
Arene-Osmium(II) Complexes in Homogeneous Catalysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
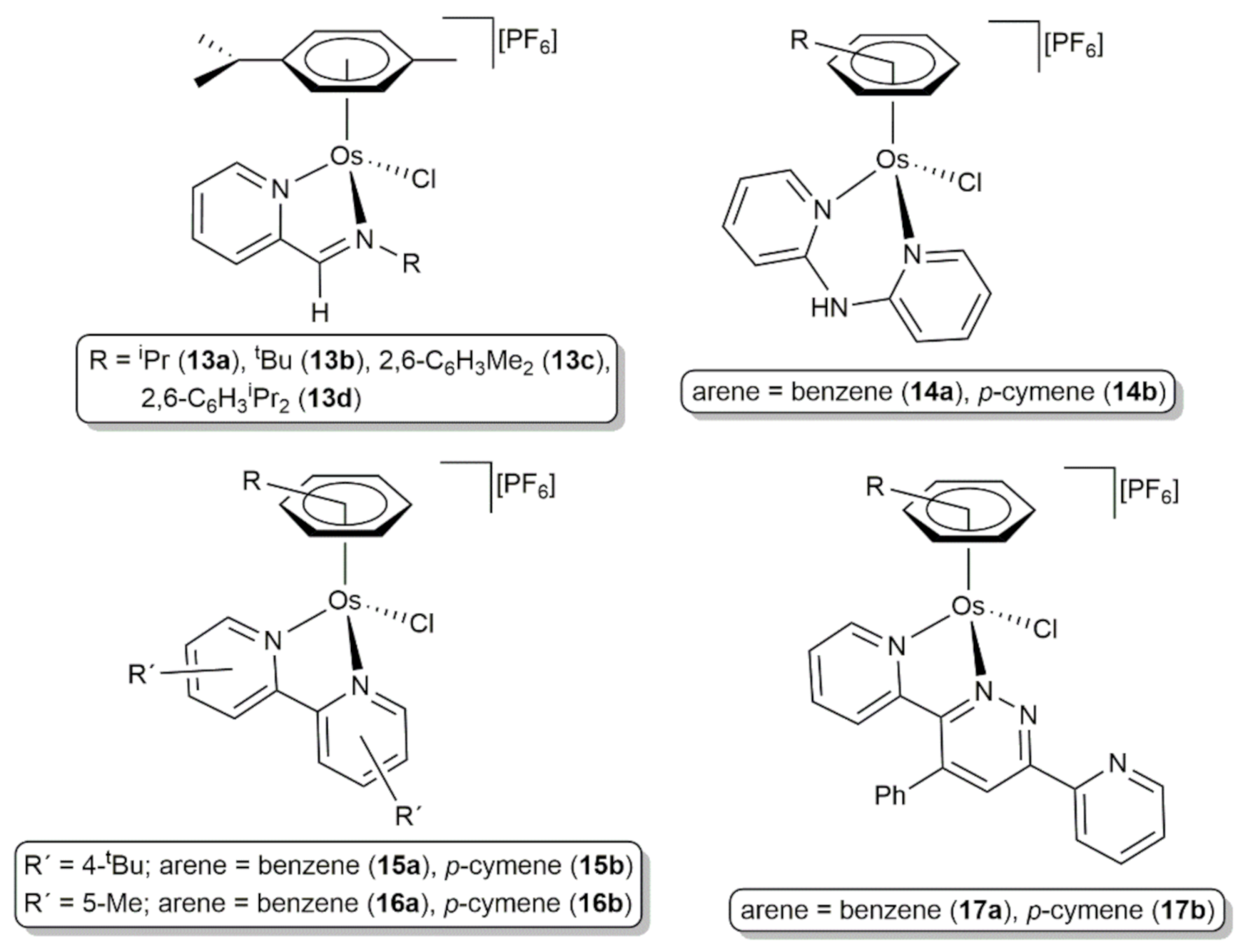{kind=link}
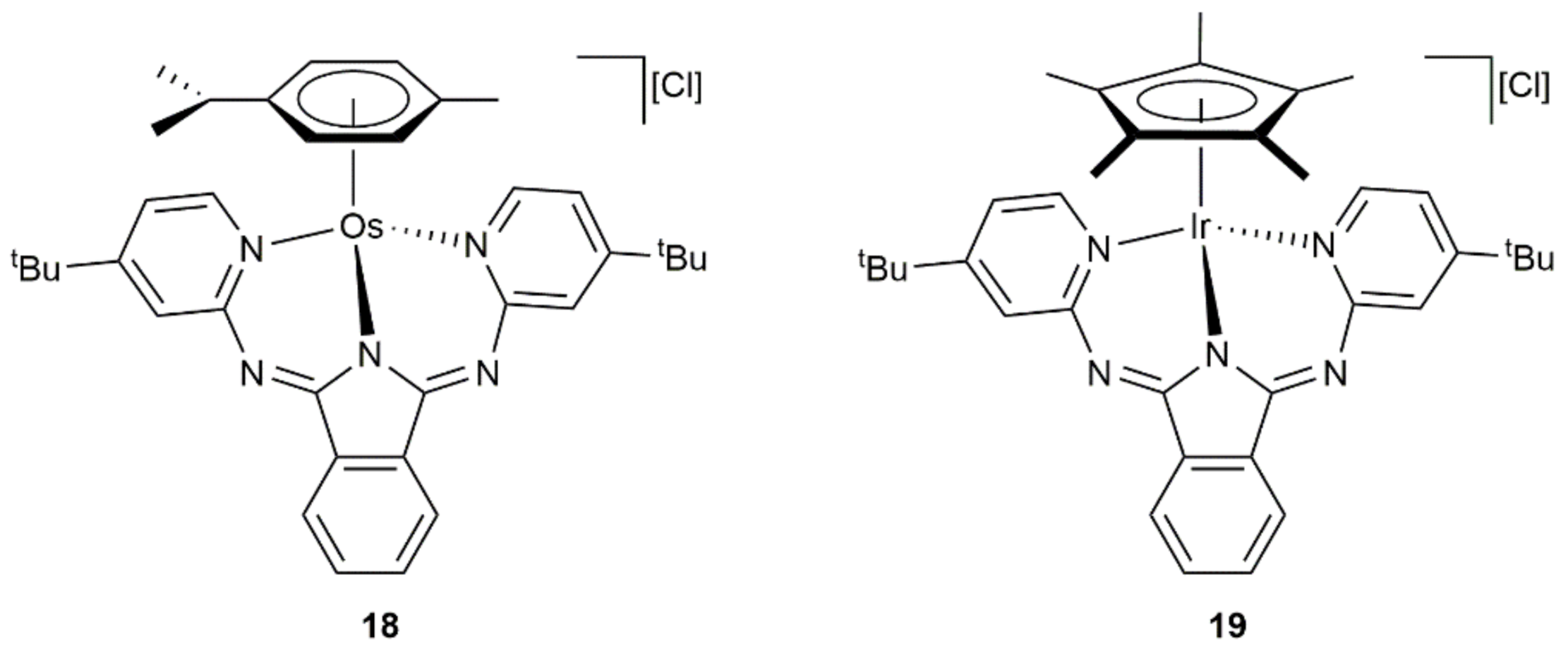{kind=link}
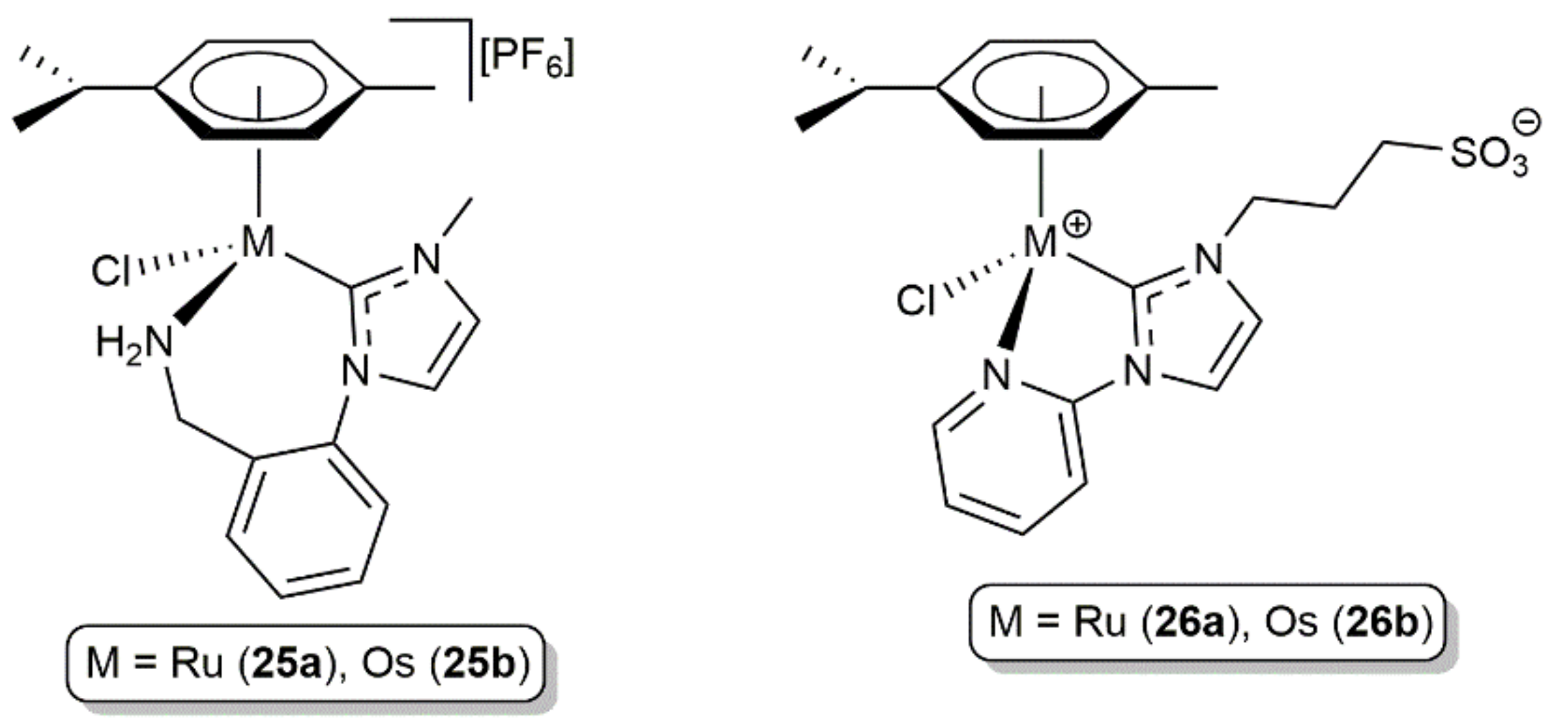{kind=link}
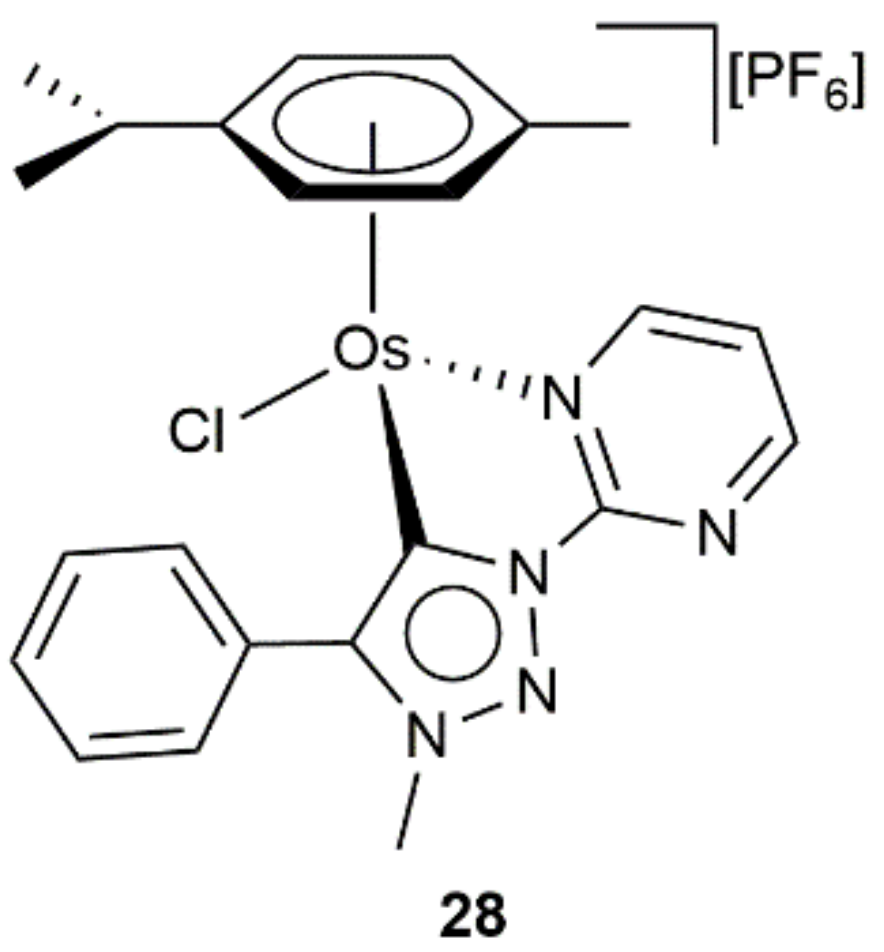{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
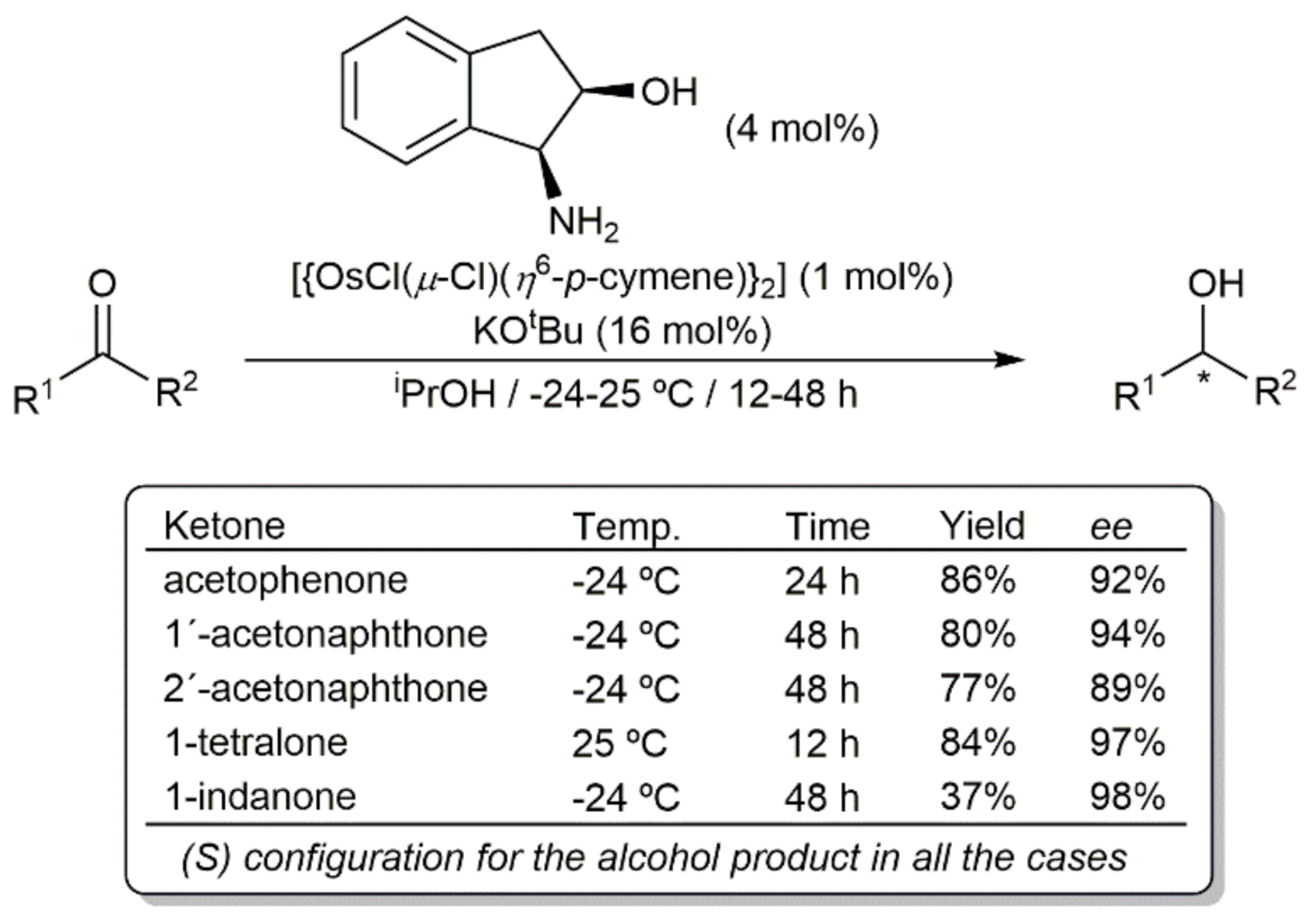{kind=link}
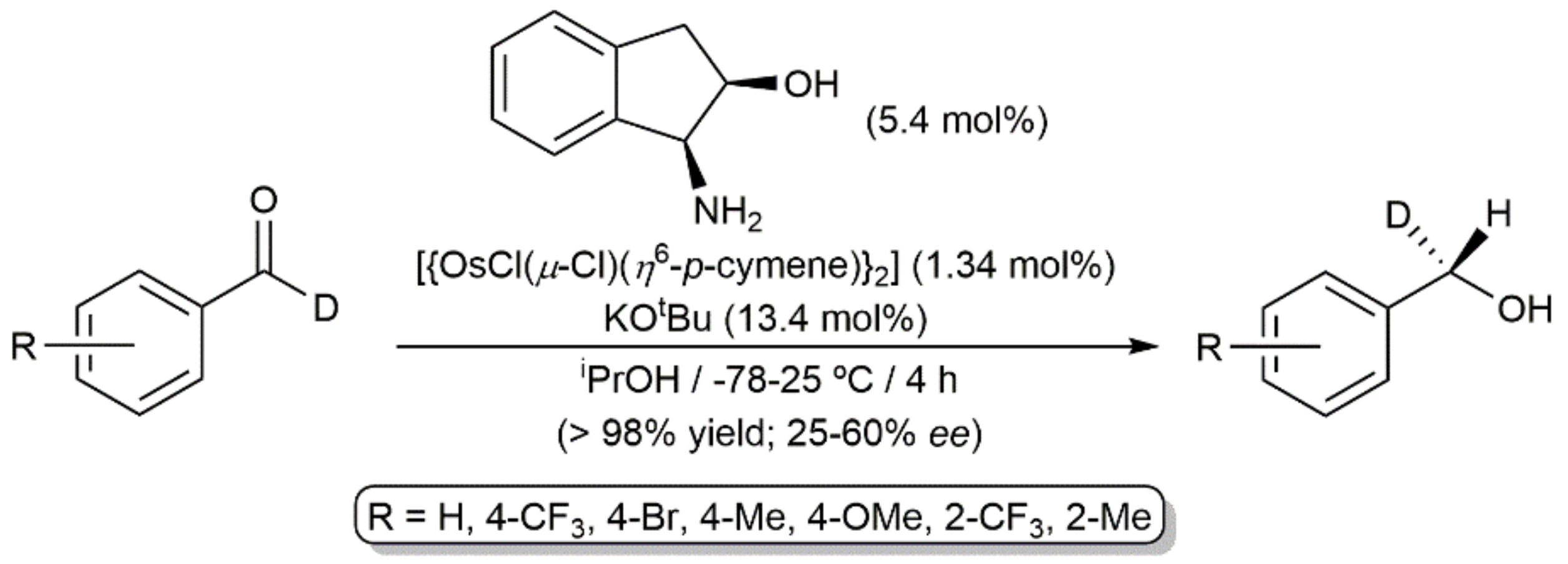{kind=link}
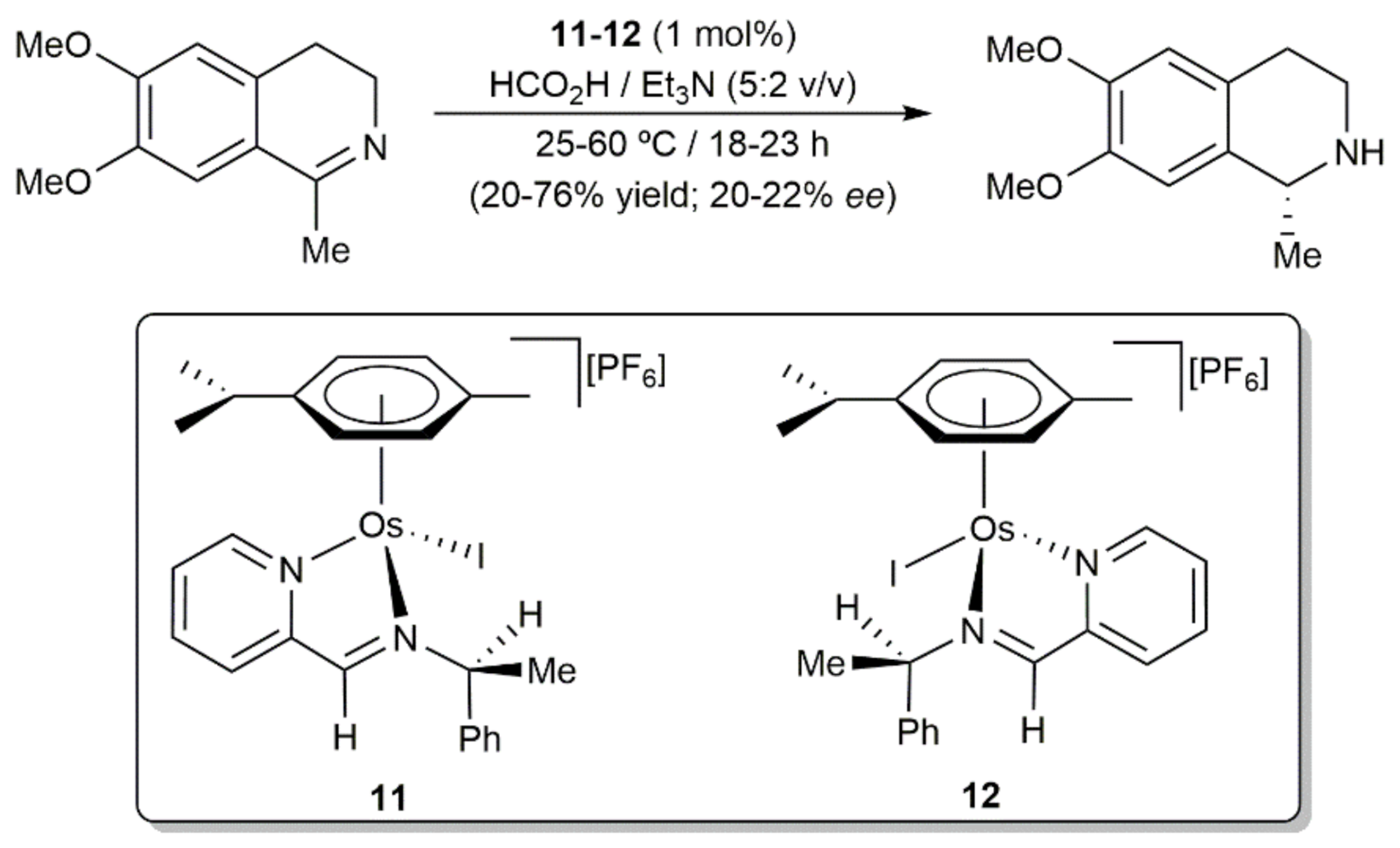{kind=link}
{kind=link}
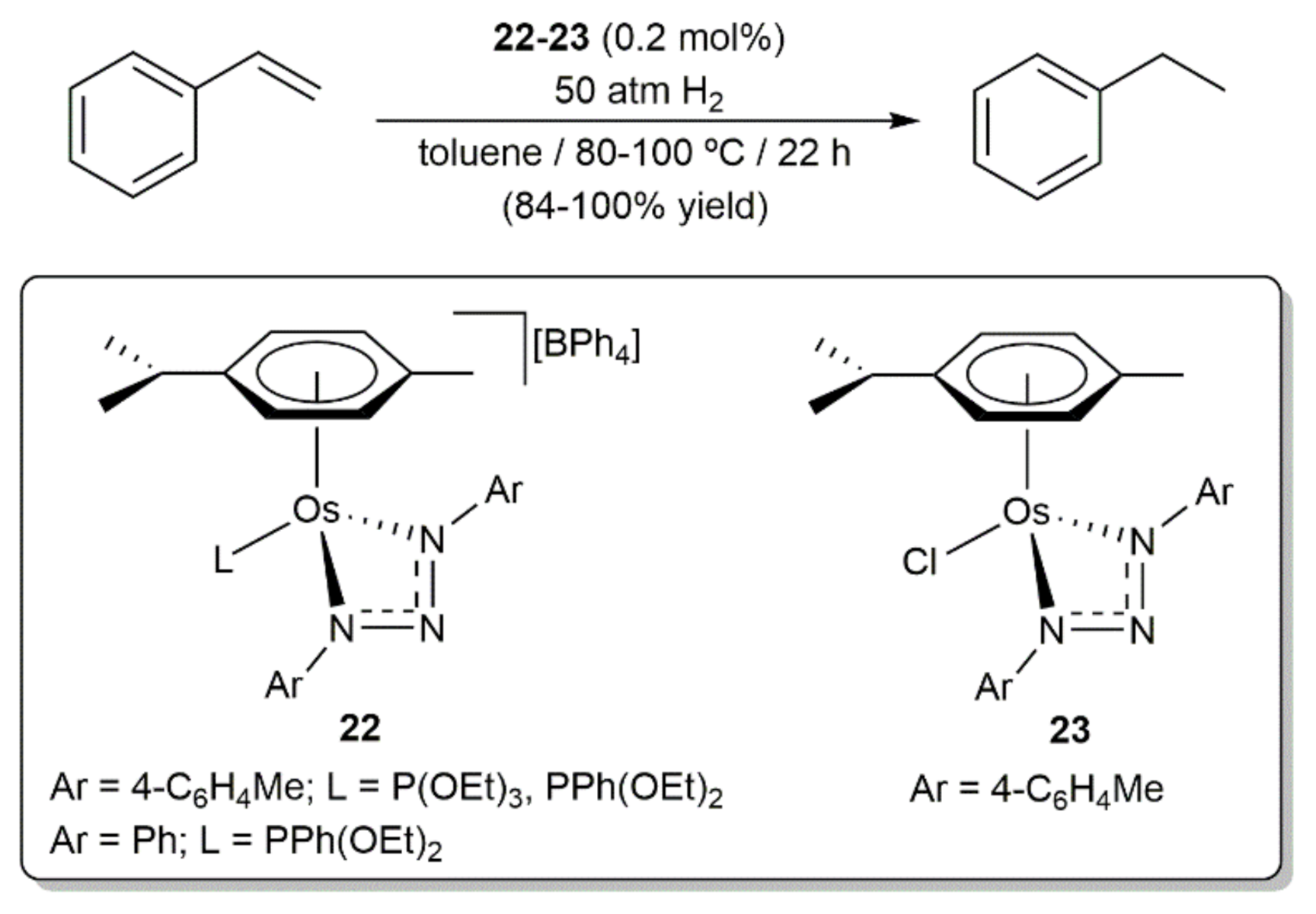{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
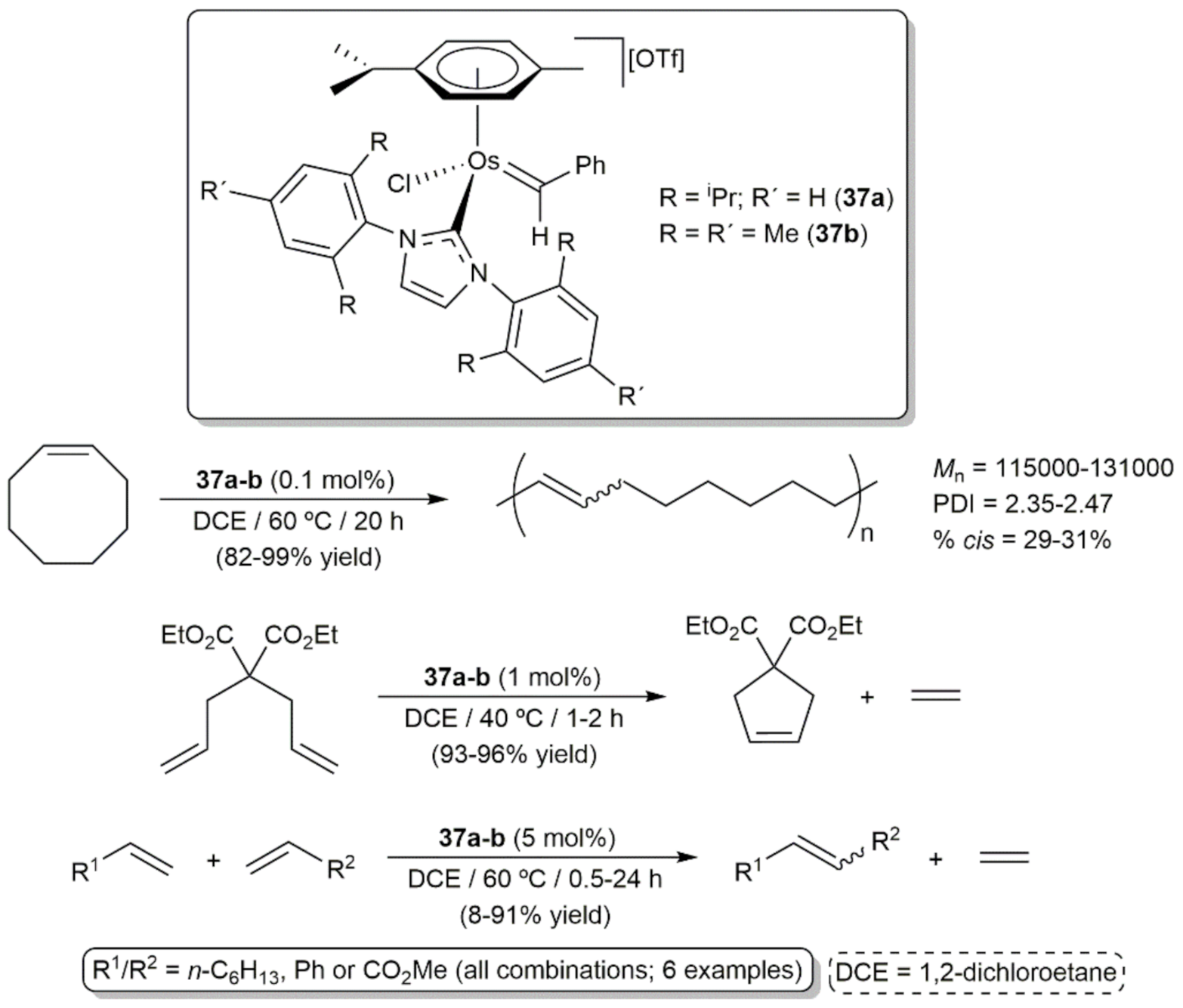{kind=link}
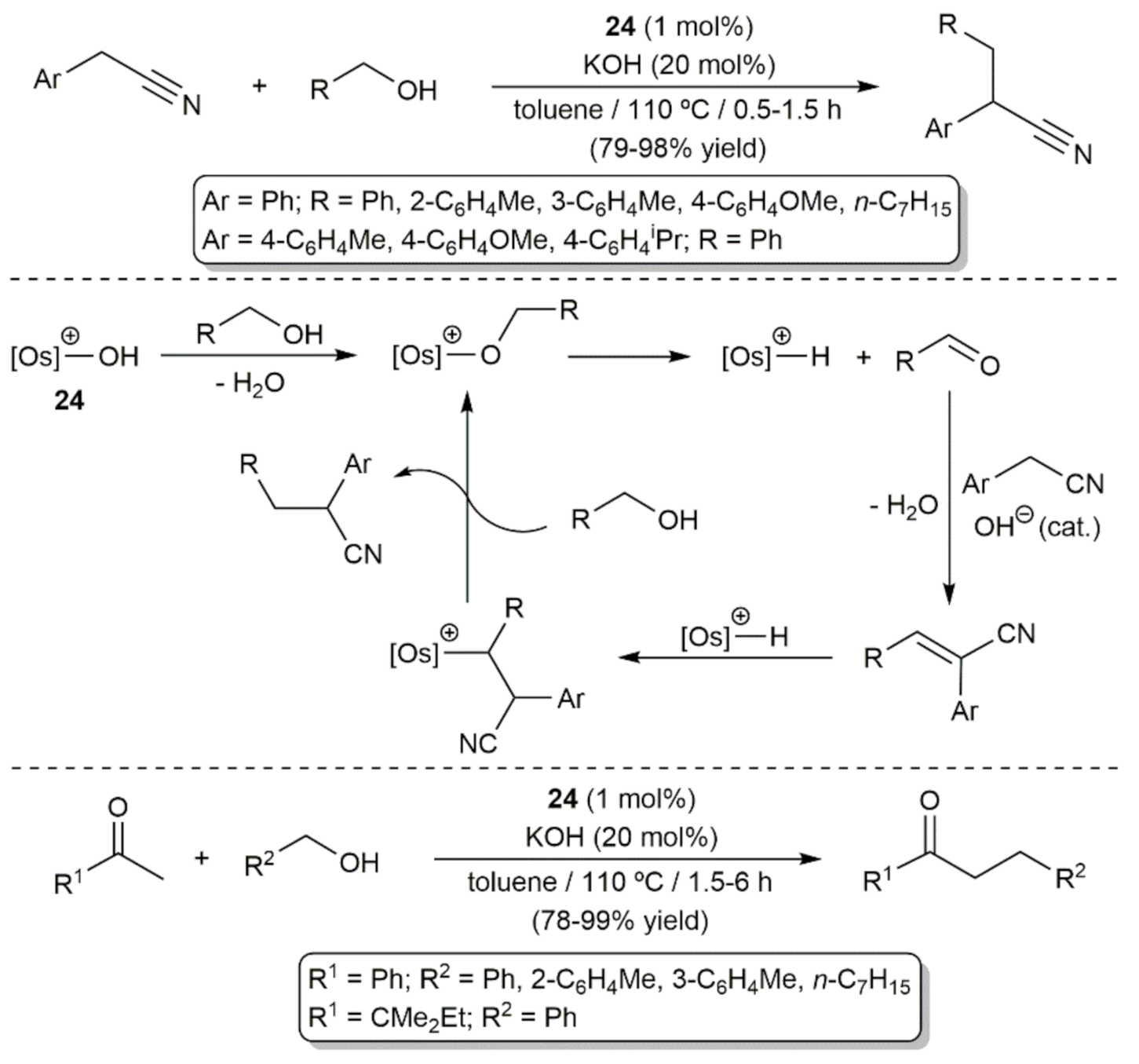{kind=link}
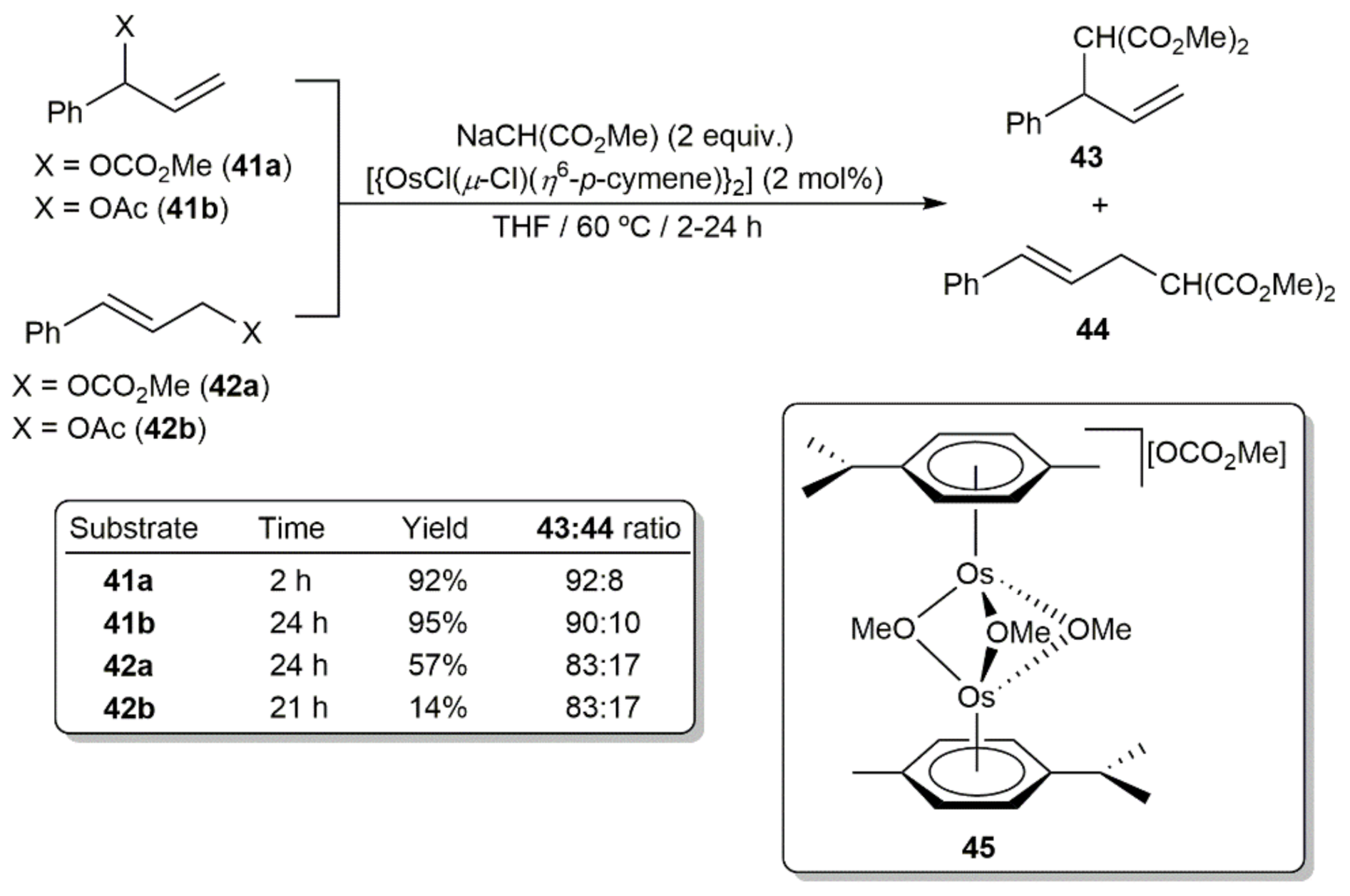{kind=link}
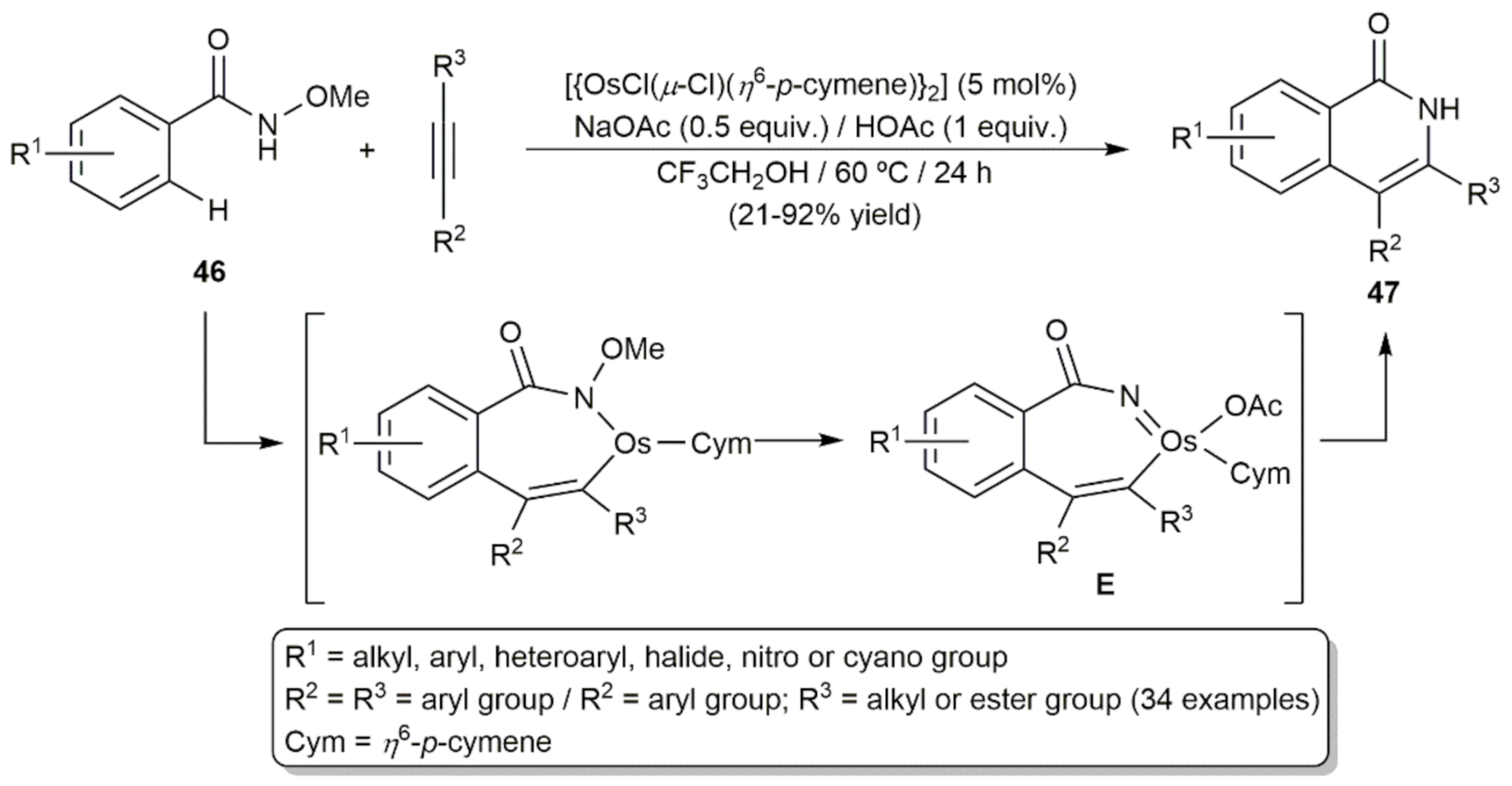{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
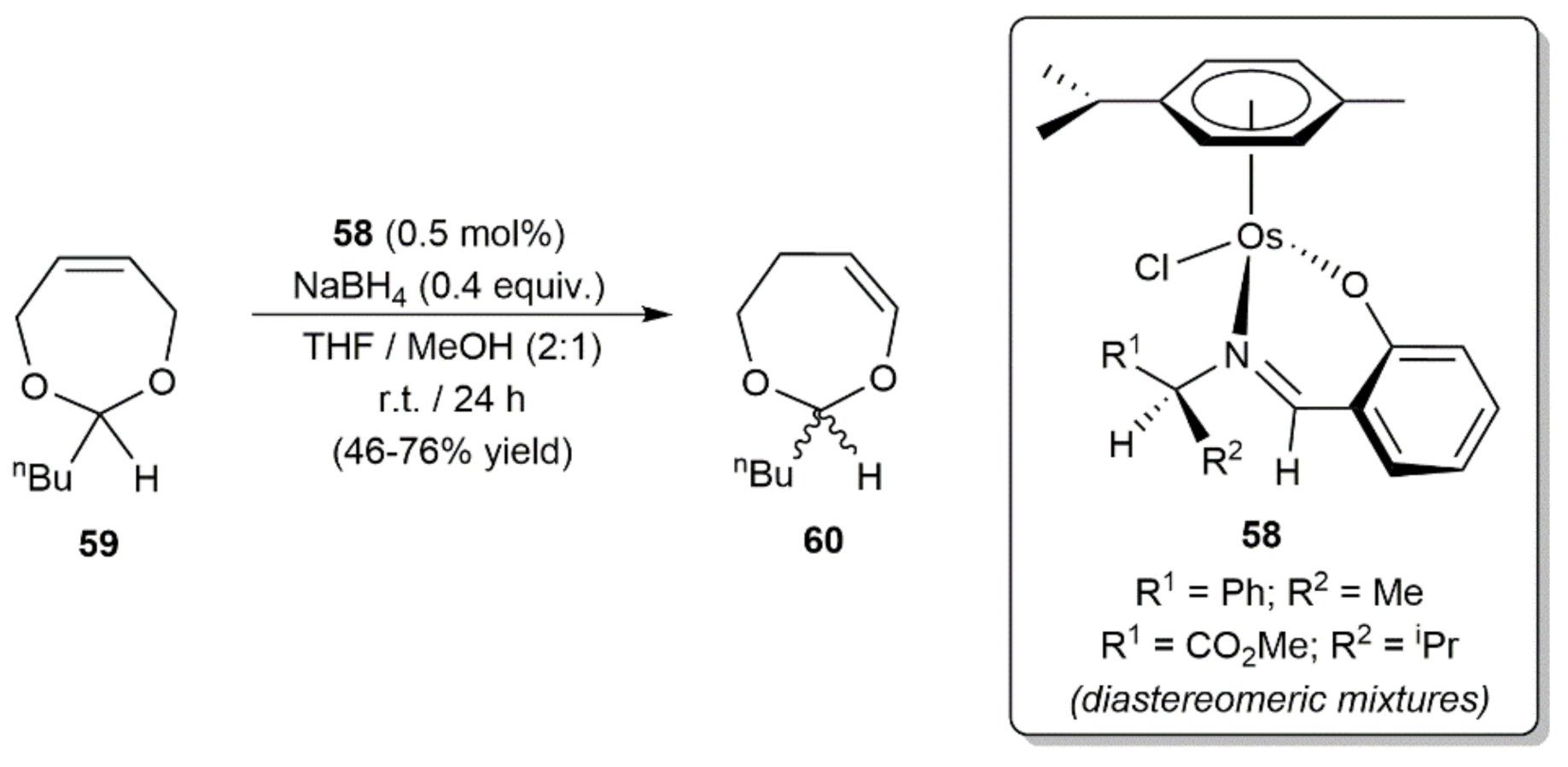
2. Transfer Hydrogenation and Hydrogenation Reactions
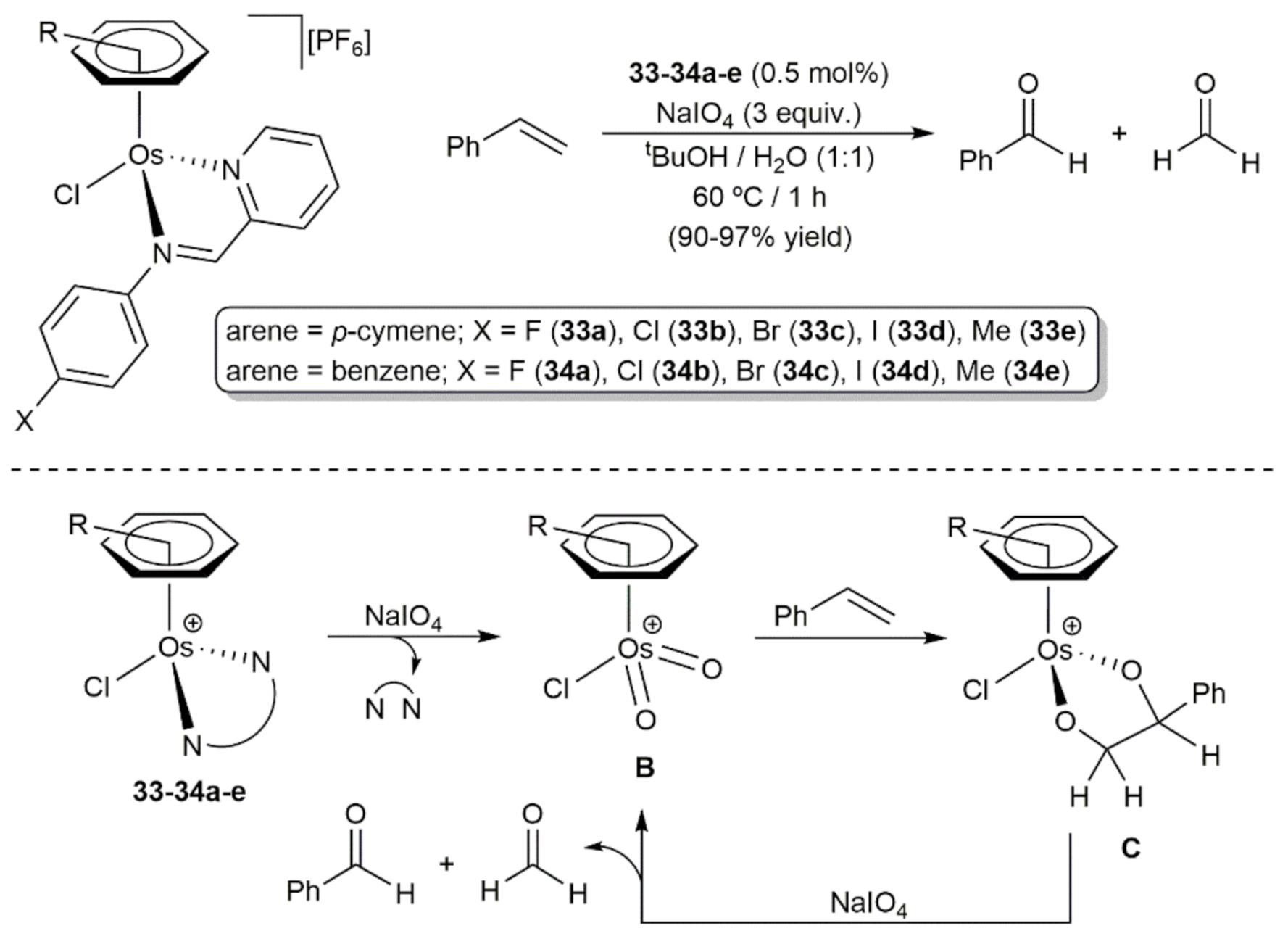
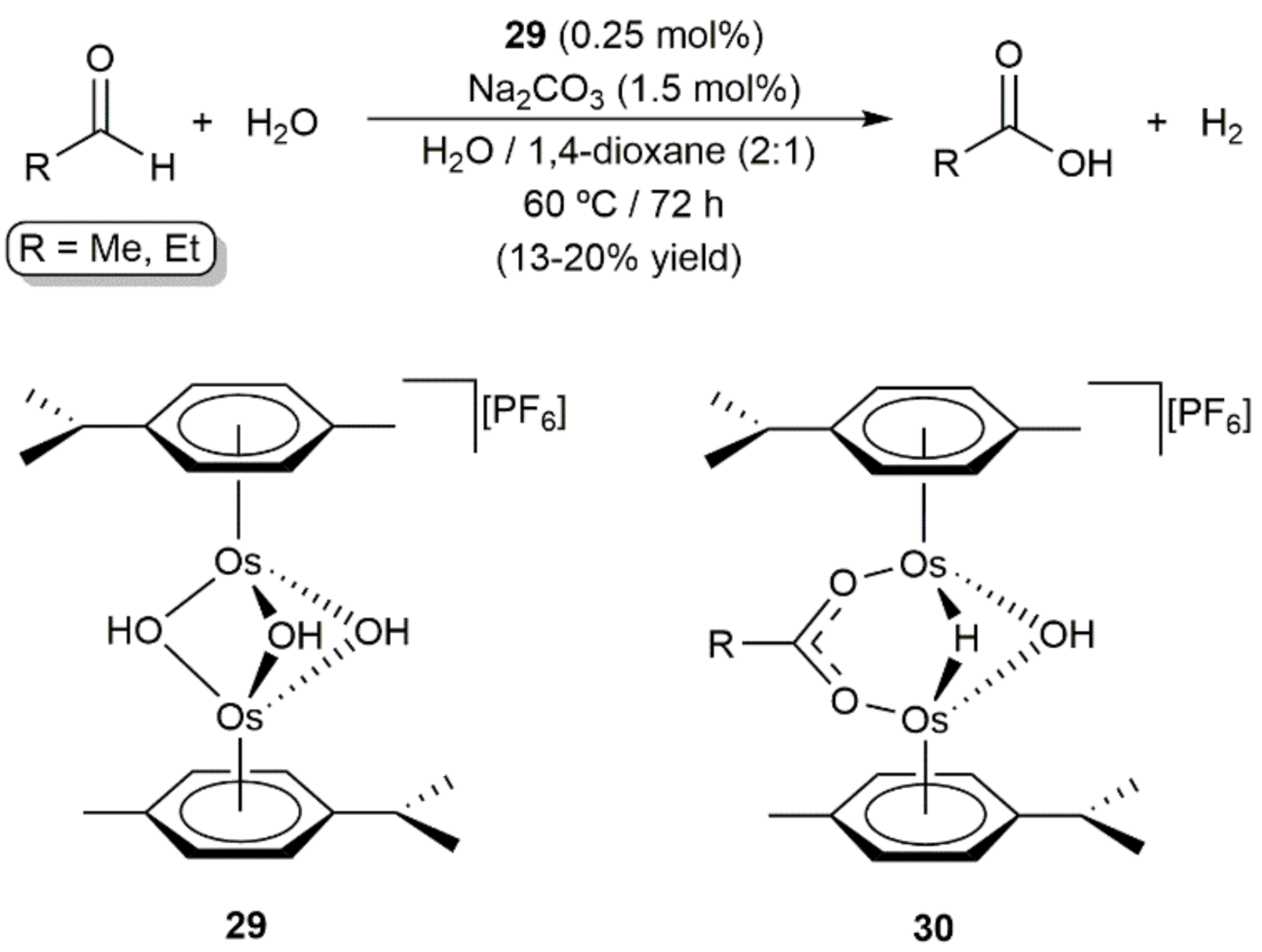
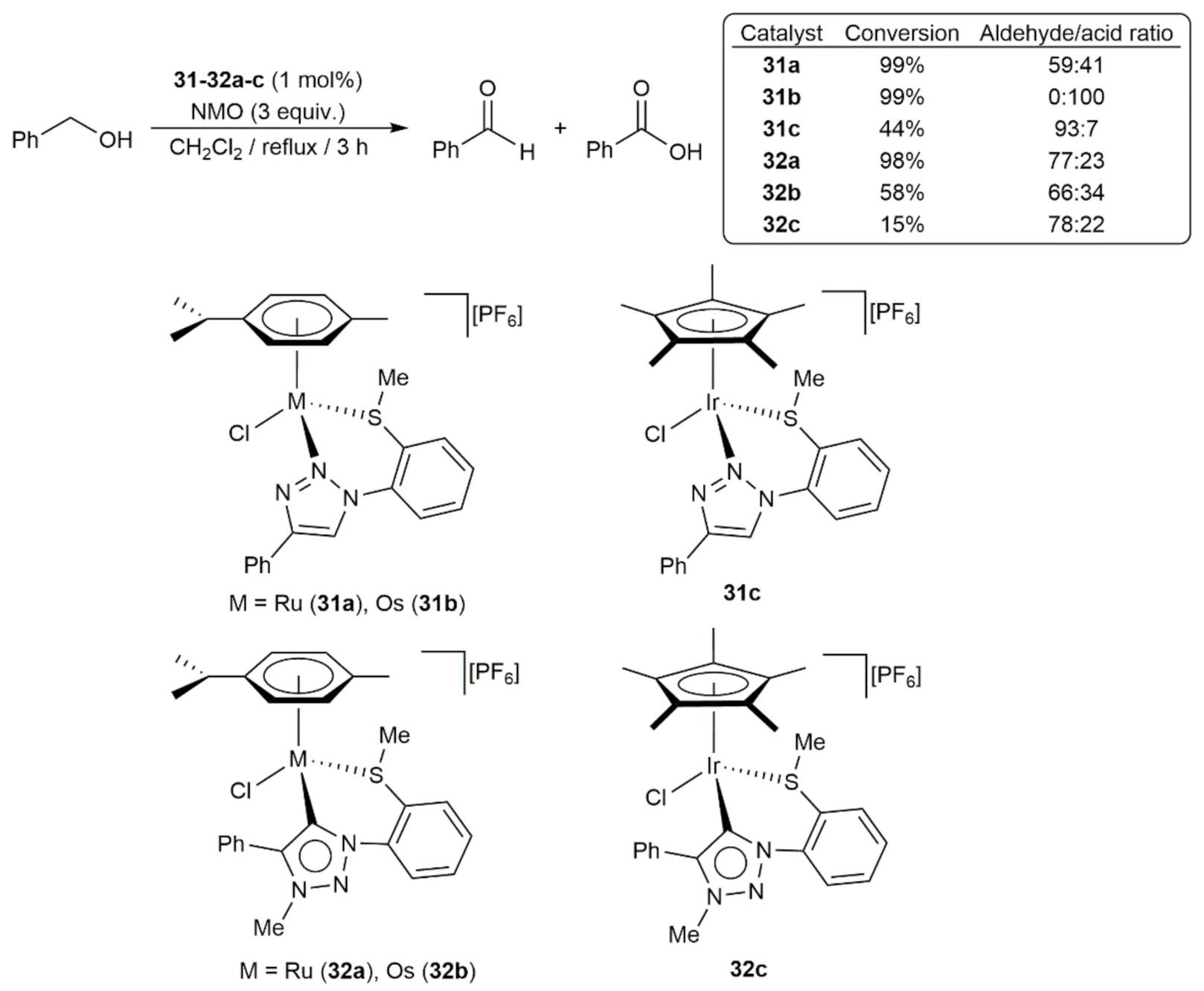
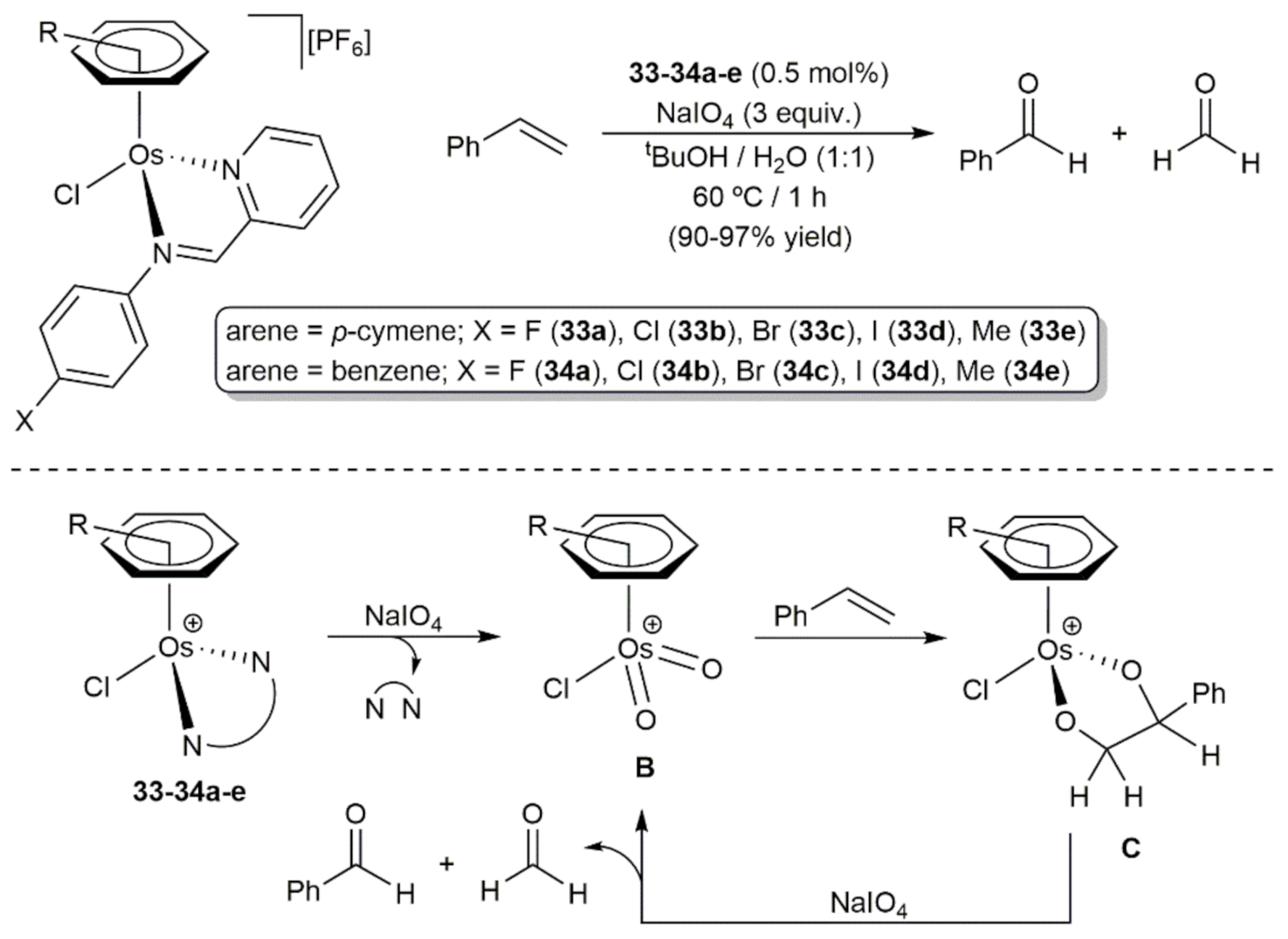
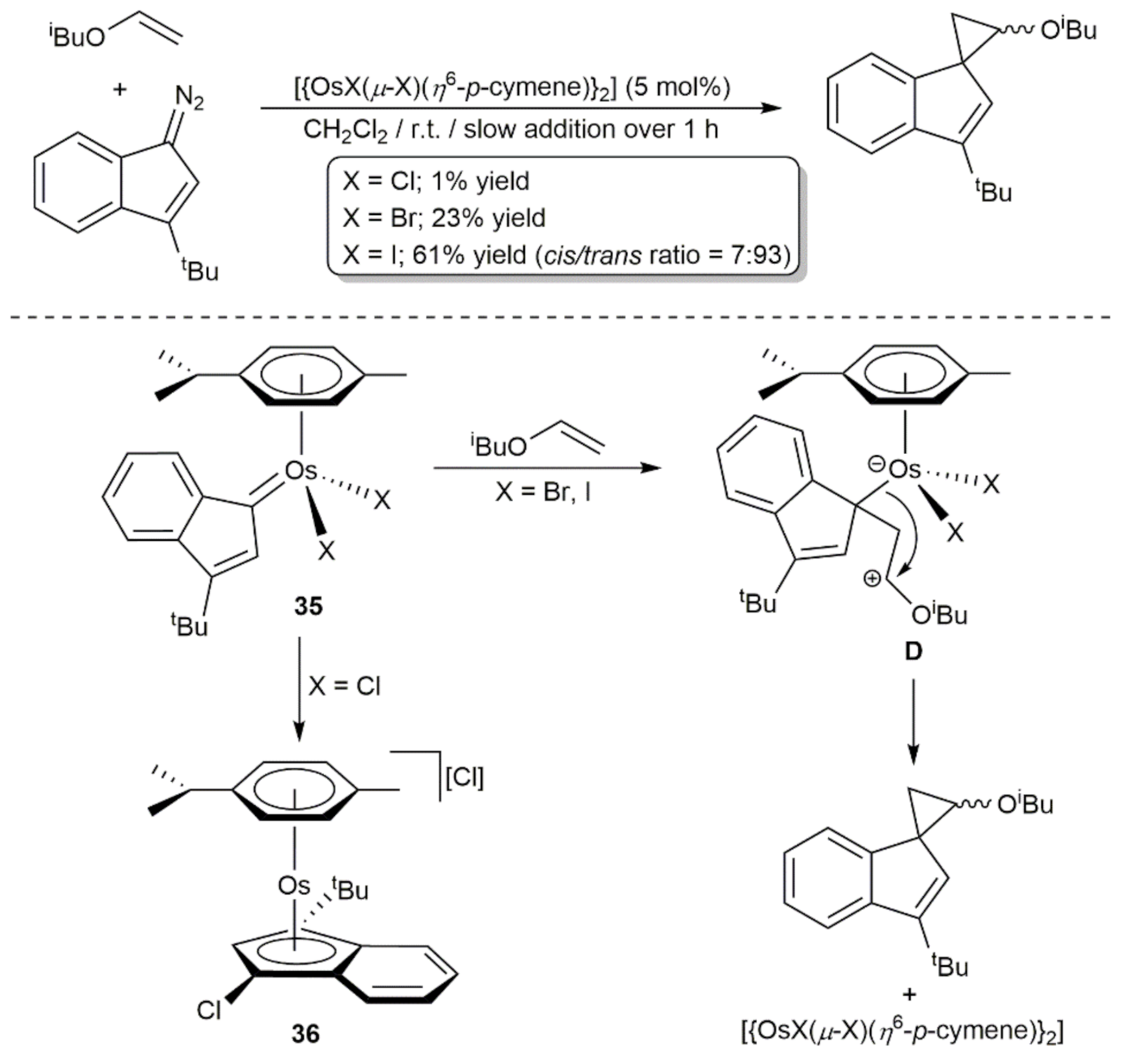
3. Oxidation Reactions
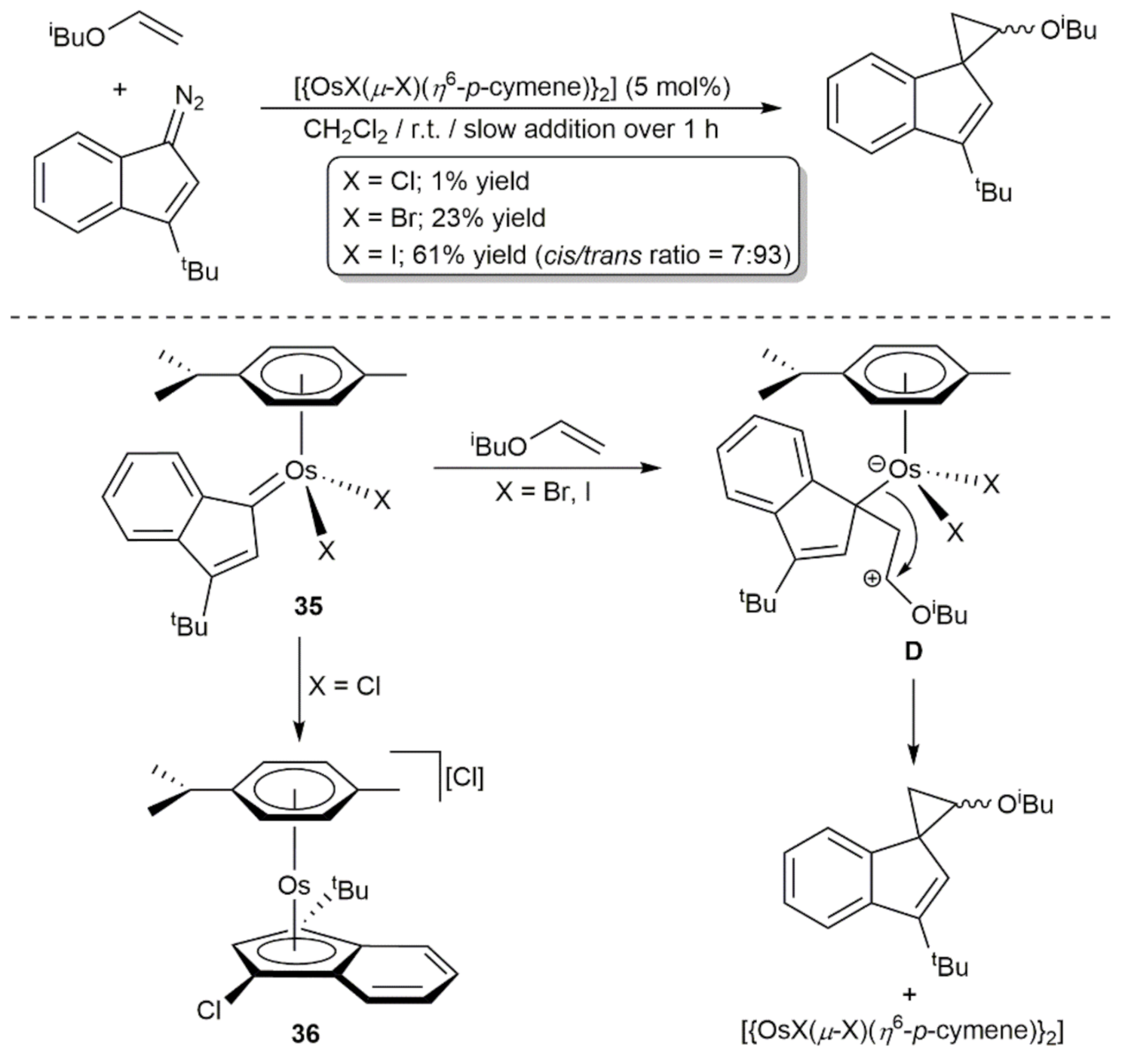
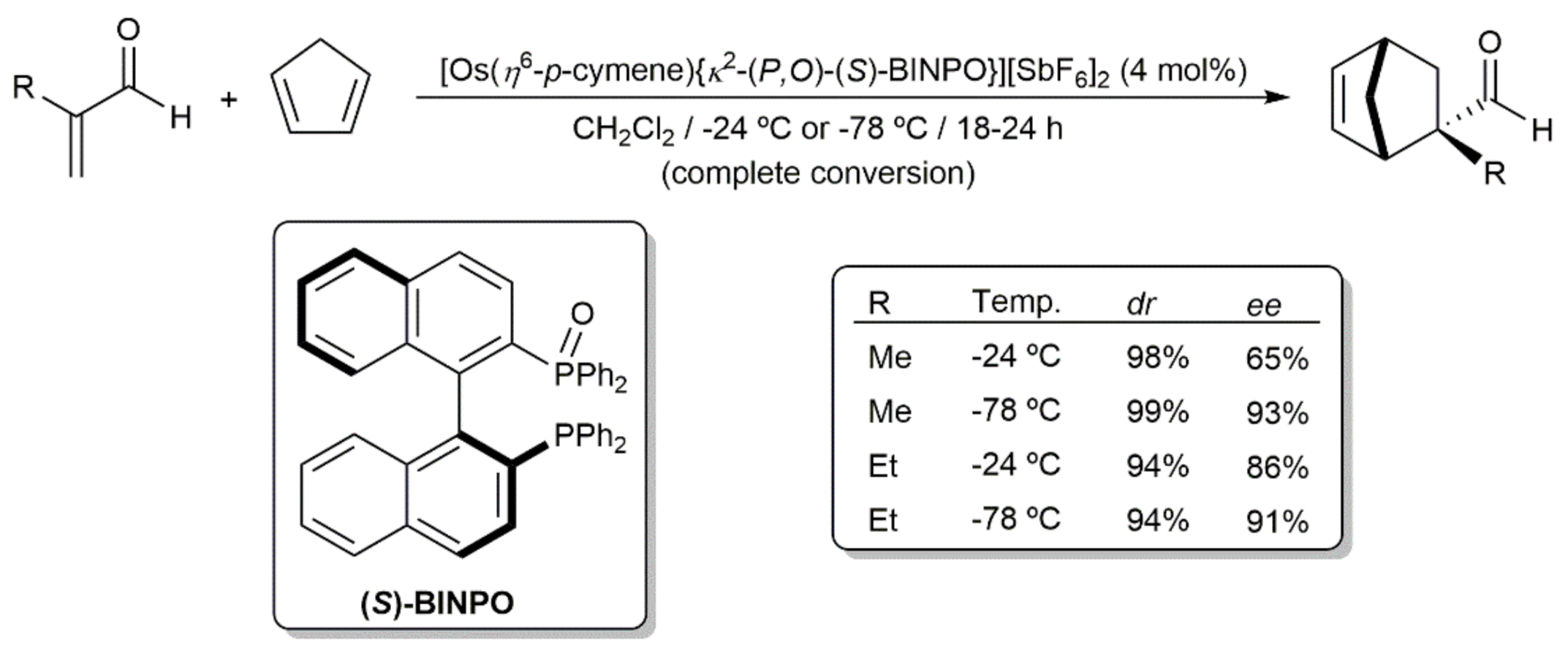
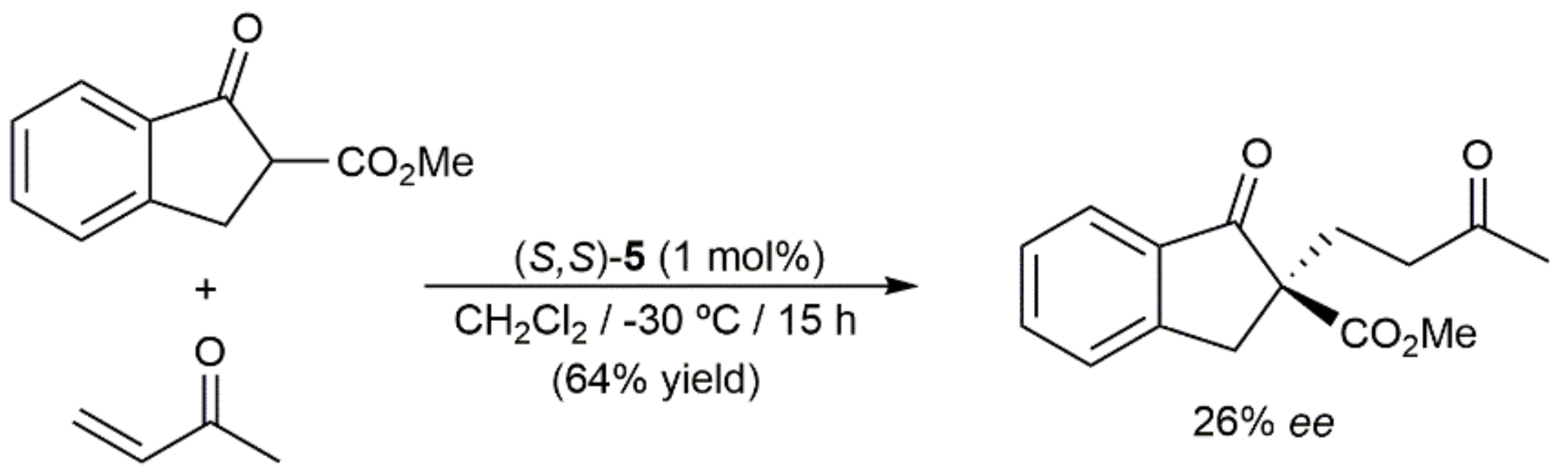
4. C-C Bond Forming Reactions
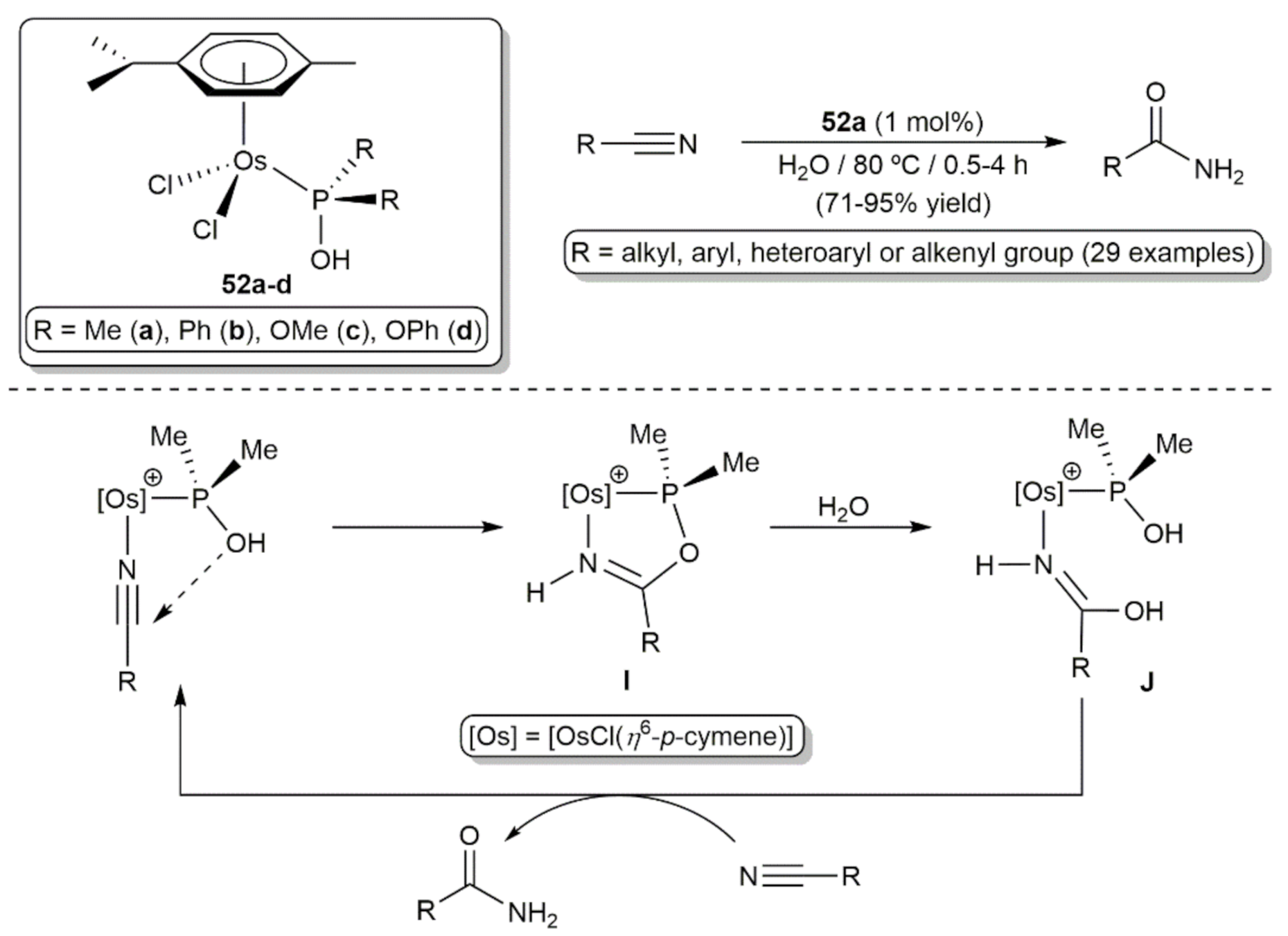
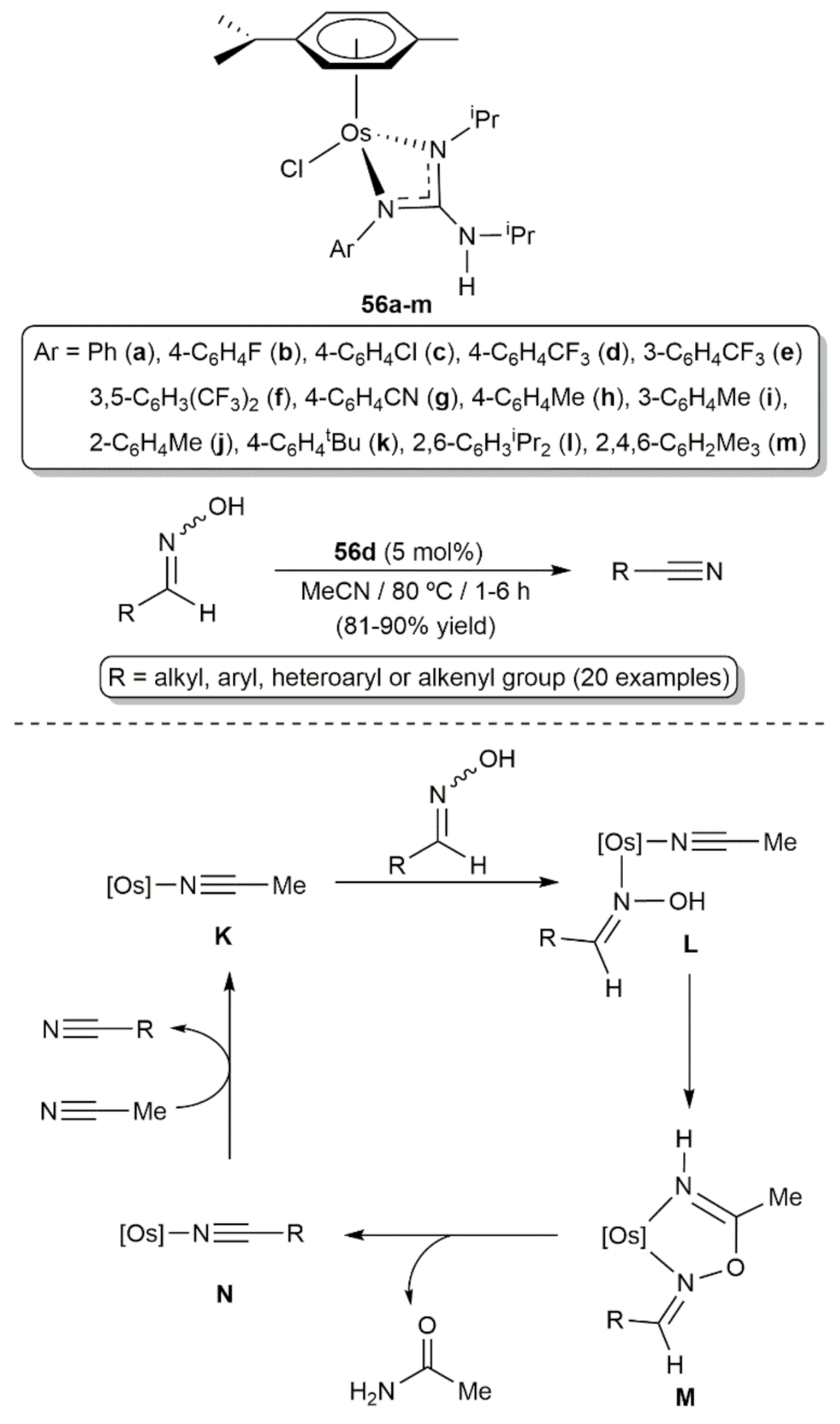
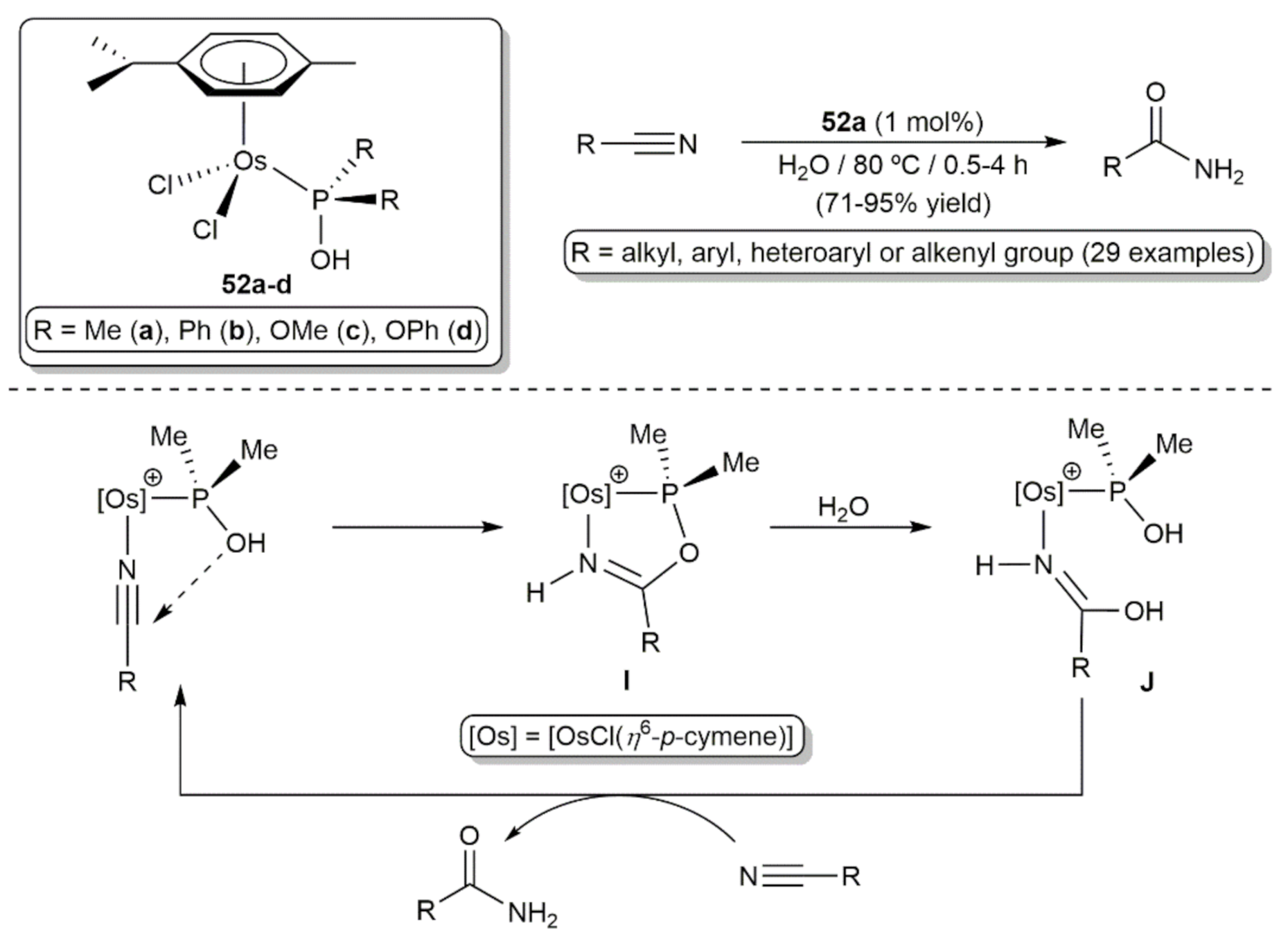
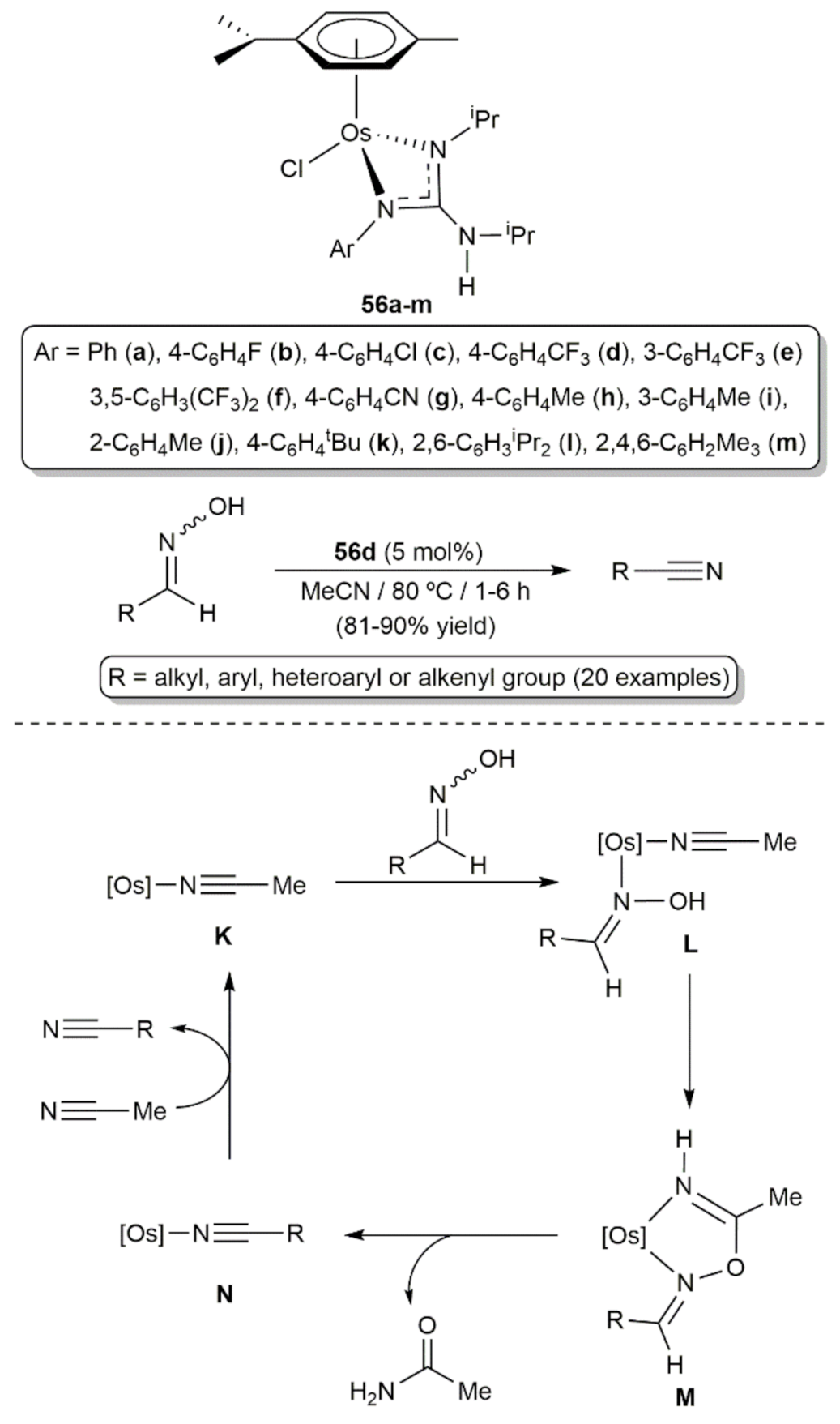
5. Nitrile Hydration Reactions
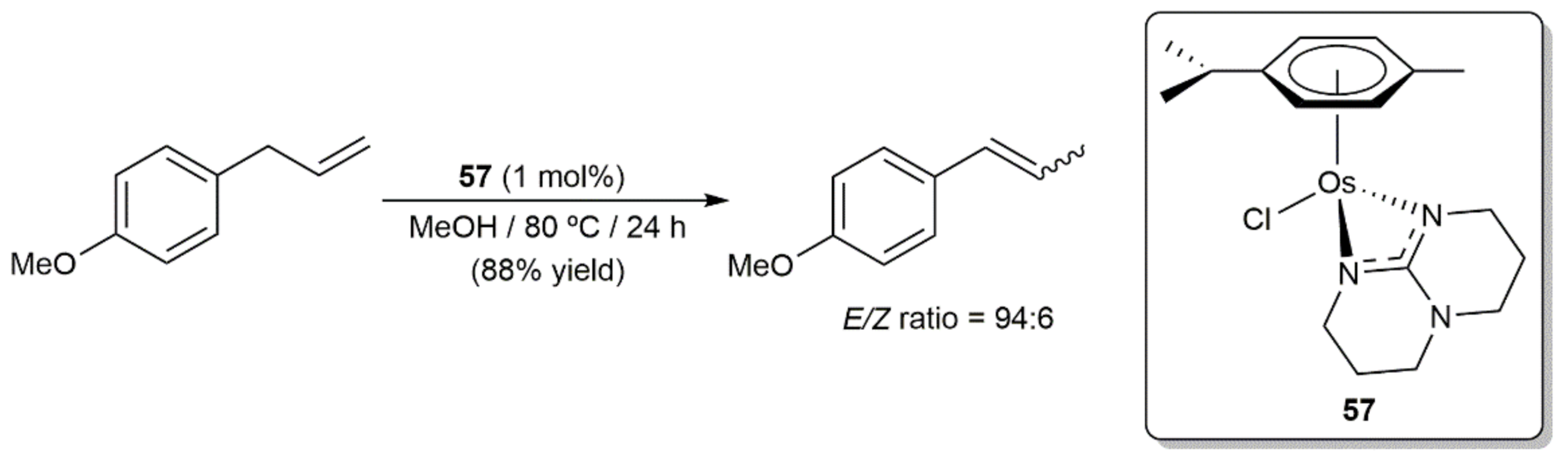
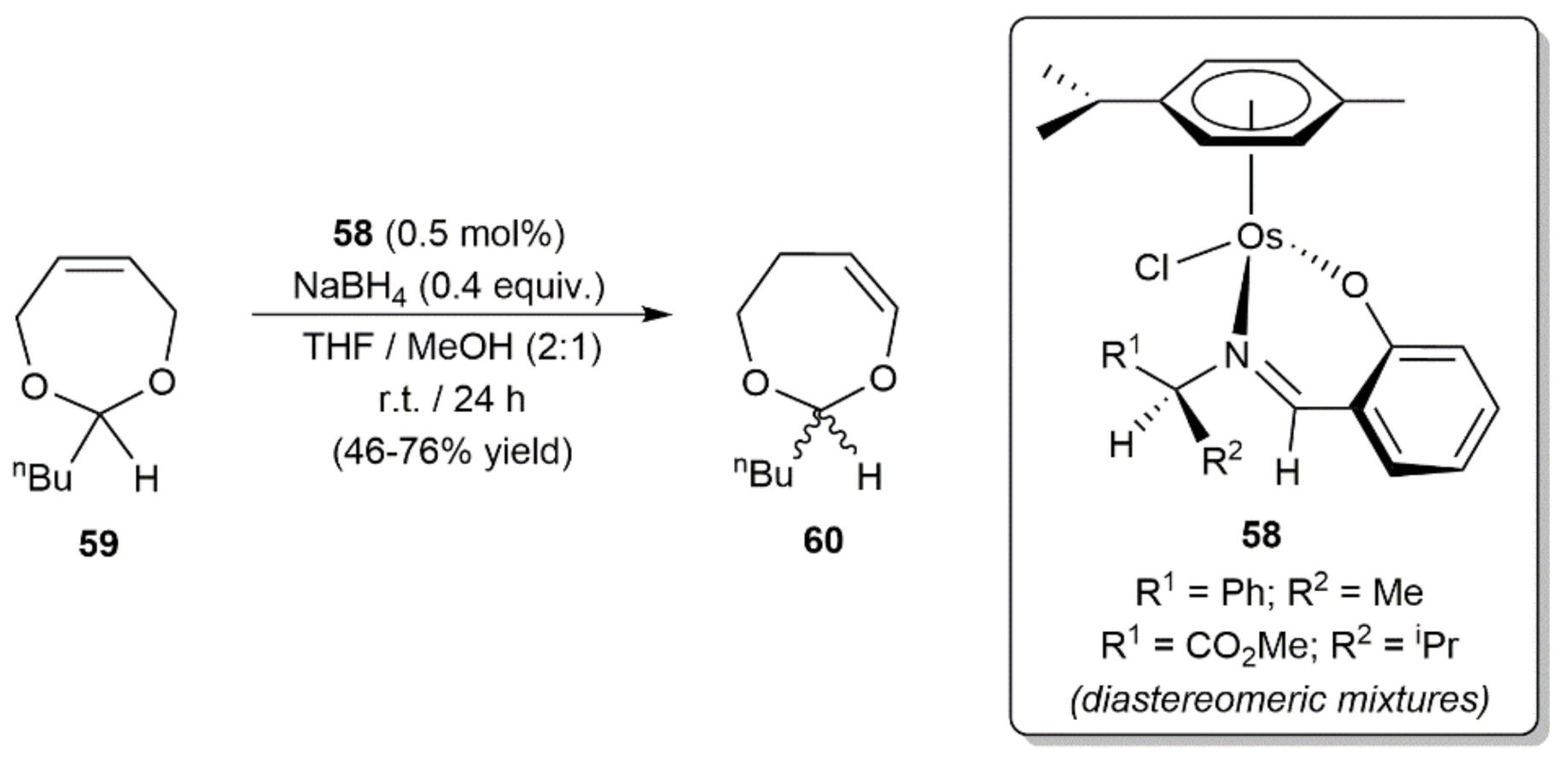
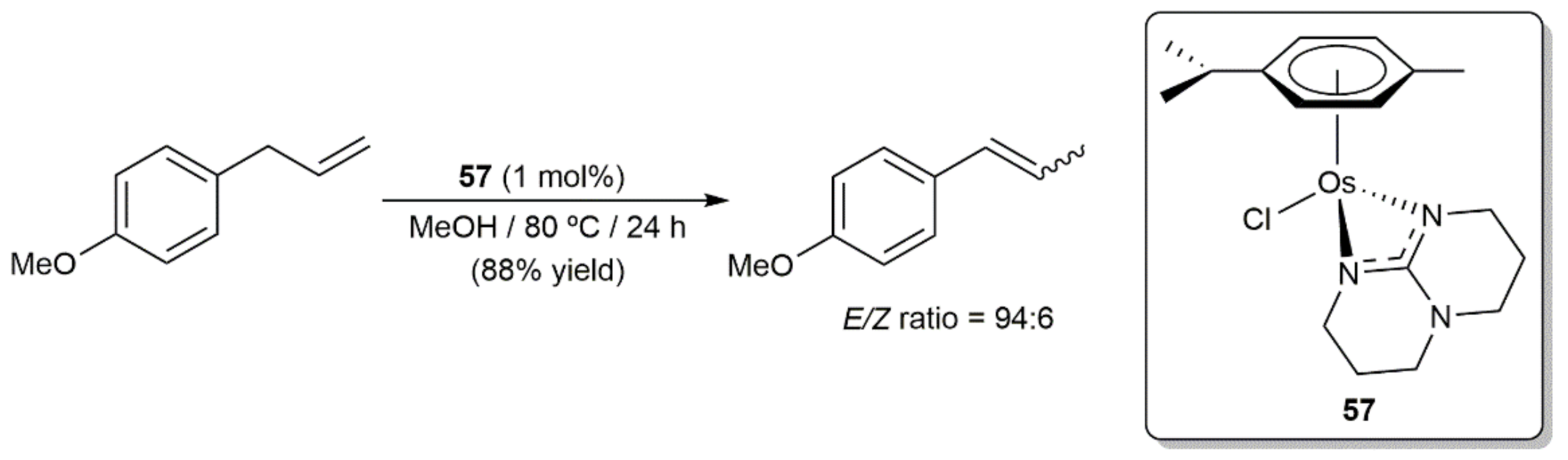
6. Other Catalytic Transformations
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seyferth, D. Bis(benzene)chromium. Its discovery by E.O. Fischer and W. Hafner and subsequent work by the research groups of E.O. Fischer, H.H. Zeiss, F. Hein, C. Elschenbroich, and others. Organometallics 2002, 21, 2800–2820. [Google Scholar] [CrossRef] [Green Version]
- Gastinger, R.G.; Klabunde, K.J. π-Arene complexes of the group VIII transition metals. Transit. Met. Chem. 1979, 4, 1–13. [Google Scholar] [CrossRef]
