
Comparative Study for the Determination of Fat-Soluble Vitamins in Rice Cereal Baby Foods Using HPLC-DAD and UHPLC-APCI-MS/MS

 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Standard Solutions
2.3. Instrumentation and Chromatographic Conditions
2.3.1. HPLC-DAD
2.3.2. UHPLC-APCI-MS/MS
2.4. Samples
2.5. Sample Preparation-Extraction of Fat-Soluble Vitamins
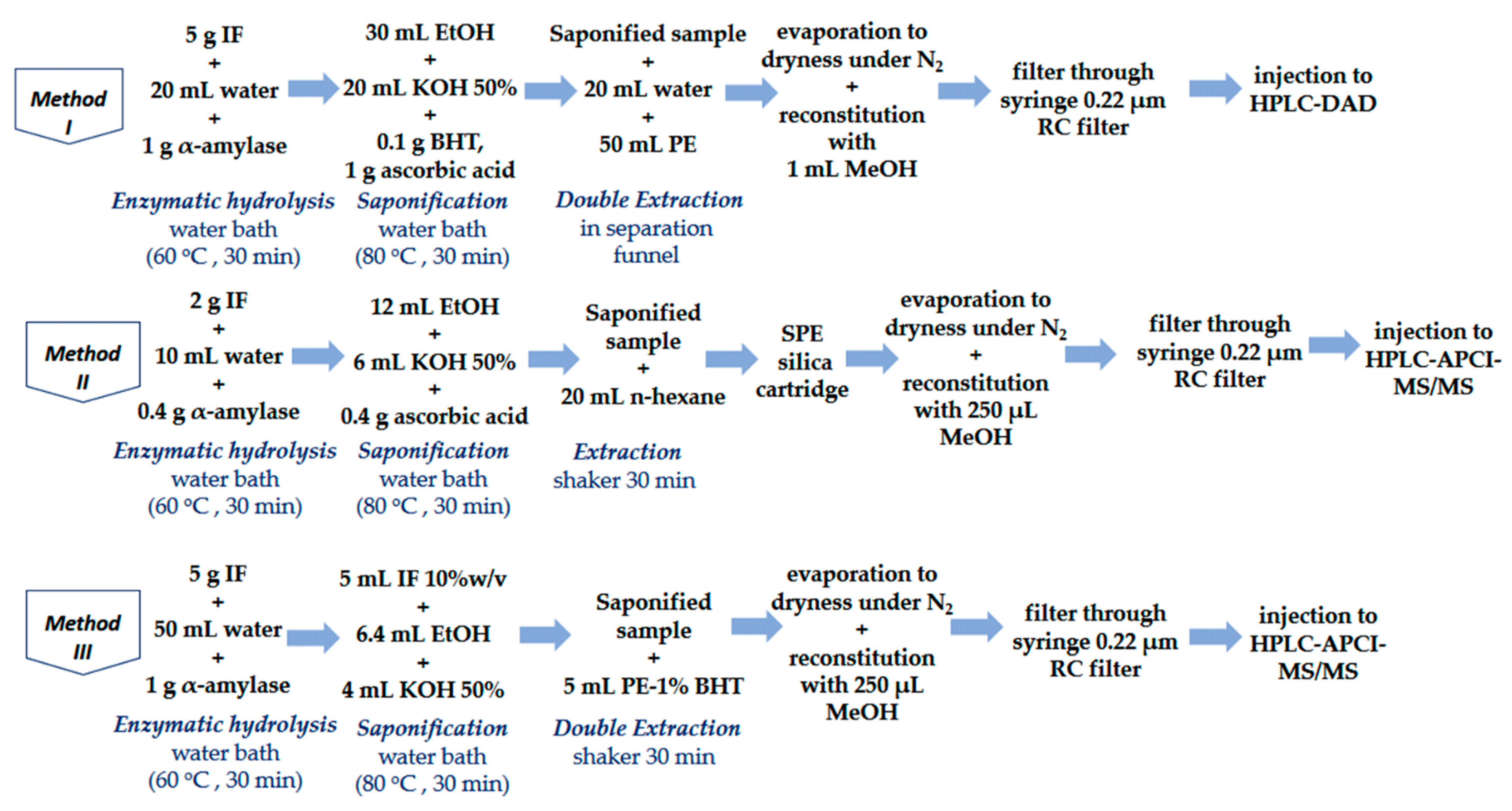
2.5.1. Method I (GB_A&E)
2.5.2. Method II (GB_D3)
2.5.3. Method III (In-House ADE)
2.6. Method Validation
2.7. Assessment of Methods
2.7.1. Measurement Uncertainty
2.7.2. External Quality Control and Real Samples
3. Results and Discussion
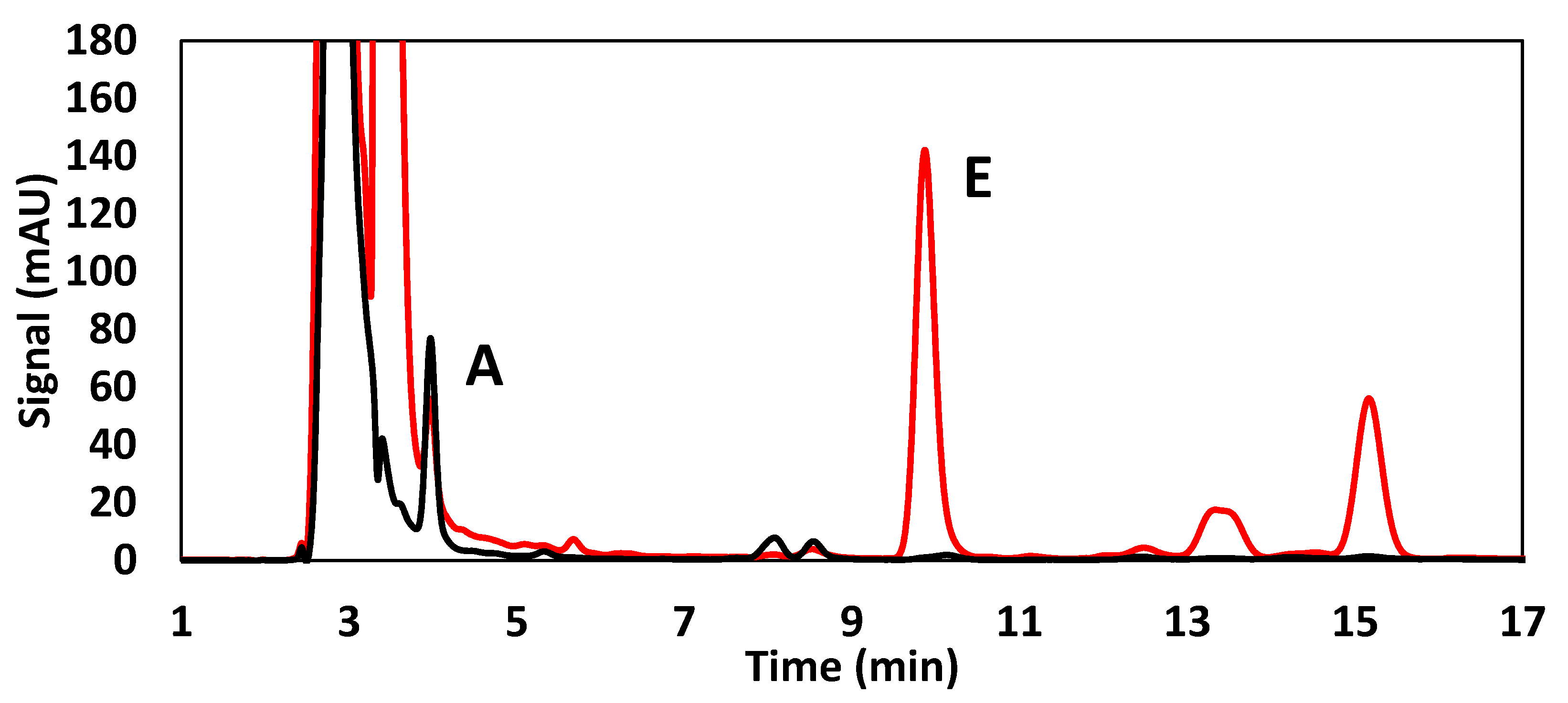
3.1. Method Optimization
3.1.1. Method I (GB_A&E)
3.1.2. Method II (GB_D3)
3.2. Method Validation
3.2.1. Linearity
3.2.2. Precision-Trueness
3.2.3. LODs and LOQs
3.2.4. Matrix Effect
3.3. Assessment of Methods
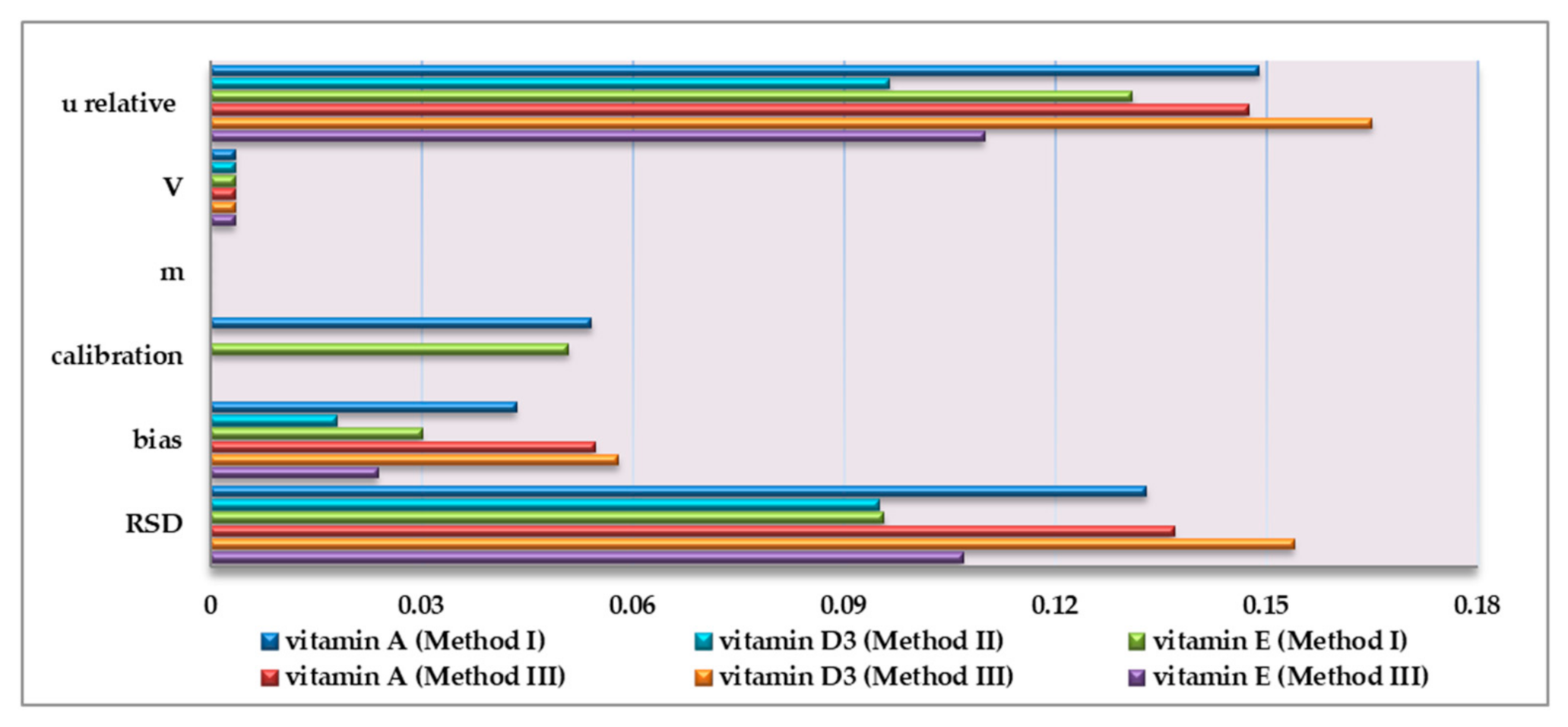
3.3.1. Measurement Uncertainty
3.3.2. External Quality Control and Real Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gentili, A.; Caretti, F. Analysis of Vitamins by Liquid Chromatography, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 9780128053928. [Google Scholar]
- Plozza, T.; Craige Trenerry, V.; Caridi, D. The simultaneous determination of vitamins A, e and β-carotene in bovine milk by high performance liquid chromatography-ion trap mass spectrometry (HPLC-MS n). Food Chem. 2012, 134, 559–563. [Google Scholar] [CrossRef]
