
Selection of Specific Nanobodies against Lupine Allergen Lup an 1 for Immunoassay Development

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
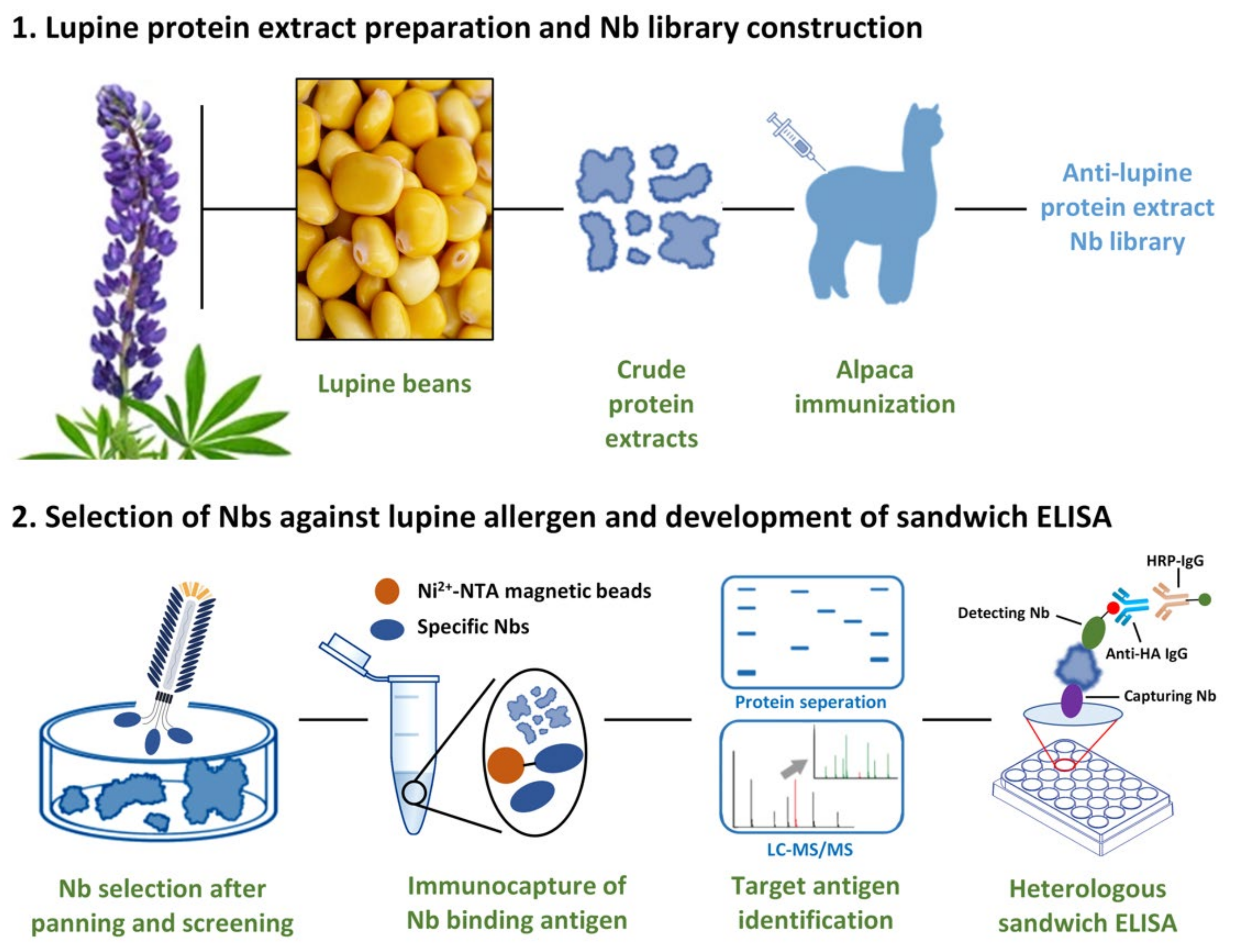
2.1. Crude Lupine Protein Extraction
2.2. Immunization and Construction of Nb Library
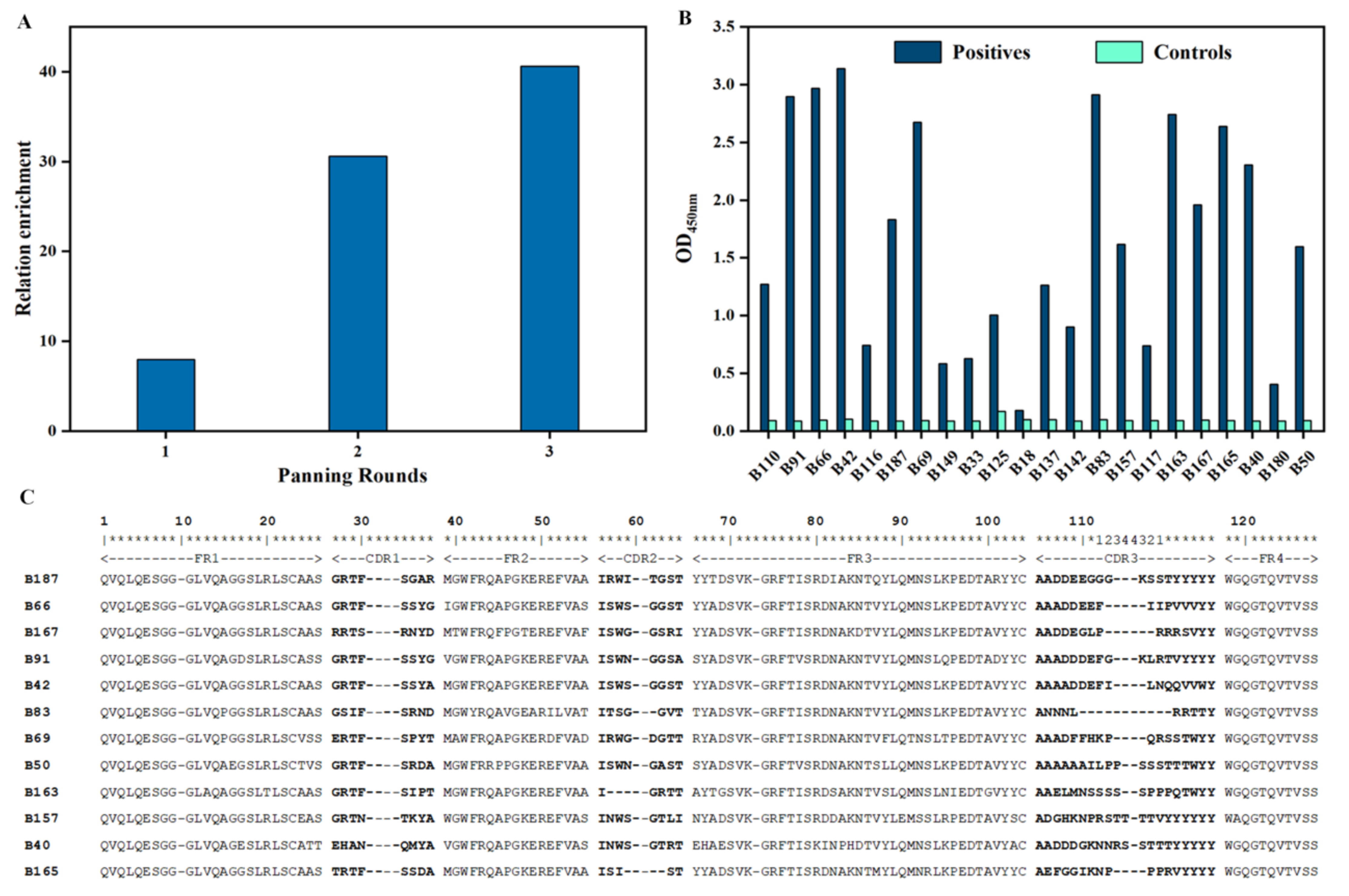
2.3. Bio-Panning
2.4. Screening of Specific Nbs
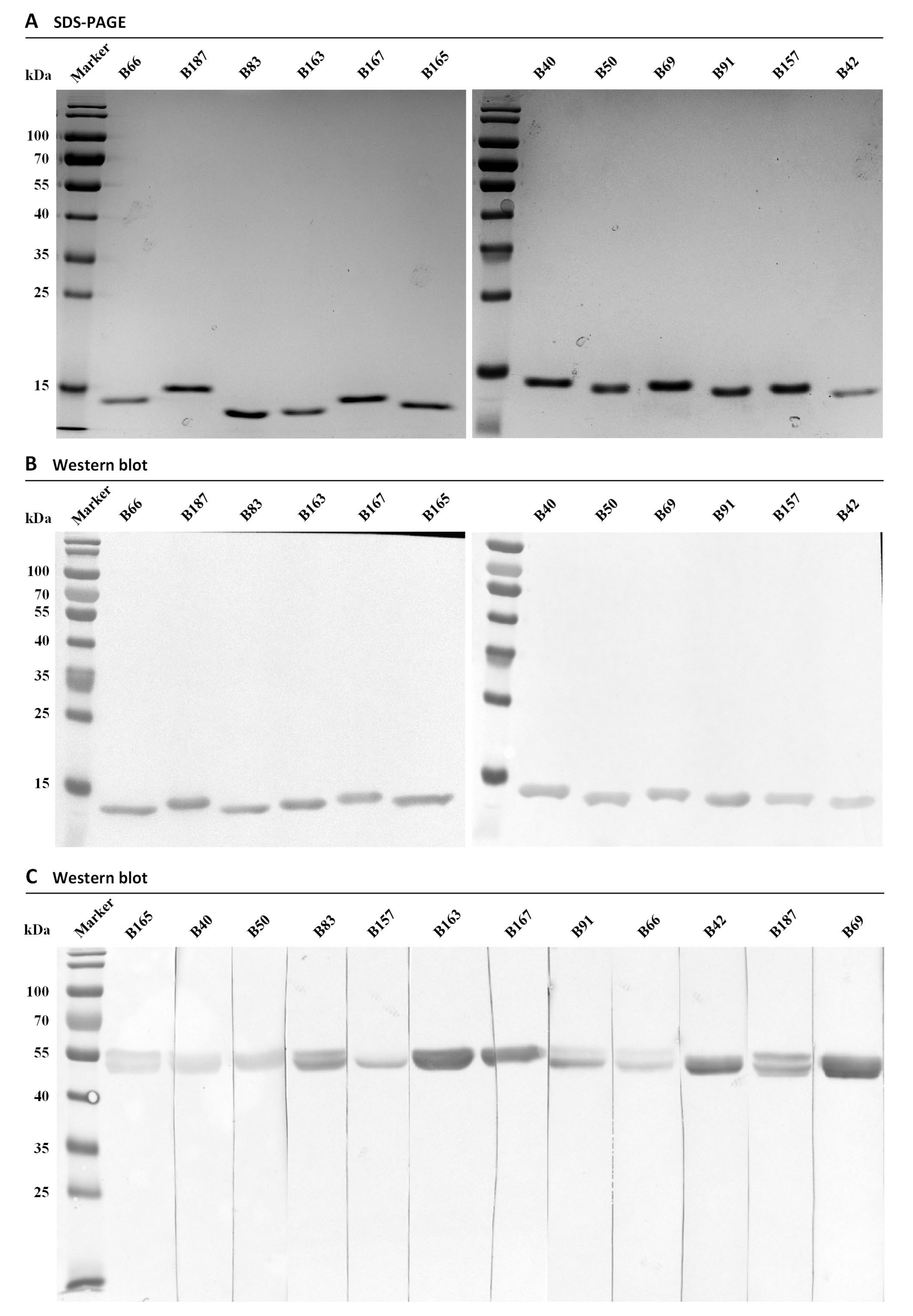
2.5. Expression and Purification of Specific Nbs
2.6. Identification of Target Antigen by Immuno-Capturing and LC-MS/MS
2.7. Characterization of Nbs (Affinity, Specificity, Thermostability)
2.7.1. Affinity
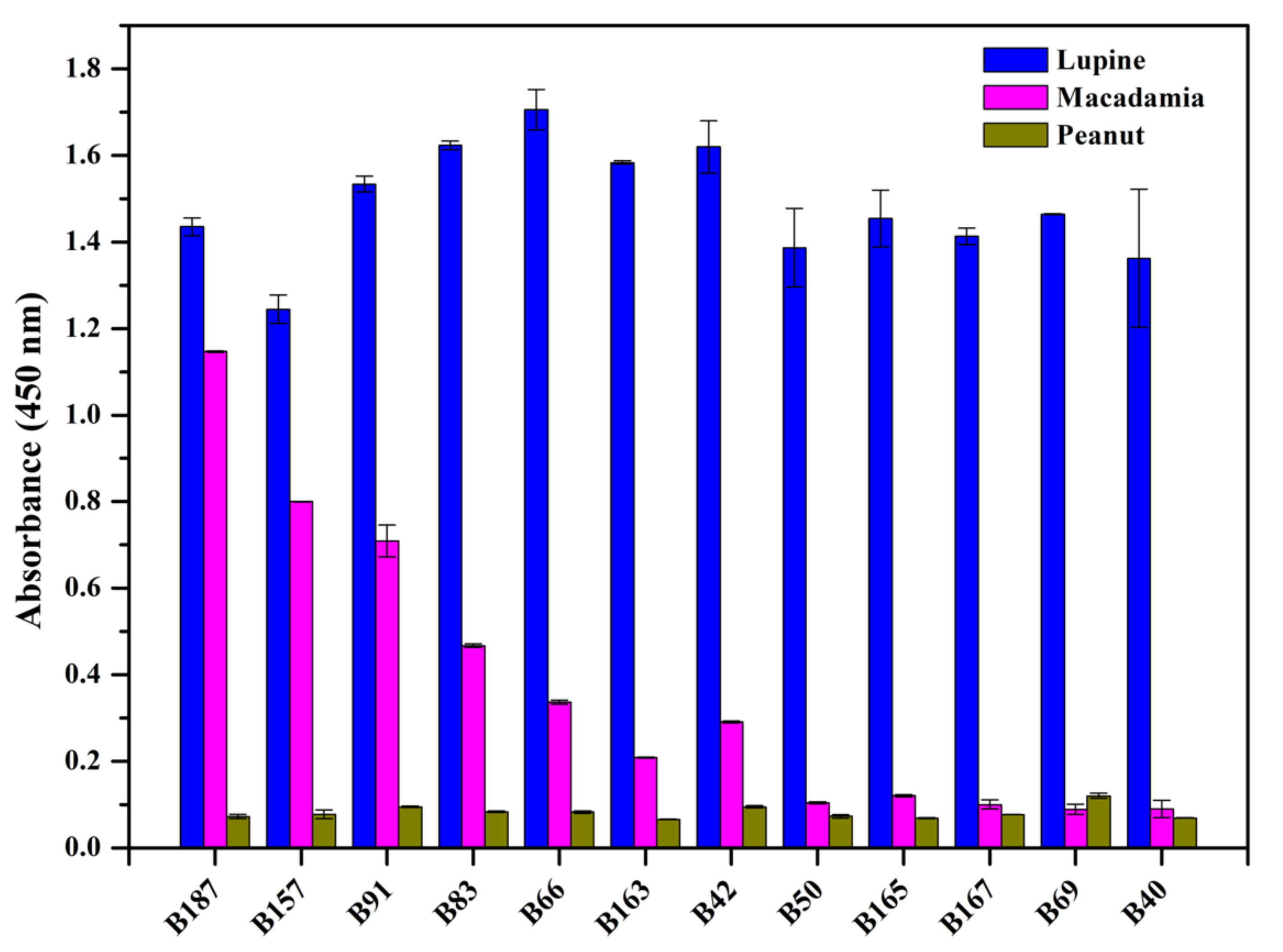
2.7.2. Specificity
2.7.3. Thermostability
2.7.4. Binding and Cross-Reaction of Nbs
2.8. Nb-Mediated Heterologous Sandwich ELISA
2.8.1. Expression and Purification of His-Tagged Nbs
2.8.2. Selection of Nb-Pair and Optimization of Concentration
2.8.3. Development of a Sandwich ELISA
2.8.4. Detection of the Spiked Sample
3. Results
3.1. Construction of an Immune Nb Library
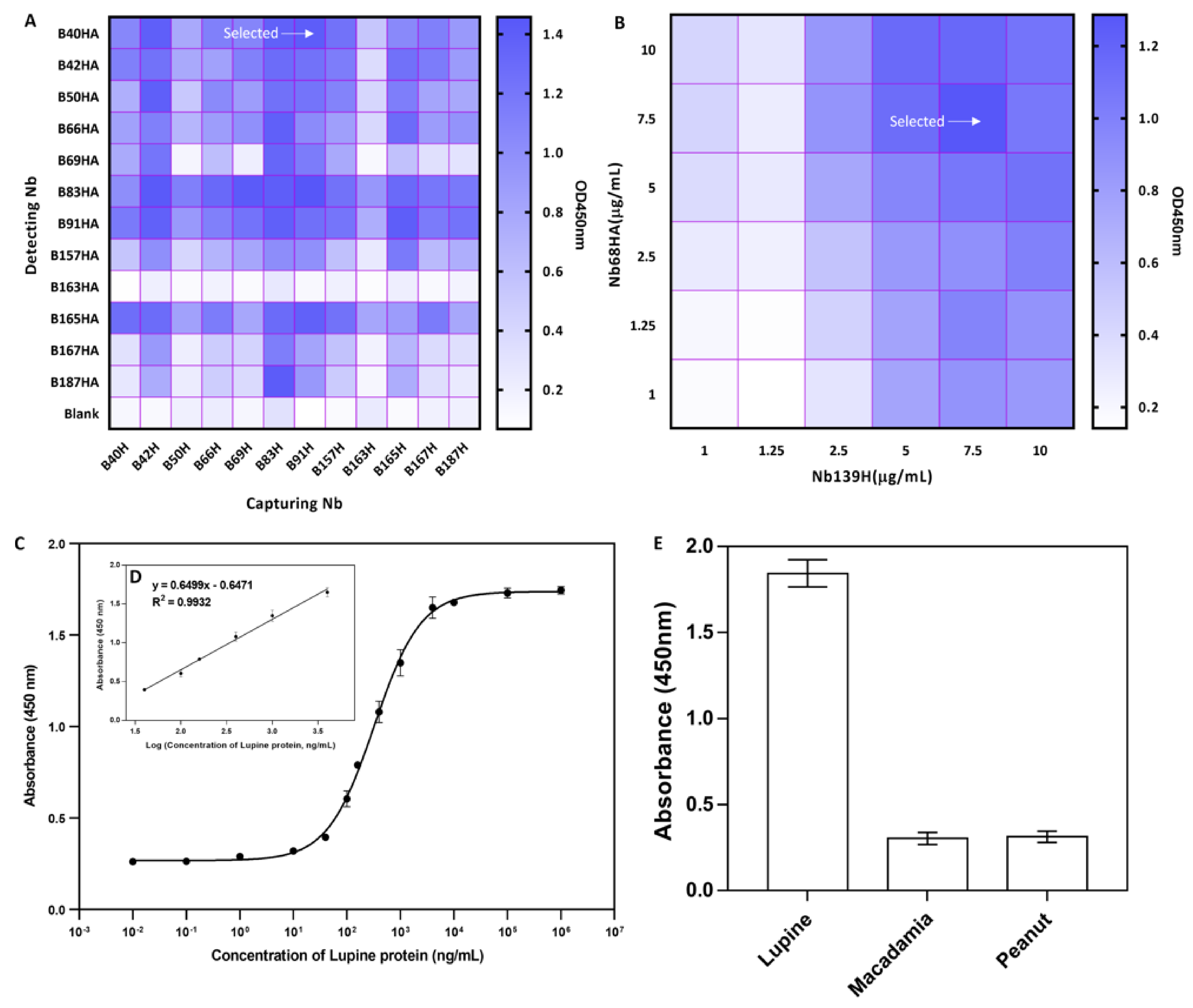
3.2. Bio-Panning and Screening of Nbs
3.3. Purification and Specificity Analysis of Nbs
3.4. Characteristics of Nbs
3.4.1. Affinity of Nbs
3.4.2. Thermostability