- Muetterties, E.L.; Bleeke, J.R.; Wucherer, E.J.; Albright, T. Structural, stereochemical, and electronic features of arene-metal complexes. Chem. Rev. 1982, 82, 499–525. [Google Scholar] [CrossRef]
- Kündig, P.E. Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis; Springer: Berlin, Germany, 2004. [Google Scholar]
- Rosillo, M.; Domínguez, G.; Pérez-Castells, J. Chromium arene complexes in organic synthesis. Chem. Soc. Rev. 2007, 36, 1589–1604. [Google Scholar] [CrossRef]
- Pampaloni, G. Aromatic hydrocarbons as ligands. Recent advances in the synthesis, the reactivity and the applications of bis(η6-arene) complexes. Coord. Chem. Rev. 2010, 254, 402–419. [Google Scholar] [CrossRef]
- Takemoto, S.; Matsuzaka, H. Recent topics on catalytic transformations of aromatic molecules via η6 arene transition metal complexes. Tetrahedron Lett. 2018, 59, 697–703. [Google Scholar] [CrossRef]
- Walton, J.W.; Wilkinson, L. π-Coordinated arene metal complexes and catalysis. Organomet. Chem. 2018, 42, 125–171. [Google Scholar] [CrossRef]
- Shvydkiy, N.V.; Perekalin, D.S. Reactions of arene replacement in transition metal complexes. Coord. Chem. Rev. 2020, 411, 213238. [Google Scholar] [CrossRef]
- Winkhaus, G.; Singer, H. Ruthen(II)-komplexe mit zweizähnigem cycloheptatrien und benzol. J. Organomet. Chem. 1967, 7, 487–491. [Google Scholar] [CrossRef]
- Le Bozec, H.; Touchard, D.; Dixneuf, P.H. Organometallic Chemistry of Arene Ruthenium and Osmium Complexes. Adv. Organomet. Chem. 1989, 29, 163–247. [Google Scholar] [CrossRef]
- Bennett, M.A. Recent advances in the chemistry of arene complexes of ruthenium(0) and ruthenium(II). Coord. Chem. Rev. 1997, 166, 225–254. [Google Scholar] [CrossRef]
- Pigge, F.C.; Coniglio, J.J. Stoichiometric applications of η6-arene ruthenium(II) complexes in organic chemistry. Curr. Org. Chem. 2001, 5, 757–784. [Google Scholar] [CrossRef]
- Geldbach, T.J.; Pregosin, P.S. η1 to η6 Ru-arene π complexation: New bonding modes and P-C cleavage chemistry. Eur. J. Inorg. Chem. 2002, 2002, 1907–1918. [Google Scholar] [CrossRef]
- Therrien, B. Functionalised η6-arene ruthenium complexes. Coord. Chem. Rev. 2009, 253, 493–519. [Google Scholar] [CrossRef]