- Nimalaratne, C.; Sun, C.; Wu, J.; Curtis, J.M.; Schieber, A. Quantification of selected fat soluble vitamins and carotenoids in infant formula and dietary supplements using fast liquid chromatography coupled with tandem mass spectrometry. Food Res. Int. 2014, 66, 69–77. [Google Scholar] [CrossRef]
- Penniston, K.L.; Tanumihardjo, S.A. The acute and chronic toxic effects of vitamin A. Am. J. Clin. Nutr. 2006, 83, 191–201. [Google Scholar] [CrossRef]
- Katsa, M.; Proestos, C. Vitamin Analysis in Juices and Nonalcoholic Beverages; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128152584. [Google Scholar]
- Ball, G.F.M. Vitamins in Foods: Analysis, Bioavailability and Stability; Taylor & Francis: Oxfordshire, UK, 2006; ISBN 9781574448047. [Google Scholar]
- Blake, C.J. Status of methodology for the determination of fat-soluble vitamins in foods, dietary supplements, and vitamin premixes. J. AOAC Int. 2007, 90, 897–910. [Google Scholar] [PubMed]
- Zhang, Y.; Zhou, W.E.; Yan, J.Q.; Liu, M.; Zhou, Y.; Shen, X.; Ma, Y.L.; Feng, X.S.; Yang, J.; Li, G.H. A review of the extraction and determination methods of thirteen essential vitamins to the human body: An update from 2010. Molecules 2018, 23, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Nollet, L.M.L. Food Analysis; Routledge: London, UK, 2013; ISBN 9781439830857. [Google Scholar]
- Perales, S.; Alegría, A.; Barbera, R.; Farré, R. Review: Determination of vitamin D in dairy products by high performance liquid chromatography. Food Sci. Technol. Int. 2005, 11, 451–462. [Google Scholar] [CrossRef]
