw Oysters and Detection by qRT-PCR and Droplet-Digital RT-PCR" Foods 10, no. 8: 1804. https://doi.org/10.3390/foods10081804
APA StylePlante, D., Bran Barrera, J. A., Lord, M., Iugovaz, I., & Nasheri, N. (2021). Development of an RNA Extraction Protocol for Norovirus from Raw Oysters and Detection by qRT-PCR and Droplet-Digital RT-PCR. Foods, 10(8), 1804. https://doi.org/10.3390/foods10081804