- Süss-Fink, G. Water-soluble arene ruthenium complexes: From serendipity to catalysis and drug design. J. Organomet. Chem. 2014, 751, 2–19. [Google Scholar] [CrossRef]
- Singh, S.K.; Pandey, D.S. Multifaceted half-sandwich arene-ruthenium complexes: Interactions with biomolecules, photoac-tivation, and multinuclearity approach. RSC Adv. 2014, 4, 1819–1840. [Google Scholar] [CrossRef]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 38, 4764–4776. [Google Scholar] [CrossRef]
- Severin, K. Supramolecular chemistry with organometallic half-sandwich complexes. Chem. Commun. 2006, 37, 3859–3867. [Google Scholar] [CrossRef] [PubMed]
- Therrien, B. Arene ruthenium cages: Boxes full of surprises. Eur. J. Inorg. Chem. 2009, 2009, 2445–2453. [Google Scholar] [CrossRef] [Green Version]
- Süss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef]
- Mishra, A.; Kang, S.C.; Chi, K.-W. Coordination-Driven Self-Assembly of Arene-Ruthenium Compounds. Eur. J. Inorg. Chem. 2013, 2013, 5222–5232. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Hartinger, C.; Dyson, P. Opening the lid on piano-stool complexes: An account of ruthenium(II)–arene complexes with medicinal applications. J. Organomet. Chem. 2014, 751, 251–260. [Google Scholar] [CrossRef]
- Singh, A.K.; Pandey, D.S.; Xu, Q.; Braunstein, P. Recent advances in supramolecular and biological aspects of arene ruthe-nium(II) complexes. Coord. Chem. Rev. 2014, 270, 31–56. [Google Scholar] [CrossRef]
- Murray, B.; Babak, M.; Hartinger, C.; Dyson, P. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Therrien, B. Arene Ruthenium Complexes in Supramolecular Chemistry. Med. Chem. 2018, 71, 379–402. [Google Scholar] [CrossRef]
- Zaki, M.; Hairat, S.; Aazam, E.S. Scope of organometallic compounds based on transition metal-arene systems as anticancer agents: Starting from the classical paradigm to targeting multiple strategies. RSC Adv. 2019, 9, 3239–3278. [Google Scholar] [CrossRef] [Green Version]
- Therrien, B. Unmasking Arene Ruthenium Building Blocks. Chem. Rec. 2021, 21, 460–468. [Google Scholar] [CrossRef]
- Delaude, L.; Demonceau, A. Retracing the evolution of monometallic ruthenium–arene catalysts for C–C bond formation. Dalton Trans. 2012, 41, 9257. [Google Scholar] [CrossRef]