- Yin, S.; Yang, Y.; Wu, L.; Li, Y.; Sun, C. Recent advances in sample preparation and analysis methods for vitamin D and its analogues in different matrices. TrAC Trends Anal. Chem. 2019, 110, 204–220. [Google Scholar] [CrossRef]
- Melfi, M.T.; Nardiello, D.; Cicco, N.; Candido, V.; Centonze, D. Simultaneous determination of water- and fat-soluble vitamins, lycopene and beta-carotene in tomato samples and pharmaceutical formulations: Double injection single run by reverse-phase liquid chromatography with UV detection. J. Food Compos. Anal. 2018, 70, 9–17. [Google Scholar] [CrossRef]
- Escrivá, A.; Esteve, M.J.; Farré, R.; Frígola, A. Determination of liposoluble vitamins in cooked meals, milk and milk products by liquid chromatography. J. Chromatogr. A 2002, 947, 313–318. [Google Scholar] [CrossRef]
- Chávez-Servín, J.L.; Castellote, A.I.; López-Sabater, M.C. Vitamins A and E content in infant milk-based powdered formulae after opening the packet. Food Chem. 2008, 106, 299–309. [Google Scholar] [CrossRef]
- Rodas Mendoza, B.; Morera Pons, S.; Castellote Bargalló, A.I.; López-Sabater, M.C. Rapid determination by reversed-phase high-performance liquid chromatography of Vitamins A and E in infant formulas. J. Chromatogr. A 2003, 1018, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Fanali, C.; D’Orazio, G.; Fanali, S.; Gentili, A. Advanced analytical techniques for fat-soluble vitamin analysis. TrAC Trends Anal. Chem. 2017, 87, 82–97. [Google Scholar] [CrossRef]