3.4.3. Cross-Reaction of Selected Nbs
3.5. Identification of Target Antigen by LC-MS/MS
3.6. Development of a Heterologous Sandwich ELISA
3.7. Cross-Reactivity of the Developed Sandwich ELISA
3.8. Analysis of Spiked Sample
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Prusinski, J. White Lupin (Lupinus albus L.)—Nutritional and Health Values in Human Nutrition—a Review. Czech J. Food Sci. 2017, 35, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Villarino, C.B.; Jayasena, V.; Coorey, R.; Chakrabarti-Bell, S.; Johnson, S.K. Nutritional, Health, and Technological Functionality of Lupin Flour Addition to Bread and Other Baked Products: Benefits and Challenges. Crit. Rev. Food Sci. Nutr. 2016, 56, 835–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, M.L.; de Las Marinas, M.D.; Fernandez, J.; Gamboa, P.M. Lupin allergy: A hidden killer in the home. Clin. Exp. Allergy 2010, 40, 1461–1466. [Google Scholar] [CrossRef]
- Soller, L.; La Vieille, S.; Chan, E.S. First reported case in Canada of anaphylaxis to lupine in a child with peanut allergy. Allergy Asthma Clin. Immunol. 2018, 14, 64. [Google Scholar] [CrossRef]