- Crochet, P.; Cadierno, V.; Cadierno-Menéndez, V. Arene-ruthenium(ii) complexes with hydrophilic P-donor ligands: Versatile catalysts in aqueous media. Dalton Trans. 2014, 43, 12447. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gupta, R.K.; Pandey, D.S. Half-sandwich arene ruthenium complexes: Synthetic strategies and relevance in catalysis. Chem. Soc. Rev. 2014, 43, 707–733. [Google Scholar] [CrossRef]
- Ackermann, L. Robust ruthenium(II)-catalyzed C-H arylations: Carboxylate assistance for the efficient synthesis of angiotensin-II-receptor blockers. Org. Process Res. Dev. 2015, 19, 260–269. [Google Scholar] [CrossRef]
- Bruneau, C.; Dixneuf, P.H. Ruthenium(II)-catalysed functionalization of C-H bonds with alkenes: Alkenylation versus alkylation. Top. Organomet. Chem. 2016, 55, 137–188. [Google Scholar]
- Manikandan, R.; Jeganmohan, M. Recent advances in the ruthenium(II)-catalyzed chelation-assisted C-H olefination of substituted aromatics, alkenes and heteroaromatics with alkenes via the deprotonation pathway. Chem. Commun. 2017, 53, 8931–8947. [Google Scholar] [CrossRef] [PubMed]
- Gichumbi, J.M.; Friedrich, H.B. Half-sandwich complexes of platinum group metals (Ir, Rh, Ru and Os) and some recent bilogical and catalytic applications. J. Organomet. Chem. 2018, 866, 123–143. [Google Scholar] [CrossRef]
- Adams, R.D.; Selegue, J.P. Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F.G.A., Abel, E.W., Eds.; Pergamon Press: Oxford, UK, 1982; Volume 4, pp. 1020–1022. [Google Scholar]
- Bennett, M.A. Comprehensive Organometallic Chemistry II; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon Press: Oxford, UK, 1995; Volume 7, pp. 556–587. [Google Scholar]
- Gimeno, J.; Cadierno, V.; Crochet, P. Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier Science: Oxford, UK, 2007; Volume 6, pp. 516–540. [Google Scholar]
- Gimeno, J.; Cadierno, V. Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier Science: Oxford, UK, 2007; Volume 6, pp. 600–619. [Google Scholar]
- Sánchez-Delgado, R.A.; Rosales, M.; Esteruelas, M.A.; Oro, L.A. Homogeneous catalysis by osmium complexes: A review. J. Mol. Catal. A Chem. 1995, 96, 231–243. [Google Scholar] [CrossRef]