- Wang, X.; Li, K.; Yao, L.; Wang, C.; Van Schepdael, A. Recent advances in vitamins analysis by capillary electrophoresis. J. Pharm. Biomed. Anal. 2018, 147, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Souverain, S.; Rudaz, S.; Veuthey, J. Matrix effect in LC-ESI-MS and LC-APCI-MS with off-line and on-line extraction procedures. J. Chromatogr. A 2004, 1058, 61–66. [Google Scholar] [CrossRef]
- Heudi, O.; Trisconi, M.J.; Blake, C.J. Simultaneous quantification of Vitamins A, D3 and E in fortified infant formulae by liquid chromatography-mass spectrometry. J. Chromatogr. A 2004, 1022, 115–123. [Google Scholar] [CrossRef]
- Hrvolová, B.; Martínez-Huélamo, M.; Colmán-Martínez, M.; Hurtado-Barroso, S.; Lamuela-Raventós, R.; Kalina, J. Development of an Advanced HPLC–MS/MS Method for the Determination of Carotenoids and Fat-Soluble Vitamins in Human Plasma. Int. J. Mol. Sci. 2016, 17, 1719. [Google Scholar] [CrossRef] [Green Version]
- Huck, C.W.; Popp, M.; Scherz, H.; Bonn, G.K. Development and evaluation of a new method for the determination of the carotenoid content in selected vegetables by HPLC and HPLC-MS-MS. J. Chromatogr. Sci. 2000, 38, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentili, A.; Caretti, F. Evaluation of a method based on liquid chromatography-diode array detector-tandem mass spectrometry for a rapid and comprehensive characterization of the fat-soluble vitamin and carotenoid profile of selected plant foods. J. Chromatogr. A 2011, 1218, 684–697. [Google Scholar] [CrossRef]
- GB 5413.9-2010 Determination of Vitamin A, D, E in Foods for Infants and Young Children, Milk and Milk Products; National Food Safety Standard; Ministry of Health, the People’s Republic of China: Beijing, China, 2010; pp. 1–10.