- Mennini, M.; Dahdah, L.; Mazzina, O.; Fiocchi, A. Lupin and Other Potentially Cross-Reactive Allergens in Peanut Allergy. Curr. Allergy Asthma Rep. 2016, 16, 84. [Google Scholar] [CrossRef]
- Jappe, U.; Vieths, S. Lupine, a source of new as well as hidden food allergens. Mol. Nutr. Food Res. 2010, 54, 113–126. [Google Scholar] [CrossRef]
- Goggin, D.E.; Mir, G.; Smith, W.B.; Stuckey, M.; Smith, P.M. Proteomic analysis of lupin seed proteins to identify conglutin Beta as an allergen, Lup an. J. Agric. Food Chem. 2008, 56, 6370–6377. [Google Scholar] [CrossRef]
- Lima-Cabello, E.; Alche, J.D.; Jimenez-Lopez, J.C. Narrow-Leafed Lupin Main Allergen beta-Conglutin (Lup an 1) Detection and Quantification Assessment in Natural and Processed Foods. Foods 2019, 8, 513. [Google Scholar] [CrossRef] [Green Version]
- Demmel, A.; Hupfer, C.; Ilg Hampe, E.; Busch, U.; Engel, K.H. Development of a real-time PCR for the detection of lupine DNA (lupinus species) in foods. J. Agric. Food Chem. 2008, 56, 4328–4332. [Google Scholar] [CrossRef] [PubMed]
- Demmel, A.; Hupfer, C.; Busch, U.; Engel, K.H. Detection of lupine (Lupinus spp) DNA in processed foods using real-time PCR. Food Control 2011, 22, 215–220. [Google Scholar] [CrossRef]