- Chelucci, G.; Baldino, S.; Baratta, W. Recent Advances in Osmium-Catalyzed Hydrogenation and Dehydrogenation Reactions. Acc. Chem. Res. 2015, 48, 363–379. [Google Scholar] [CrossRef]
- Chelucci, G.; Pinna, G.A.; Pinna, G.; Solinas, M.; Sechi, B. Osmium complexes in catalysis of olefin hydrogenation and isomerization. Chin. J. Catal. 2016, 37, 1824–1836. [Google Scholar] [CrossRef]
- Chelucci, G. Ruthenium and osmium complexes in C-C bond-forming reactions by borrowing hydrogen catalysis. Coord. Chem. Rev. 2017, 331, 1–36. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Seo, C.S.G.; Morris, R.H. Catalytic Homogeneous Asymmetric Hydrogenation: Successes and Opportunities. Organometallics 2019, 38, 47–65. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Dell’Acqua, A.; Tin, S.; de Vries, J.G. Metal-catalysed selective transfer hydrogenation of α,β-unsaturated carbonyl compounds to allylic alcohols. Green Chem. 2020, 22, 3323–3357. [Google Scholar] [CrossRef]
- Blacker, A.J. The Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp. 1215–1244. [Google Scholar]
- Ikariya, T.; Blacker, A.J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid−Triethylamine Mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Accounts Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
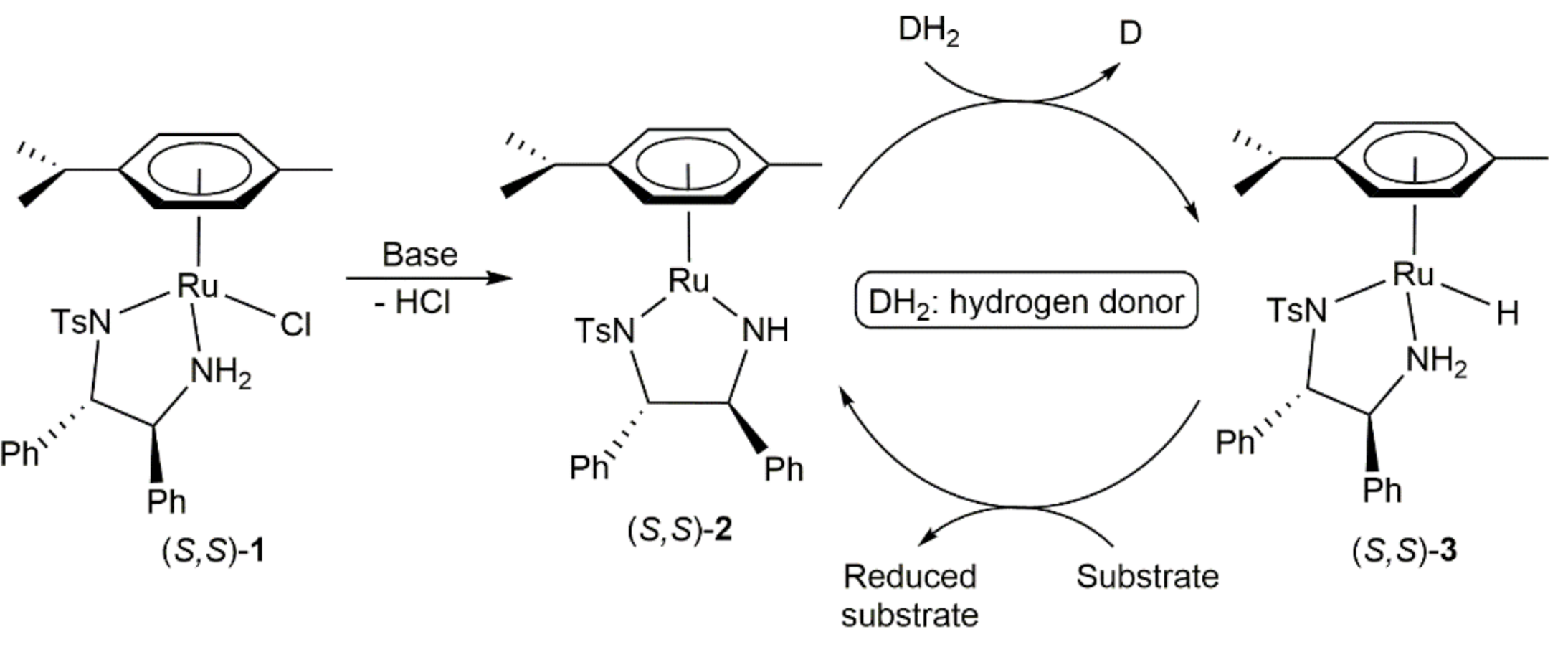
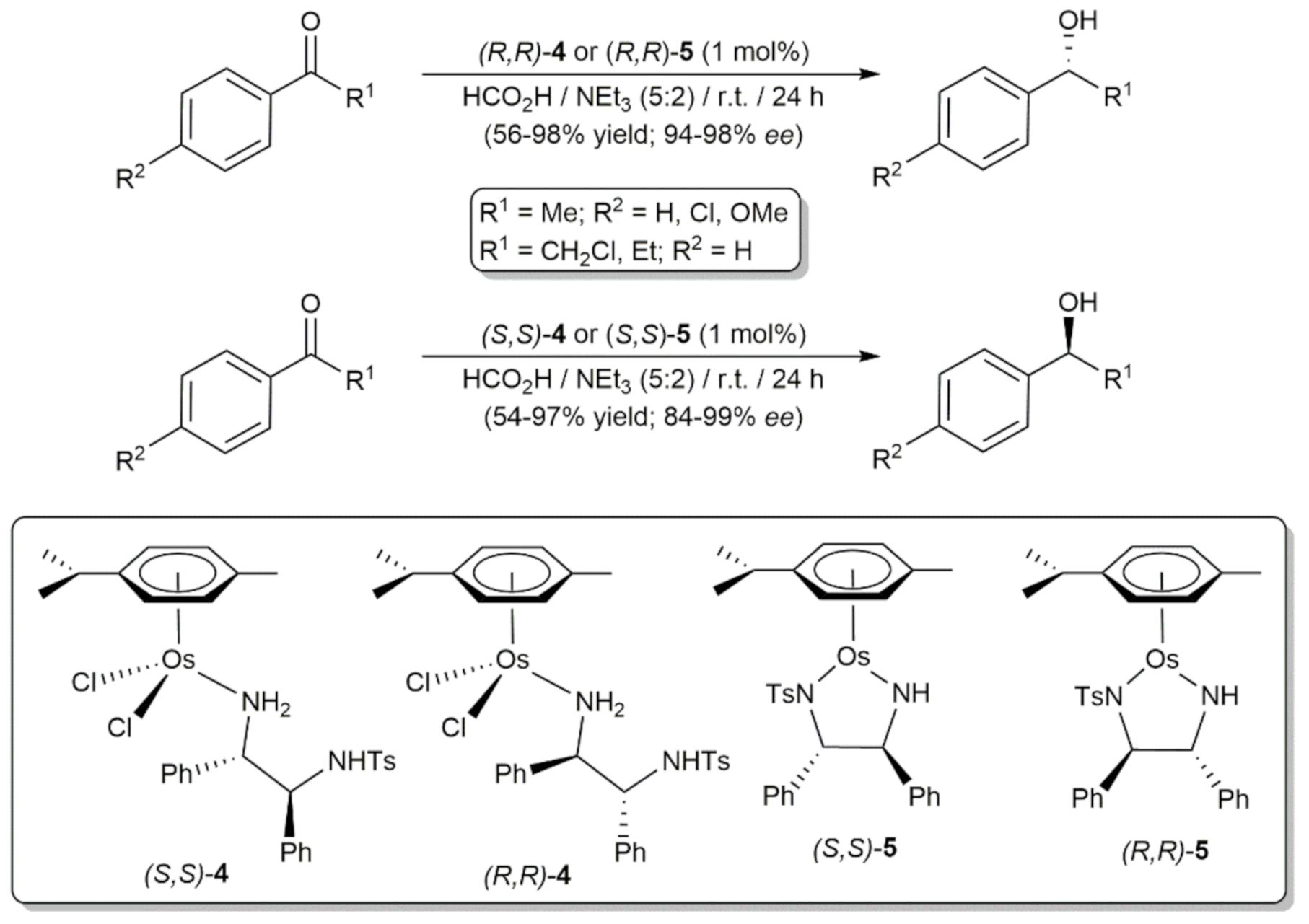
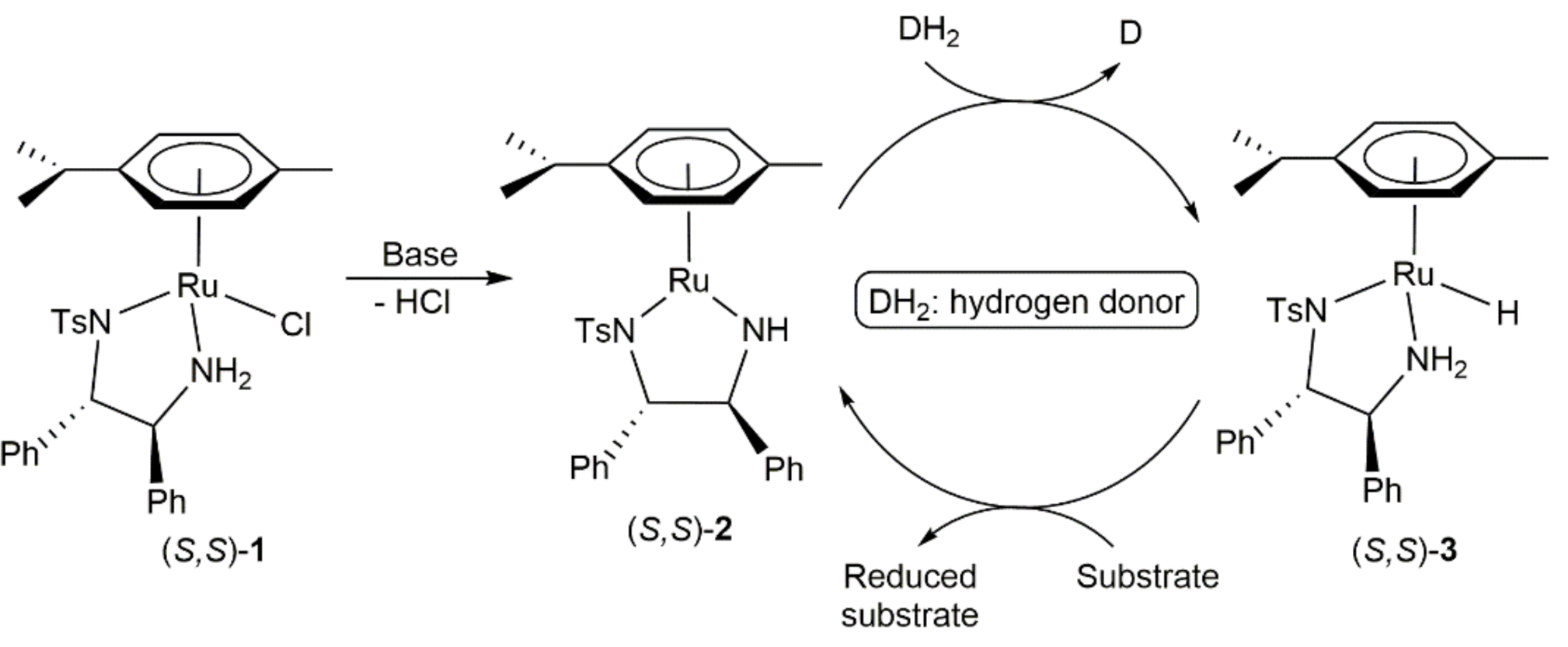
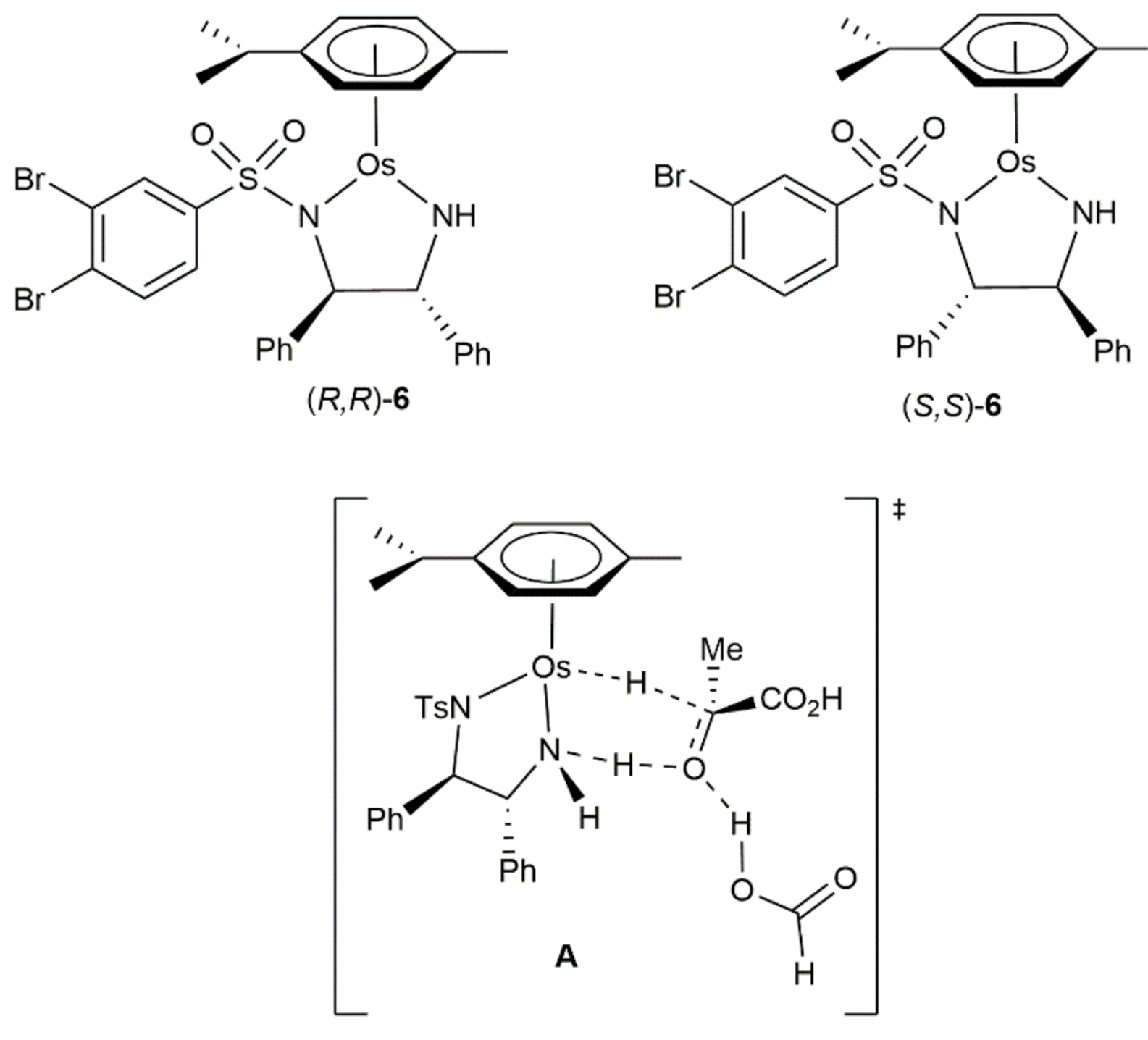
- Coverdale, J.; Sanchez-Cano, C.; Clarkson, G.J.; Soni, R.; Wills, M.; Sadler, P.J. Easy To Synthesize, Robust Organo-osmium Asymmetric Transfer Hydrogenation Catalysts. Chem. A Eur. J. 2015, 21, 8043–8046. [Google Scholar] [CrossRef] [Green Version]
- Coverdale, J.P.C.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G.J.; Habtemariam, A.; Wills, M.; Sadler, P.J. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347–354. [Google Scholar] [CrossRef]
- Bolitho, E.M.; Coverdale, J.P.C.; Bridgewater, H.E.; Clarkson, G.J.; Quinn, P.D.; Sanchez-Cano, C.; Sadler, P.J. Tracking reactions of asymmetric organo-osmium transfer hydrogenation catalysts in cancer cells. Angew. Chem. Int. Ed. 2021, 60, 6462–6472. [Google Scholar] [CrossRef]