- McMahon, A.; Christiansen, S.; Shine, L.; Loi, C.; Dowell, D. Simultaneous determination of 13-cis and all-trans vitamin a palmitate (retinyl palmitate), vitamin a acetate (retinyl acetate), and total vitamin e (a-tocopherol and DL-a-tocopherol acetate) in infant formula and adult nutritionals by normal phase HPLC. J. AOAC Int. 2013, 96, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, D.L.; Black, C.K.; Denison, J.E.; Seipelt, C.T. Simultaneous Determination of Vitamins D2 and D3 by LC-MS/MS in Infant Formula and Adult Nutritionals: First Action 2011.13. J. AOAC Int. 2012, 95, 177–186. [Google Scholar] [CrossRef]
- EN 12823-1:2014 Determination of Vitamin A by High Performance Liquid Chromatography. Part 1: Measurement of All-Trans-retinol and 13-Cis-Retinol. 2014. Available online: https://standards.iteh.ai/catalog/standards/cen/ebb9809c-1c7d-4e44-a2d5-3aab12a12c7f/en-12823-1-2014 (accessed on 16 March 2021).
- 2002/657/EC Commission Decision 2002/657/EC, implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 8–36. [CrossRef]
- GB 5009.82-2016 Determination of Vitamin A, D, E in Food; National Food Safety Standard; Ministry of Health, the People’s Republic of China: Beijing, China, 2016; pp. 1–40.
- Katsa, M.; Proestos, C.; Komaitis, E. Determination of fat soluble vitamins A and E in infant formulas by HPLC-DAD. Curr. Res. Nutr. Food Sci. 2016, 4. [Google Scholar] [CrossRef]
- Eurachem. The Fitness for Purpose of Analytical Methods A Laboratory Guide to Method Validation and Related Topics; Eurachem: Windsor, UK, 2014; p. 70. [Google Scholar]
- Eurachem. Quantifying Uncertainty in Analytical Measurements. J. Bangladesh Acad. Sci. 2018, 41, 145–163. [Google Scholar] [CrossRef]
- Taverniers, I.; De Loose, M.; Van Bockstaele, E. Trends in quality in the analytical laboratory. I. Traceability and measurement uncertainty of analytical results. TrAC Trends Anal. Chem. 2004, 23, 480–490. [Google Scholar] [CrossRef]
- Konieczka, P.; Namieśnik, J. Estimating uncertainty in analytical procedures based on chromatographic techniques. J. Chromatogr. A 2010, 1217, 882–891. [Google Scholar] [CrossRef] [PubMed]
- AOAC INTERNATIONAL. Guidelines for Standard Method Performance Requirements: Appendix F. AOAC Off. Methods Anal. 2016, 9, 1–18. [Google Scholar]
- Dasenaki, M.E.; Bletsou, A.A.; Hanafi, A.H.; Thomaidis, N.S. Liquid chromatography–tandem mass spectrometric methods for the determination of spinosad, thiacloprid and pyridalyl in spring onions and estimation of their pre-harvest interval values. Food Chem. 2016, 213, 395–401. [Google Scholar] [CrossRef]
- European Commission Guidance. Document for competent authorities for the control of compliance with EU legislation on: Regulation (EU) No 1169/2011 of the European Parliament and of the Council of 25 October 2011 on the provision of food information to consumers, amending. Eur. Comm. 2012, 1, 1–15. [Google Scholar]