- Koeberl, M.; Sharp, M.F.; Tian, R.K.; Buddhadasa, S.; Clarke, D.; Roberts, J. Lupine allergen detecting capability and cross-reactivity of related legumes by ELISA. Food Chem. 2018, 256, 105–112. [Google Scholar] [CrossRef]
- Holden, L.; Moen, L.H.; Sletten, G.B.; Dooper, M.M. Novel polyclonal-monoclonal-based ELISA utilized to examine lupine (Lupinus species) content in food products. J. Agric. Food Chem. 2007, 55, 2536–2542. [Google Scholar] [CrossRef]
- Villa, C.; Moura, M.B.M.V.; Costa, J.; Mafra, I. Immunoreactivity of Lupine and Soybean Allergens in Foods as Affected by Thermal Processing. Foods 2020, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Cabanillas, B.; Jappe, U.; Novak, N. Allergy to Peanut, Soybean, and Other Legumes: Recent Advances in Allergen Characterization, Stability to Processing and IgE Cross-Reactivity. Mol. Nutr. Food Res. 2018, 62, 1700446. [Google Scholar] [CrossRef]
- Hassanzadeh-Ghassabeh, G.; Devoogdt, N.; Pauw, P.D.; Vincke, C.; Muyldermans, S. Nanobodies and their potential applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Wu, S.; Wang, Y.; Lin, J.; Sun, Y.; Zhang, C.; Gu, J.; Yang, F.; Lv, H.; Ji, X.; et al. Unbiased Immunization Strategy Yielding Specific Nanobodies against Macadamia Allergen of Vicilin-like Protein for Immunoassay Development. J. Agric. Food Chem. 2021, 69, 5178–5188. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.S.; Colwell, L.J. Comparative analysis of nanobody sequence and structure data. Proteins 2018, 86, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S.; Baral, T.N.; Retamozzo, V.C.; De Baetselier, P.; De Genst, E.; Kinne, J.; Leonhardt, H.; Magez, S.; Nguyen, V.K.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 2009, 128, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting Nanobodies’ Singular Traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Nadal, P.; Canela, N.; Katakis, I.; O’Sullivan, C.K. Extraction, Isolation, and Characterization of Globulin Proteins from Lupinus albus. J. Agric. Food Chem. 2011, 59, 2752–2758. [Google Scholar] [CrossRef]
- Muranyi, I.S.; Otto, C.; Pickardt, C.; Koehler, P.; Schweiggert-Weisz, U. Microscopic characterisation and composition of proteins from lupin seed (Lupinus angustifolius L.) as affected by the isolation procedure. Food Res. Int. 2013, 54, 1419–1429. [Google Scholar] [CrossRef]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.; Wohlkonig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havli, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2007, 1, 2856–2860. [Google Scholar] [CrossRef]
- Magnusson, B.; Örnemark, U. (Eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics; LGC (Teddington) Ltd.: Teddington, UK, 2014; Volume 10. [Google Scholar]
- Fontanari, G.G.; Martins, J.M.; Kobelnik, M.; Pastre, I.A.; Areas, J.A.G.; Batistuti, J.P.; Fertonani, F.L. Thermal studies on protein isolates of white lupin seeds (Lupinus albus). J. Anal. Calorim. 2012, 108, 141–148. [Google Scholar] [CrossRef] [Green Version]
- De Meyer, T.; Eeckhout, D.; De Rycke, R.; De Buck, S.; Muyldermans, S.; Depicker, A. Generation of VHH antibodies against the Arabidopsis thaliana seed storage proteins. Plant. Mol. Biol. 2014, 84, 83–93. [Google Scholar] [CrossRef]
- Li, Z.; Pinto Torres, J.E.; Goossens, J.; Vertommen, D.; Caljon, G.; Sterckx, Y.G.; Magez, S. An Unbiased Immunization Strategy Results in the Identification of Enolase as a Potential Marker for Nanobody-Based Detection of Trypanosoma evansi. Vaccines 2020, 8, 415. [Google Scholar] [CrossRef] [PubMed]
- Guillamon, E.; Rodriguez, J.; Burbano, C.; Muzquiz, M.; Pedrosa, M.M.; Cabanillas, B.; Crespo, J.F.; Sancho, A.I.; Mills, E.N.; Cuadrado, C. Characterization of lupin major allergens (Lupinus albus L.). Mol. Nutr. Food Res. 2010, 54, 1668–1676. [Google Scholar] [CrossRef]