- Ngo, A.H.; Do, L.H. Structure–activity relationship study of half-sandwich metal complexes in aqueous transfer hydrogenation catalysis. Inorg. Chem. Front. 2020, 7, 583–591. [Google Scholar] [CrossRef]
- Infante-Tadeo, S.; Rodríguez-Fanjul, V.; Habtemariam, A.; Pizarro, A.M. Osmium(II) tethered half-sandwich complexes: pH-dependent aqueous speciation and transfer hydrogenation in cells. Chem. Sci. 2021, in press. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X. Mechanistic insights into asymmetric transfer hydrogenation of pyruvic acid catalysed by chiral osmium complexes with formic assisted proton transfer. Chem. Commun. 2019, 55, 9633–9636. [Google Scholar] [CrossRef]
- Faller, J.W.; Lavoie, A.R. Enantioselective Routes to Both Enantiomers of Aryl Alcohols with a Single Catalyst Antipode: Ru and Os Transfer Hydrogenation Catalysts. Org. Lett. 2001, 3, 3703–3706. [Google Scholar] [CrossRef]
- Palmer, M.; Walsgrove, A.T.; Wills, M. (1R,2S)-(+)-cis-1-Amino-2-indanol: An Effective Ligand for Asymmetric Catalysis of Transfer Hydrogenations of Ketones. J. Org. Chem. 1997, 62, 5226–5228. [Google Scholar] [CrossRef]
- Faller, J.W.; Lavoie, A.R. Enantioselective Syntheses of Nonracemic Benzyl-α-dAlcohols via Catalytic Transfer-Hydrogenation with Ru, Os, Rh, and Ir Catalysts. Organometallics 2002, 21, 3493–3495. [Google Scholar] [CrossRef]
- Carmona, D.; Lamata, M.P.; Viguri, F.; Dobrinovich, I.; Lahoz, F.J.; Oro, L.A. On the Sense of the Enantioselection in Hydrogen Transfer Reactions from 2-Propanol to Ketones. Adv. Synth. Catal. 2002, 344, 499–502. [Google Scholar] [CrossRef]
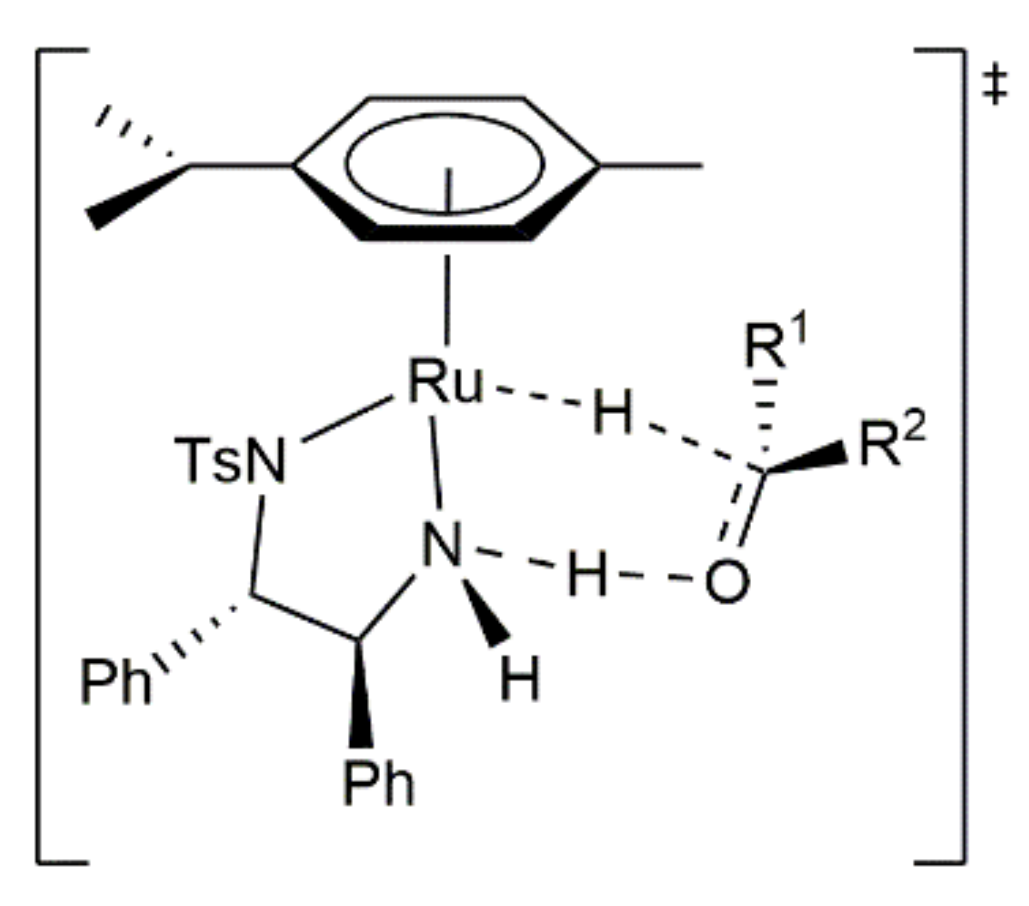
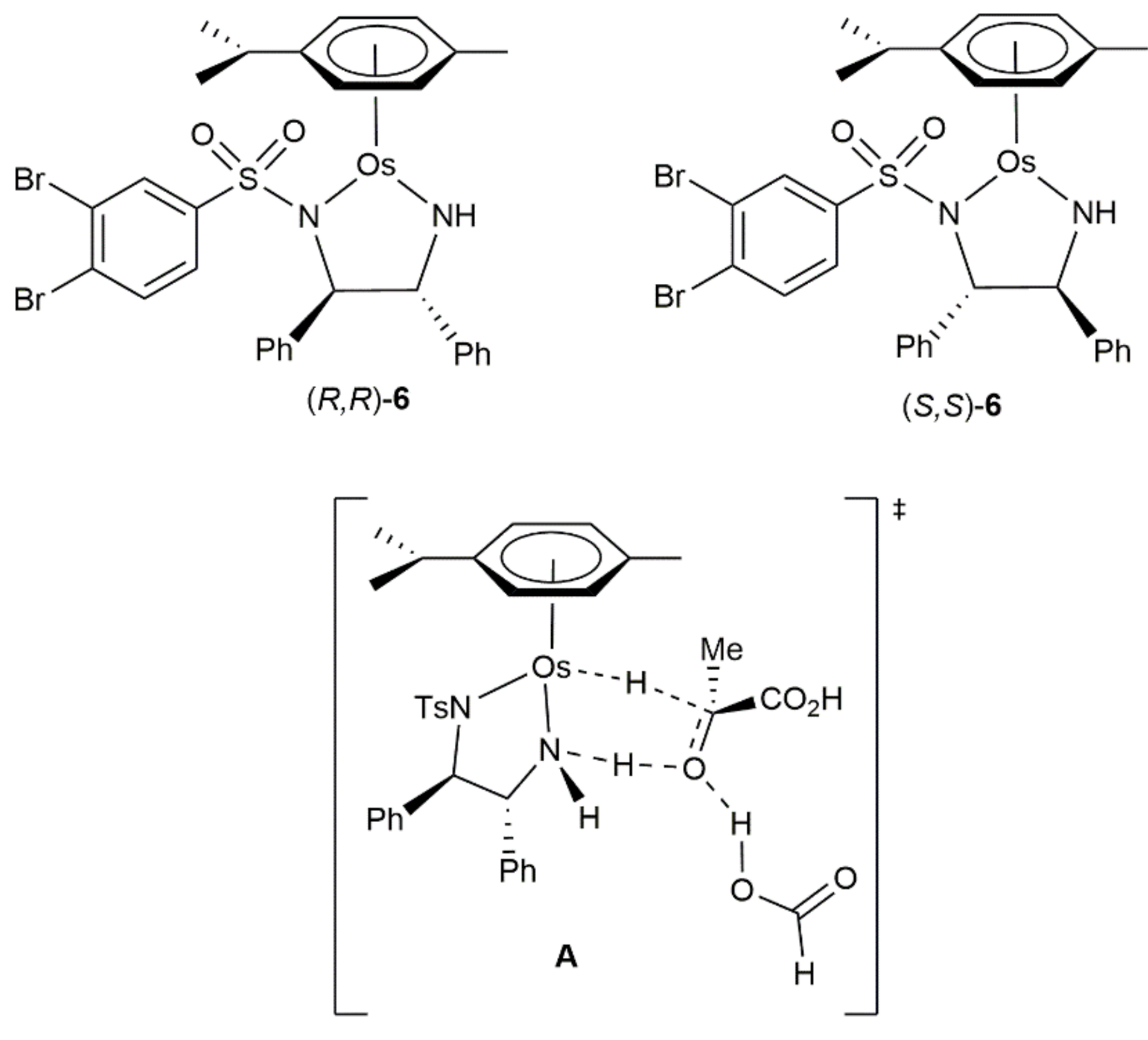
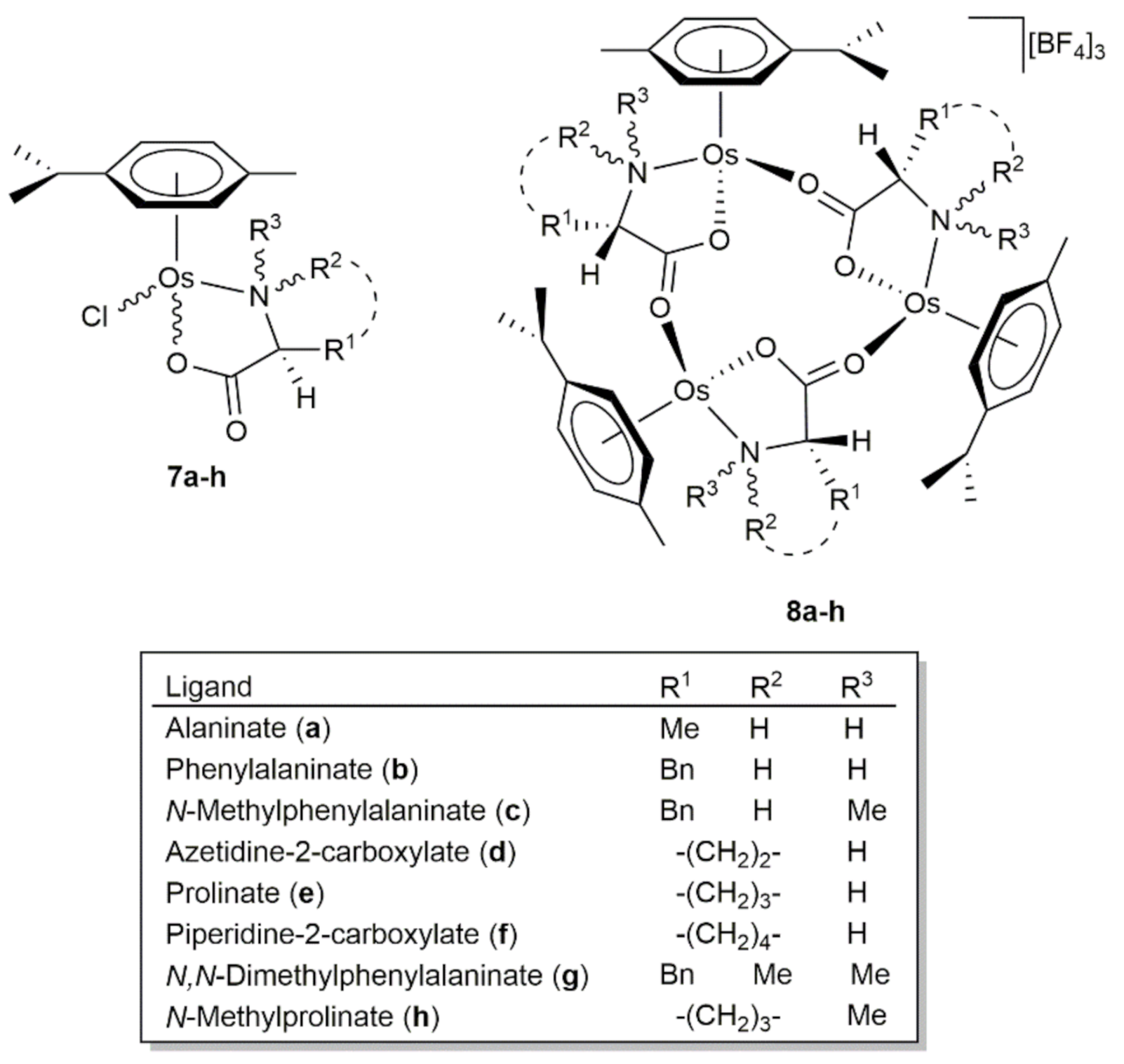
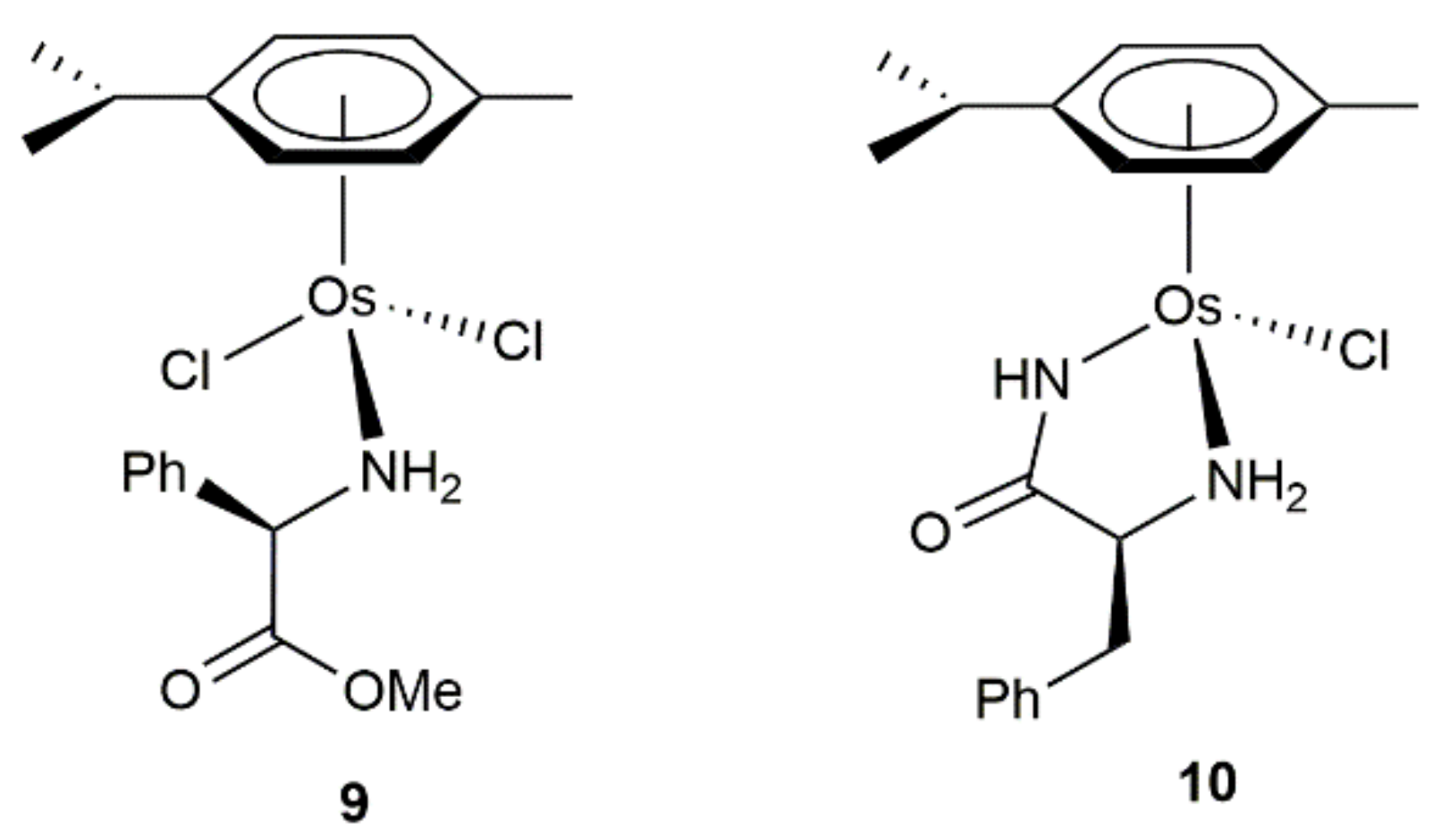
- Carmona, D.; Lahoz, F.J.; García-Orduña, P.; Oro, L.A.; Lamata, M.P.; Viguri, F. Half-sandwich complexes of osmium(II) with l-α-amino carboxylate ligands as asymmetric transfer hydrogenation catalysts. On the origin of the enantioselectivity. Organometallics 2012, 31, 3333–3345. [Google Scholar] [CrossRef]
- Sarfraz, R.A.; Kazi, T.G.; Iqbal, S.; Afridi, H.I.; Jamali, M.K.; Jalbani, N.; Arain, M.B. Synthesis, structure determination and chemoselective catalytic studies of amino acids complexes of osmium(II). Appl. Organomet. Chem. 2008, 22, 187–192. [Google Scholar] [CrossRef]