- Gentili, A.; Caretti, F.; Bellante, S.; Ventura, S.; Canepari, S.; Curini, R. Comprehensive profiling of carotenoids and fat-soluble vitamins in milk from different animal species by LC-DAD-MS/MS hyphenation. J. Agric. Food Chem. 2013, 61, 1628–1639. [Google Scholar] [CrossRef]
- Mathiasson, L.; Turner, C.; Berg, H.; Dahlberg, L.; Theobald, A.; Anklam, E.; Ginn, R.; Sharman, M.; Ulberth, F.; Gabernig, R. Development of methods for the determination of vitamins A, E and β-carotene in processed foods based on supercritical fluid extraction: A collaborative study Development of methods for the determination of vitamins A, E and β-carotene in processed f. Food Addit. Contam. 2002, 19, 632–646. [Google Scholar] [CrossRef]
- Phinney, K.W.; Rimmer, C.A.; Brown Thomas, J.; Sander, L.C.; Sharpless, K.E.; Wise, S.A. Isotope Dilution Liquid Chromatography-Mass Spectrometry Methods for Fat-and Water-Soluble Vitamins in Nutritional Formulations. Anal. Chem. 2011, 83, 92–98. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Parent Ion (m/z) | Quantifier Ion (m/z) | CE (eV) | Qualifier Ion (m/z) | CE (eV) | Tube Lens | Retention Time (min) |
|---|---|---|---|---|---|---|---|
| Retinol | 269.2 | 91.4 | 33 | 105.2 | 11 | 52 | 0.85 |
| Retinyl acetate | 269.2 | 91.4 | 33 | 105.2 | 11 | 52 | 1 |
| Cholecalciferol | 385.2 | 259.1 | 13 | 367.2 | 11 | 53 | 1.8 |
| Cholecalciferol-d3 | 388.4 | 370 | 11 | 259.1 | 11 | 68 | 1.8 |
| α-Tocopherol | 431.1 | 165.2 | 20 | 137.2 | 35 | 75 | 2 |
| α-Toco-pheryl acetate | 473.4 | 207.1 | 17 | 165.2 | 21 | 97 | 2.5 |
| α-Tocopherol-d6 | 437.4 | 171.1 | 22 | 143.1 | 35 | 65 | 2 |
| Compound | Method | Concentration Range (mg L−1) | Calibration Curve | Coefficient of Determination (r2) |
|---|---|---|---|---|
| Retinol | I | 0.88–50.0 | y = (90.1 ± 1.2) x + (35.3 ± 26.8) | 0.9996 |
| III | 0.600–30.0 | y = (326.9 ± 6.6) × 103 x − (17.1 ± 9.2) × 104 | 0.997 | |
| α-Tocopherol | I | 9.25–500 | y = (0.11 ± 8.02) × 102 x − (6.4 ± 18.2) | 0.9996 |
| III | 7.00–300 | y = (4.73 ± 0.11) x − (25.2 ± 14.8) | 0.996 | |
| Cholecalciferol | II | 0.005–2.56 | y = (76 ± 1.11) × 10−4 x + (16.25 ± 1.15) × 10−2 | 0.9998 |
| III | 0.008–0.480 | y = (69.9 ± 3.4) × 10−4 x + (3.1 ± 77.7) × 10−3 | 0.998 |
| Compound | Method | Concentration Range (mg kg−1) | Calibration Curve | Coefficient of Determination (r2) |
|---|---|---|---|---|
| Retinol | I | 0.750–10.0 | y = (360.0 ± 10.6) x + (88.7 ± 58.1) | 0.995 |
| III | 0.300–15.0 | y = (47.9 ± 1.8) × 104 x + (31.7 ± 12.2) × 104 | 0.99 | |
| α-Tocopherol | I | 9.00–100 | y = (36.3 ± 1.0) x + (80.7 ± 81.6) | 0.995 |
| III | 3.50–150 | y = (9.05 ± 0.23) x − (51.9 ± 15.7) | 0.995 | |
| Cholecalciferol | II | 0.008–0.160 | y = (55.2 ± 2.4) × 10−3 x + (46.4 ± 31.1) × 10−2 | 0.992 |
| III | 0.008–0.240 | y = (10.9 ± 6.6) × 10−3 x + (57.1 ± 76.1) × 10−2 | 0.998 |
| Compound | Method | Concentration Range (mg L−1) | Calibration Curve | Coefficient of Determination (r2) |
|---|---|---|---|---|
| Retinol | III | 0.600–30.0 | y = (145.7 ± 3.8) × 103 x + (44.8 ± 2.6) × 104 | 0.995 |