- Cabello-Hurtado, F.; Keller, J.; Ley, J.; Sanchez-Lucas, R.; Jorrin-Novo, J.V.; Ainouche, A. Proteomics for exploiting diversity of lupin seed storage proteins and their use as nutraceuticals for health and welfare. J. Proteom. 2016, 143, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Costa, J.; Mafra, I. Lupine allergens: Clinical relevance, molecular characterization, cross-reactivity, and detection strategies. Compr. Rev. Food Sci. Food Saf. 2020, 19, 3886–3915. [Google Scholar] [CrossRef] [PubMed]
- Scarafoni, A.; Ronchi, A.; Duranti, M. A real-time PCR method for the detection and quantification of lupin flour in wheat flour-based matrices. Food Chem. 2009, 115, 1088–1093. [Google Scholar] [CrossRef]
- Holden, L.; Faeste, C.K.; Egaas, E. Quantitative sandwich ELISA for the determination of lupine (Lupinus spp.) in foods. J. Agric. Food Chem. 2005, 53, 5866–5871. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Food Sample | Spiked Concentration | Detected Concentration | Recovery 1 | CV 2 |
|---|---|---|---|---|
| μg/mL | μg/mL | %, n = 3 | % | |
| Skim Milk | 0 | ND 3 | — 4 | — |
| 0.04 | 0.04 ± 0.0148 | 108.45 | 2.42 | |