- Fu, Y.; Soni, R.; Romero, M.J.; Pizarro, A.M.; Salassa, L.; Clarkson, G.J.; Hearn, J.M.; Habtemariam, A.; Wills, M.; Sadler, P.J. Mirror-Image Organometallic Osmium Arene Iminopyridine Halido Complexes Exhibit Similar Potent Anticancer Activity. Chem. A Eur. J. 2013, 19, 15199–15209. [Google Scholar] [CrossRef] [Green Version]
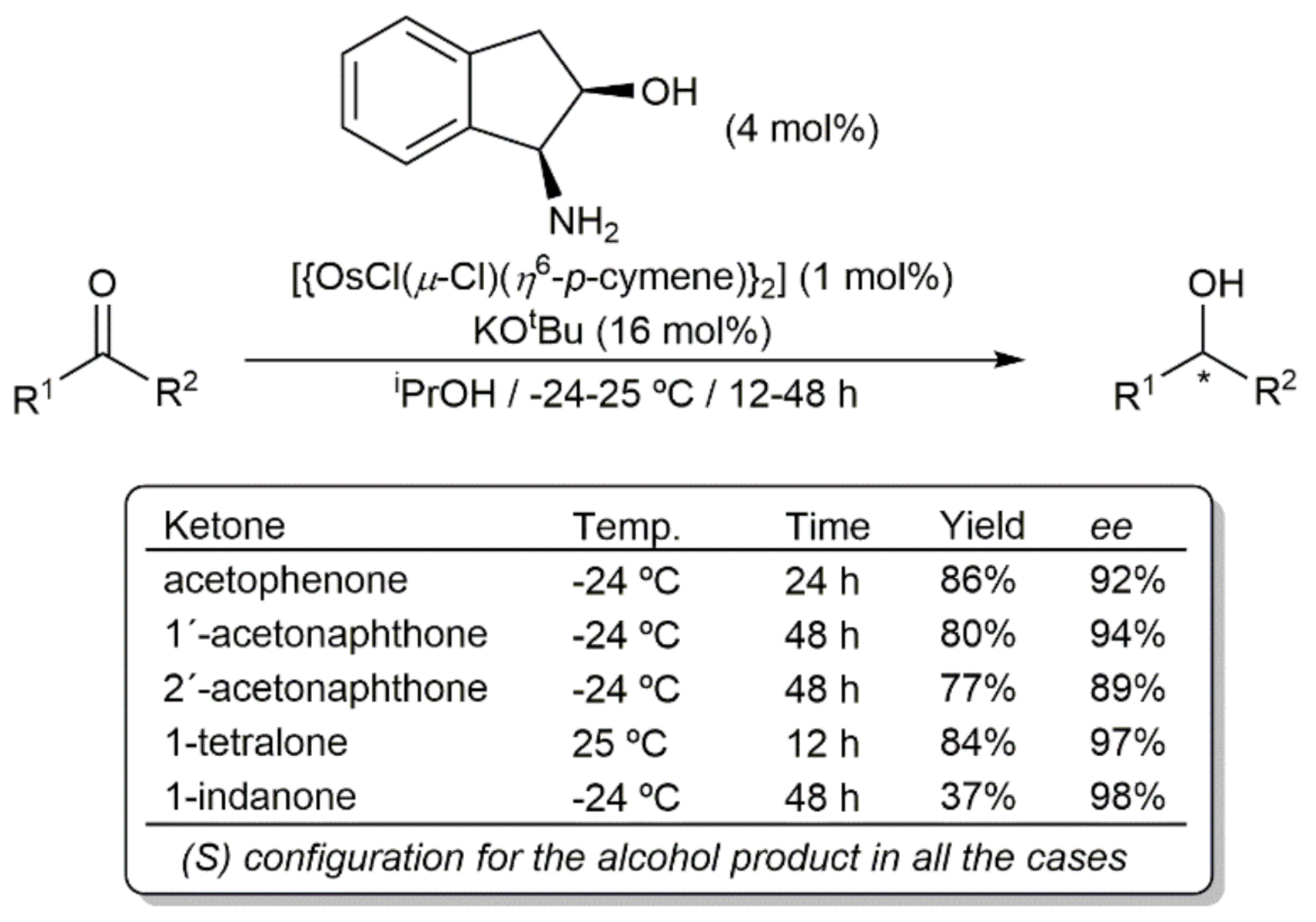
- Gichumbi, J.; Omondi, B.; Friedrich, H. Half-Sandwich Osmium(II) Complexes with Bidentate N,N-Chelating Ligands and Their Use in the Transfer Hydrogenation of Ketones. Eur. J. Inorg. Chem. 2017, 2017, 915–924. [Google Scholar] [CrossRef]
- Müller, A.L.; Bleith, T.; Roth, T.; Wadepohl, H.; Gade, L.H. Iridium Half-Sandwich Complexes with Di- and Tridentate Bis(pyridylimino)isoindolato Ligands: Stoichiometric and Catalytic Reactivity. Organometallics 2014, 34, 2326–2342. [Google Scholar] [CrossRef]
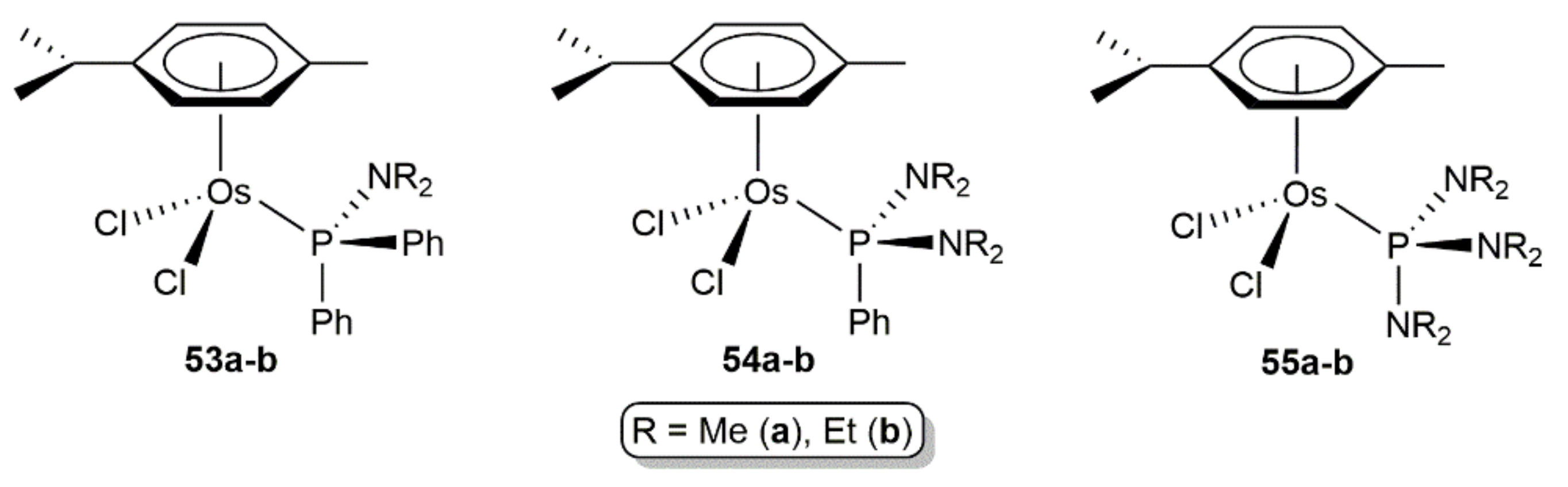
- Schreiber, D.F.; O.´Connor, C.; Grave, C.; Müller-Bunz, H.; Scopelliti, R.; Dyson, P.J.; Phillips, A.D. Synthesis, characterization, and reactivity of the first osmium β-diketiminato complexes and application in catalysis. Organometallics 2013, 32, 7345–7356. [Google Scholar] [CrossRef]
- Albertin, G.; Antoniutti, S.; Castro, J.; Paganelli, S. Preparation and reactivity of p-cymene complexes of ruthenium and osmium incorporating 1,3-triazenide ligands. J. Organomet. Chem. 2010, 695, 2142–2152. [Google Scholar] [CrossRef]
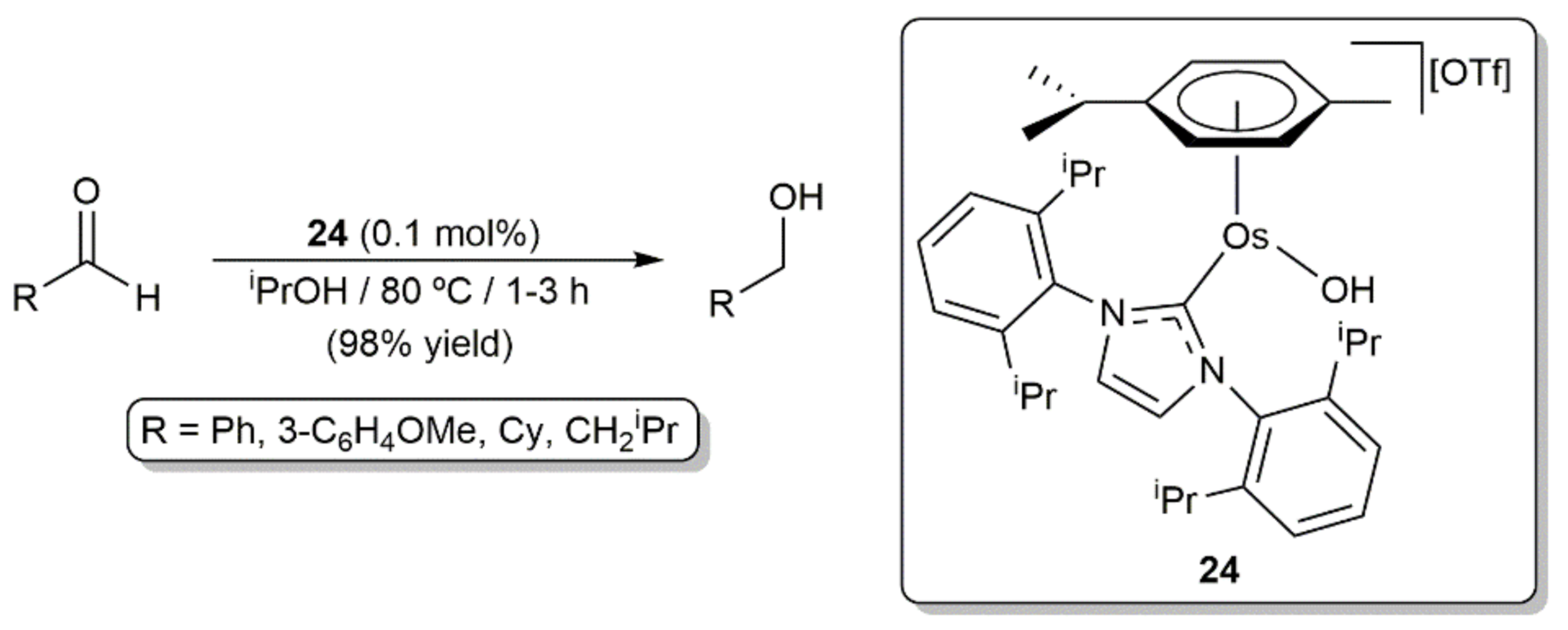
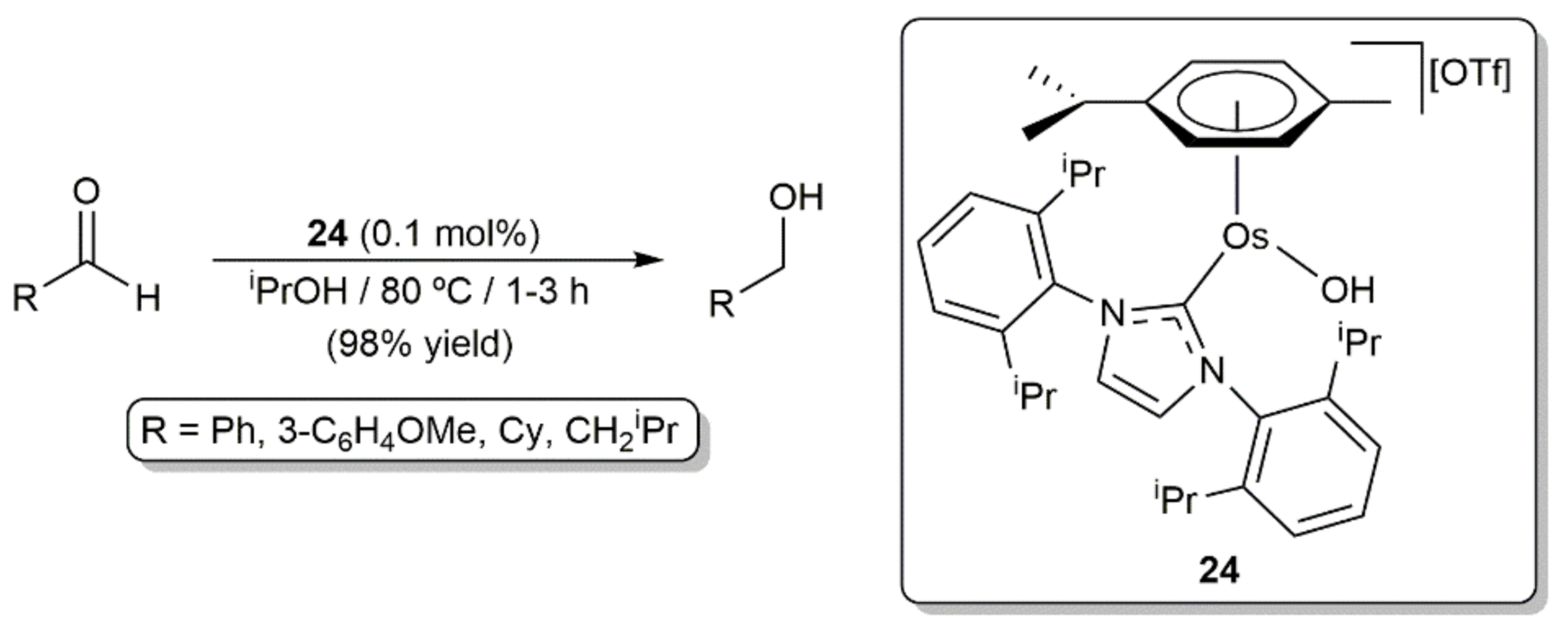
- Castarlenas, R.; Esteruelas, M.A.; Oñate, E. Preparation, X-ray structure, and reactivity of an osmium-hydroxo complex stabi-lized by an N-heterocyclic carbene ligand: A base-free catalytic precursor for hydrogen transfer from 2-propanol to aldehydes. Organometallics 2008, 27, 3240–3247. [Google Scholar] [CrossRef]
- Wylie, W.N.; Lough, A.J.; Morris, R.H. Mechanistic Investigation of the Hydrogenation of Ketones Catalyzed by a Ruthenium(II) Complex Featuring an N-Heterocyclic Carbene with a Tethered Primary Amine Donor: Evidence for an Inner Sphere Mechanism. Organometallics 2011, 30, 1236–1252. [Google Scholar] [CrossRef]