| α-Tocopherol | III | 7.00–300 | y = (4.81 ± 0.15) x − (44.1 ± 21.6) | 0.992 |
| Cholecalciferol | II | 0.008–0.160 | y = (62.8 ± 3.1) × 10−3 x + (0.355 ± 0.406) | 0.99 |
| III | 0.008–0.480 | y = (59.5 ± 3.3) × 10−4 x + (16.8 ± 7.5) × 10−2 | 0.98 |
| Compound | Fortification Level | Method I | Method III | ||||
|---|---|---|---|---|---|---|---|
| Mean Concentration (mg kg−1) (n = 12) | Mean Recovery (%) | % RSDR | Mean Concentration (mg kg−1) (n = 12) | Mean Recovery (%) | % RSDR | ||
| Retinol | low | 1.49 | 85.0 | 7.3 | 1.80 | 103 | 13.2 |
| medium | 3.69 | 105 | 13.3 | 3.43 | 98.1 | 13.7 | |
| high | 7.48 | 107 | 10.4 | 7.42 | 106 | 11.4 | |
| α-Tocopherol | low | 16.2 | 92.5 | 12.7 | 17.8 | 102 | 13.6 |
| medium | 35.7 | 102 | 9.57 | 36.4 | 104 | 10.7 | |
| high | 77.4 | 103 | 10.3 | 78.9 | 105 | 6.4 | |
| Compound | Fortification level | Method II | Method III | ||||
|---|---|---|---|---|---|---|---|
| Mean Concentration (μg kg−1) (n = 12) | Mean Recovery (%) | % RSDR | Mean Concentration (μg kg−1) (n = 12) | Mean Recovery (%) | % RSDR | ||
| Cholecalciferol | low | 42.1 | 105 | 10.3 | 38.2 | 95.5 | 12.5 |
| medium | 84.6 | 106 | 9.5 | 76.2 | 95.2 | 14.4 | |
| high | 154 | 96.4 | 13 | 158 | 98.5 | 10.9 | |
| Method | Retinol | α-Tocopherol | Cholecalciferol | |||
|---|---|---|---|---|---|---|
| LOD (mg L−1) | LOQ (mg L−1) | LOD (mg L−1) | LOQ (mg L−1) | LOD (μg L−1) | LOQ (μg L−1) | |
| I | 0.98 | 2.9 | 5.4 | 16 | - | - |
| II | - | - | - | - | 5 | 15 |
| III | 0.43 | 1.3 | 1.9 | 5.7 | 5 | 15 |
| Method | Retinol | α-Tocopherol | Cholecalciferol | |||
|---|---|---|---|---|---|---|
| LOD (mg kg−1) | LOQ (mg kg−1) | LOD (mg kg−1) | LOQ (mg kg−1) | LOD (μg kg−1) | LOQ (μg kg−1) | |
| I | 0.36 | 1.2 | 0.74 | 2.4 | - | - |
| II | - | - | - | - | 7.7 | 23 |
| III | 0.3 | 0.91 | 0.31 | 0.92 | 9.2 | 28 |
| Compound | Method | MF | %ME |
|---|---|---|---|
| Retinol | III | 0.5 | −47 |
| α-Tocopherol | III | 1 | 1.7 |
| Cholecalciferol | II | 1.1 | 14 |
| III | 0.8 | −15 |
| Method I | Method I | Method II | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | Retinol (mg kg−1) | α-Tocopherol (mg kg−1) | Cholecalciferol (μg kg−1) | ||||||
| urandom (RSD), % | 0.073 | 0.133 | 0.104 | 0.127 | 0.0957 | 0.103 | 0.103 | 0.095 | 0.095 |
| ubias, % | 0.0376 | 0.0435 | 0.0462 | 0.0272 | 0.0301 | 0.0283 | 0.0419 | 0.018 | 0.018 |
| ucalibration, % | 0.147 | 0.0542 | 0.0284 | 0.12 | 0.0509 | 0.0225 | - | - | - |
| um, % (10−7) | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 |
| uV, % | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 |
| urelative, % | 0.16 | 0.15 | 0.12 | 0.18 | 0.13 | 0.11 | 0.11 | 0.1 | 0.1 |
| urelative (k = 2), % | 0.33 | 0.3 | 0.24 | 0.35 | 0.26 | 0.23 | 0.22 | 0.19 | 0.19 |
| Mean concentration | 1.49 | 3.7 | 7.5 | 16.2 | 35.7 | 77 | 42 | 85 | 154 |
| U (k = 2), mg kg−1 & μg kg−1 | 0.48 | 1.1 | 1.8 | 5.7 | 9.4 | 18 | 9.4 | 16 | 22 |
| Method III | |||||||||
| Parameter | Retinol (mg kg−1) | a-Tocopherol (mg kg−1) | Cholecalciferol (μg kg−1) | ||||||