| 0.4 | 0.38 ± 0.0151 | 97.25 | 1.23 | |
| 4 | 3.45 ± 0.0592 | 86.25 | 3.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Zhang, C.; Yang, F.; Lin, J.; Wang, Y.; Wu, S.; Sun, Y.; Zhang, B.; Lv, H.; Ji, X.; et al. Selection of Specific Nanobodies against Lupine Allergen Lup an 1 for Immunoassay Development. Foods 2021, 10, 2428. https://doi.org/10.3390/foods10102428
Hu Y, Zhang C, Yang F, Lin J, Wang Y, Wu S, Sun Y, Zhang B, Lv H, Ji X, et al. Selection of Specific Nanobodies against Lupine Allergen Lup an 1 for Immunoassay Development. Foods. 2021; 10(10):2428. https://doi.org/10.3390/foods10102428
Chicago/Turabian StyleHu, Yaozhong, Chuan Zhang, Feier Yang, Jing Lin, Yi Wang, Sihao Wu, Ying Sun, Bowei Zhang, Huan Lv, Xuemeng Ji, and et al. 2021. "Selection of Specific Nanobodies against Lupine Allergen Lup an 1 for Immunoassay Development" Foods 10, no. 10: 2428. https://doi.org/10.3390/foods10102428
APA StyleHu, Y., Zhang, C., Yang, F., Lin, J., Wang, Y., Wu, S., Sun, Y., Zhang, B., Lv, H., Ji, X., Lu, Y., Muyldermans, S., & Wang, S. (2021). Selection of Specific Nanobodies against Lupine Allergen Lup an 1 for Immunoassay Development. Foods, 10(10), 2428. https://doi.org/10.3390/foods10102428