| urandom (RSD), % | 0.132 | 0.137 | 0.114 | 0.13 | 0.107 | 0.0639 | 0.125 | 0.154 | 0.109 |
| ubias, % | 0.0667 | 0.0547 | 0.0263 | 0.052 | 0.0238 | 0.0188 | 0.061 | 0.058 | 0.0385 |
| ucalibration, % | - | - | - | - | - | - | - | - | - |
| um,% (10−7) | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 |
| uV, % | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 | 0.0036 |
| urelative, % | 0.15 | 0.15 | 0.12 | 0.14 | 0.11 | 0.07 | 0.14 | 0.17 | 0.12 |
| urelative (k = 2), % | 0.3 | 0.3 | 0.23 | 0.28 | 0.22 | 0.13 | 0.28 | 0.33 | 0.23 |
| Mean concentration | 1.8 | 3.4 | 7.4 | 17.8 | 36.4 | 79 | 38 | 76 | 158 |
| U (k = 2), mg kg−1 & μg kg−1 | 0.53 | 1 | 1.7 | 5 | 8 | 11 | 11 | 25 | 36 |
| Compound | Assigned Value (mg kg−1) | Concentration (z-Score) | ||
|---|---|---|---|---|
| (mg kg −1) | ||||
| Method I | Method II | Method III | ||
| Retinol | 4.26 | 4.69 (0.67) | - | 4.53 (0.42) |
| α-Tocopherol | 154 | 107 (−1.78) | - | 128 (−0.98) |
| Cholecalciferol | 0.118 | - | 0.0900(−1.60) | 0.0916 (−1.51) |
| Rice Cereal | Farine Lactée | |||||
| Method | Label Value | Tolerance limits | Concentration (n = 3) | Label Value | Tolerance limits | Concentration (n = 3) |
| I | 3.50 | 2.45-5.25 | 2.95 ± 0.15 | 3.90 | 2.73-5.85 | 4.12 ± 0.23 |
| III | 3.01 ± 0.45 | 4.23 ± 0.40 | ||||
| I | 35.0 | 24.5-52.5 | 42.4 ± 2.0 | 34.00 | 23.8-51.0 | 40.9 ± 2.6 |
| III | 43.6 ± 3.1 | 39.8 ± 4.0 | ||||
| II | 70.0 | 50.0-110 | 73.4 ± 4.3 | 68.00 | 47.6-102 | 69.8 ± 3.1 |
| III | 84.0 ± 3.5 | 64.4 ± 5.0 | ||||
| Biscuit Cream | Fruit cream with 3 fruits | |||||
| Method | Label Value | Tolerance limits | Concentration (n = 3) | Label Value | Tolerance limits | Concentration (n = 3) |
| I | 4.15 | 2.91-6.23 | 4.65 ± 0.19 | 3.40 | 2.38-5.10 | 4.09 ± 0.24 |
| III | 4.82 ± 0.29 | 4.27 ± 0.0.35 | ||||
| I | 30.0 | 21.0-45.0 | 36.7 ± 3.2 | 37.0 | 25.9-55.5 | 40.7 ± 2.5 |
| III | 35.8 ± 3.1 | 40.4 ± 2.9 | ||||
| II | 69.0 | 48.3-104 | 69.8 ± 3.6 | 75.00 | 52.5-113 | 79.0 ±4.0 |
| III | 64.4 ± 5.0 | 73.2 ± 4.4 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsa, M.; Papalouka, N.; Mavrogianni, T.; Papagiannopoulou, I.; Kostakis, M.; Proestos, C.; Thomaidis, N.S. Comparative Study for the Determination of Fat-Soluble Vitamins in Rice Cereal Baby Foods Using HPLC-DAD and UHPLC-APCI-MS/MS. Foods 2021, 10, 648. https://doi.org/10.3390/foods10030648
Katsa M, Papalouka N, Mavrogianni T, Papagiannopoulou I, Kostakis M, Proestos C, Thomaidis NS. Comparative Study for the Determination of Fat-Soluble Vitamins in Rice Cereal Baby Foods Using HPLC-DAD and UHPLC-APCI-MS/MS. Foods. 2021; 10(3):648. https://doi.org/10.3390/foods10030648
Chicago/Turabian StyleKatsa, Maria, Natalia Papalouka, Theodora Mavrogianni, Irene Papagiannopoulou, Marios Kostakis, Charalampos Proestos, and Nikolaos S. Thomaidis. 2021. "Comparative Study for the Determination of Fat-Soluble Vitamins in Rice Cereal Baby Foods Using HPLC-DAD and UHPLC-APCI-MS/MS" Foods 10, no. 3: 648. https://doi.org/10.3390/foods10030648