- Castañón, E.B.; Kaposi, M.; Reich, R.M.; Kühn, F.E. Water-soluble transition metal complexes of ruthenium(II), osmium(II), rhodium(III) and iridium(III) with chelating N-heterocyclic carbene ligands in hydrogenation and transfer hydrogenation catalysis. Dalton Trans. 2018, 47, 2318–2329. [Google Scholar] [CrossRef] [PubMed]
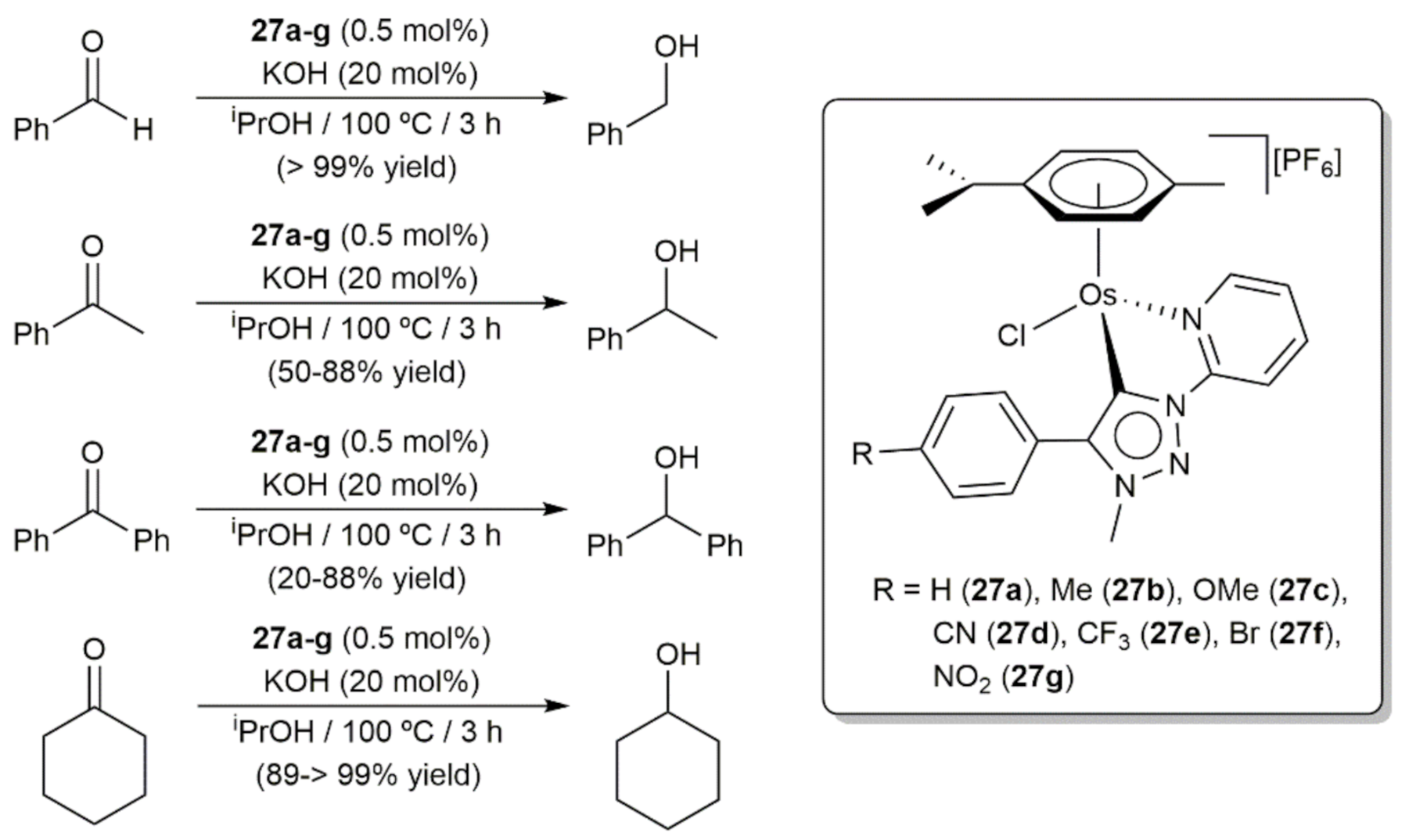
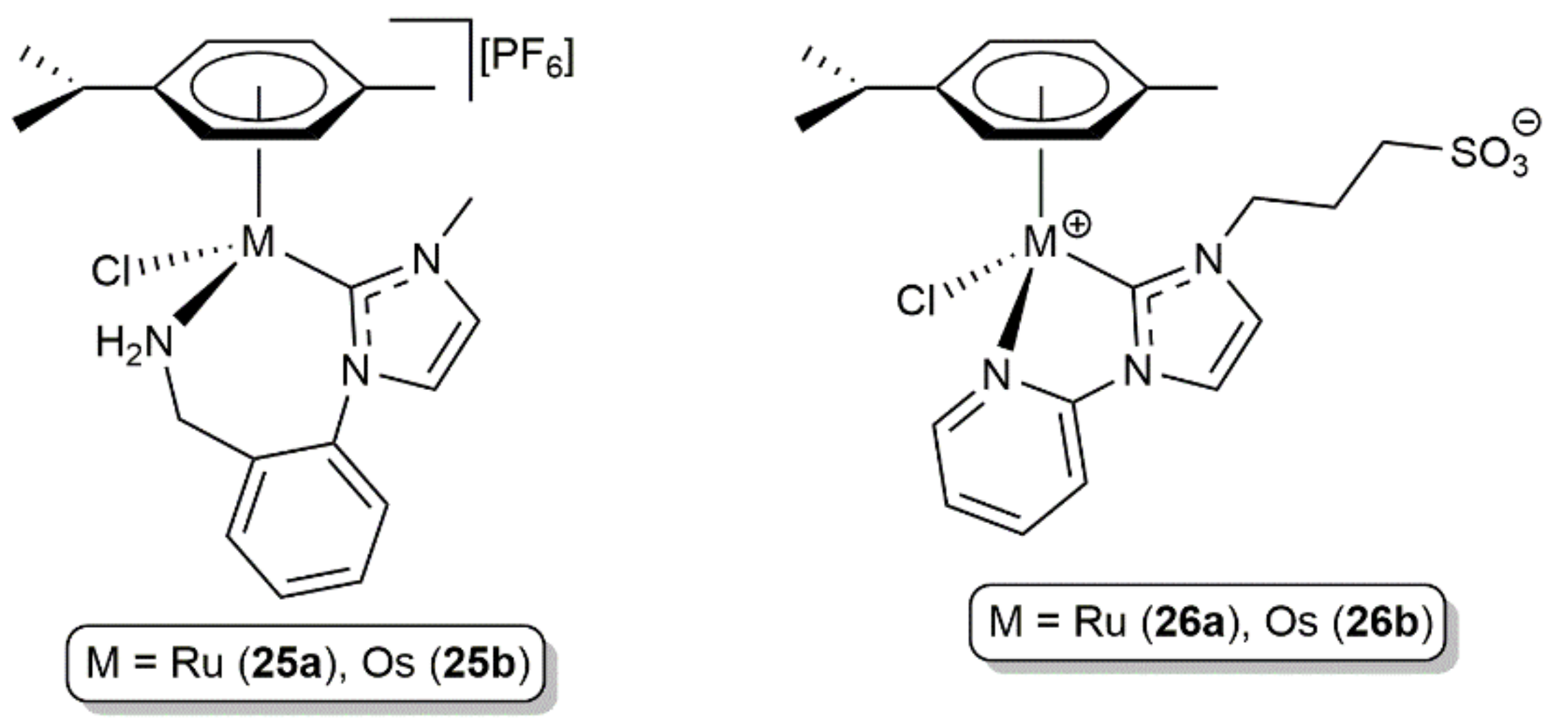
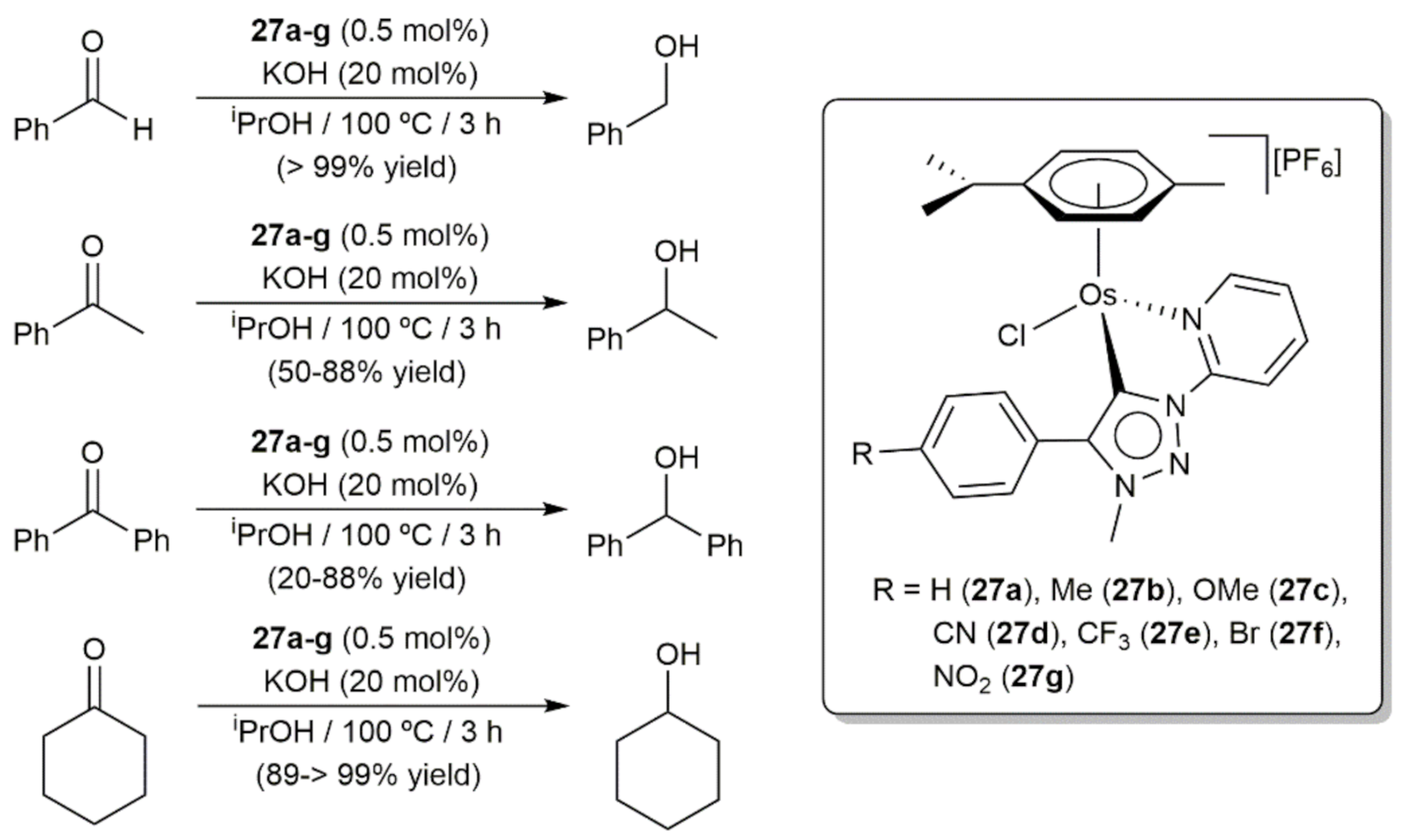
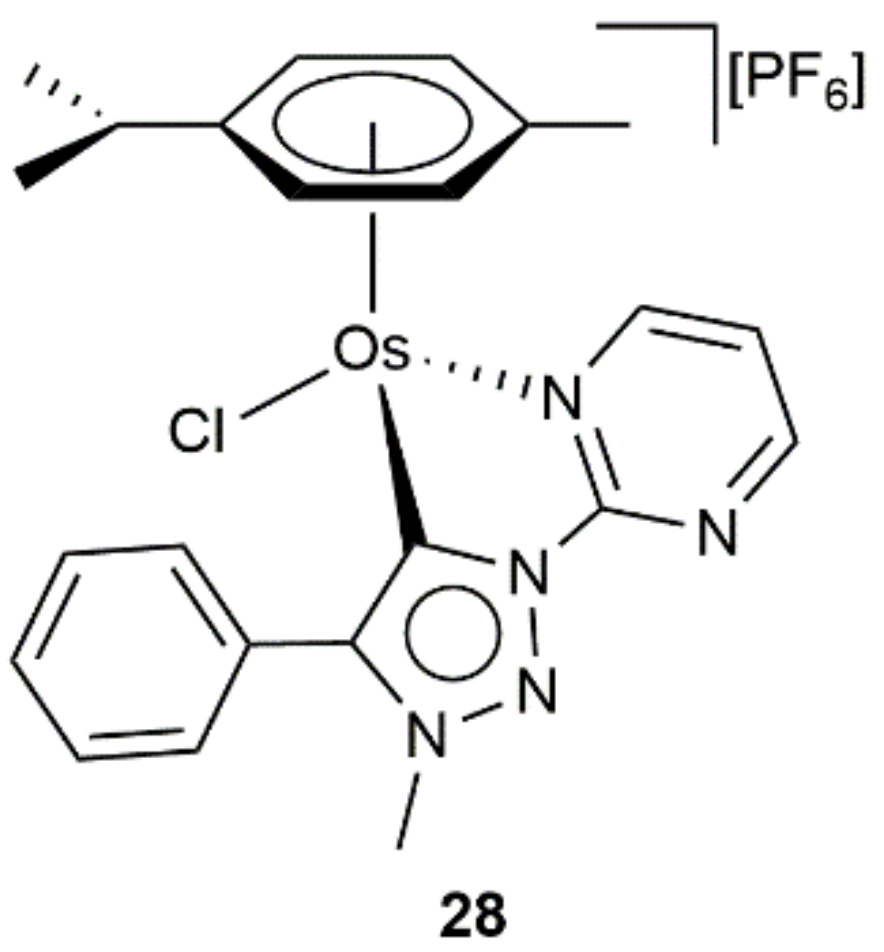
- Bolje, A.; Hohloch, S.; van der Meer, M.; Košmrlj, J.; Sarkar, B. RuII, OsII, and IrIII complexes with chelating pyridyl-mesoionic carbene ligands: Structural characterization and applications in transfer hydrogenation catalysis. Chem. Eur. J. 2015, 21, 6756–6764. [Google Scholar] [CrossRef]
- Bolje, A.; Hohloch, S.; Košmrlj, J.; Sarkar, B. RuII, IrIII and OsII mesoionic carbene complexes: Efficient catalysts for transfer hydrogenation of selected functionalities. Dalton Trans. 2016, 45, 15983–15993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Muñiz, K. The development of asymmetric deamination of alkenes with imido-osmium reagents. New J. Chem. 2005, 29, 1371–1385. [Google Scholar] [CrossRef]
- Christie, S.D.R.; Warrington, A.D. Osmium and palladium: Complementary metals in alkene activation and oxidation. Synthesis 2008, 1325–1341. [Google Scholar] [CrossRef]
- Pilgrim, B.S.; Donohoe, T.J. Osmium-Catalyzed Oxidative Cyclization of Dienes and Their Derivatives. J. Org. Chem. 2013, 78, 2149–2167. [Google Scholar] [CrossRef]
- Shul´pin, G.B.; Vinogradov, M.M.; Shul´pina, L.S. Oxidative functionalization of C-H compounds induced by extremely effi-cient osmium catalysts. Catal. Sci. Technol. 2018, 8, 4287–4313. [Google Scholar] [CrossRef]
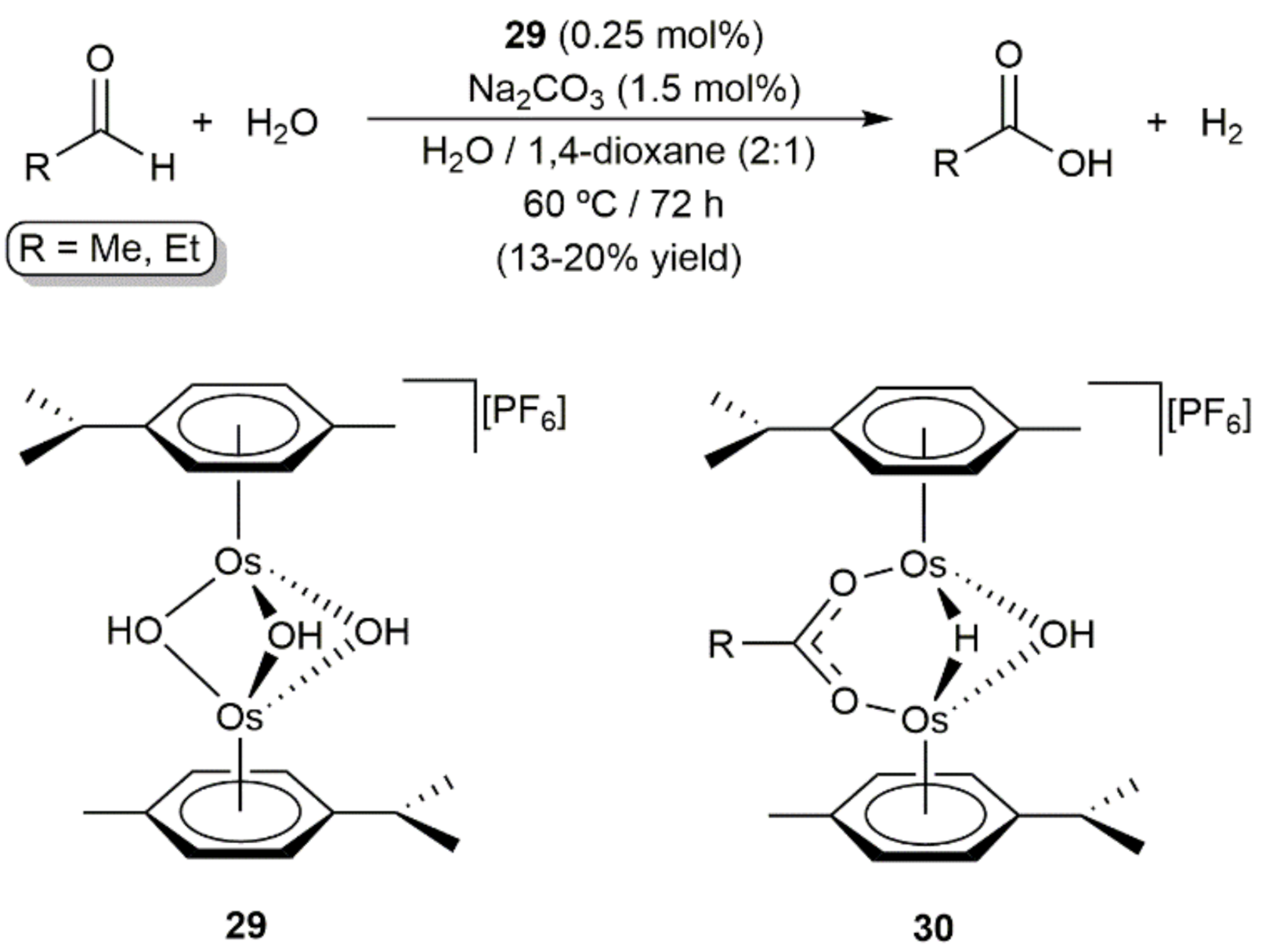
- Cabeza, J.A.; Smith, A.J.; Adams, H.; Maitlis, P.M. The reactions of the tri-μ-hydroxo-bis[η6-p-cymeneosmium(II)] cation with aldehydes and acids and the homogeneously catalysed oxidation of acetaldehyde and propionaldehyde with water. X-ray structure of [(p-MeC6H4CHMe2)2Os2(μ-HCO2)(μ-OH)(μ-H)]][PF6]. J. Chem. Soc. Dalton Trans. 1986, 17, 1155–1160. [Google Scholar] [CrossRef]
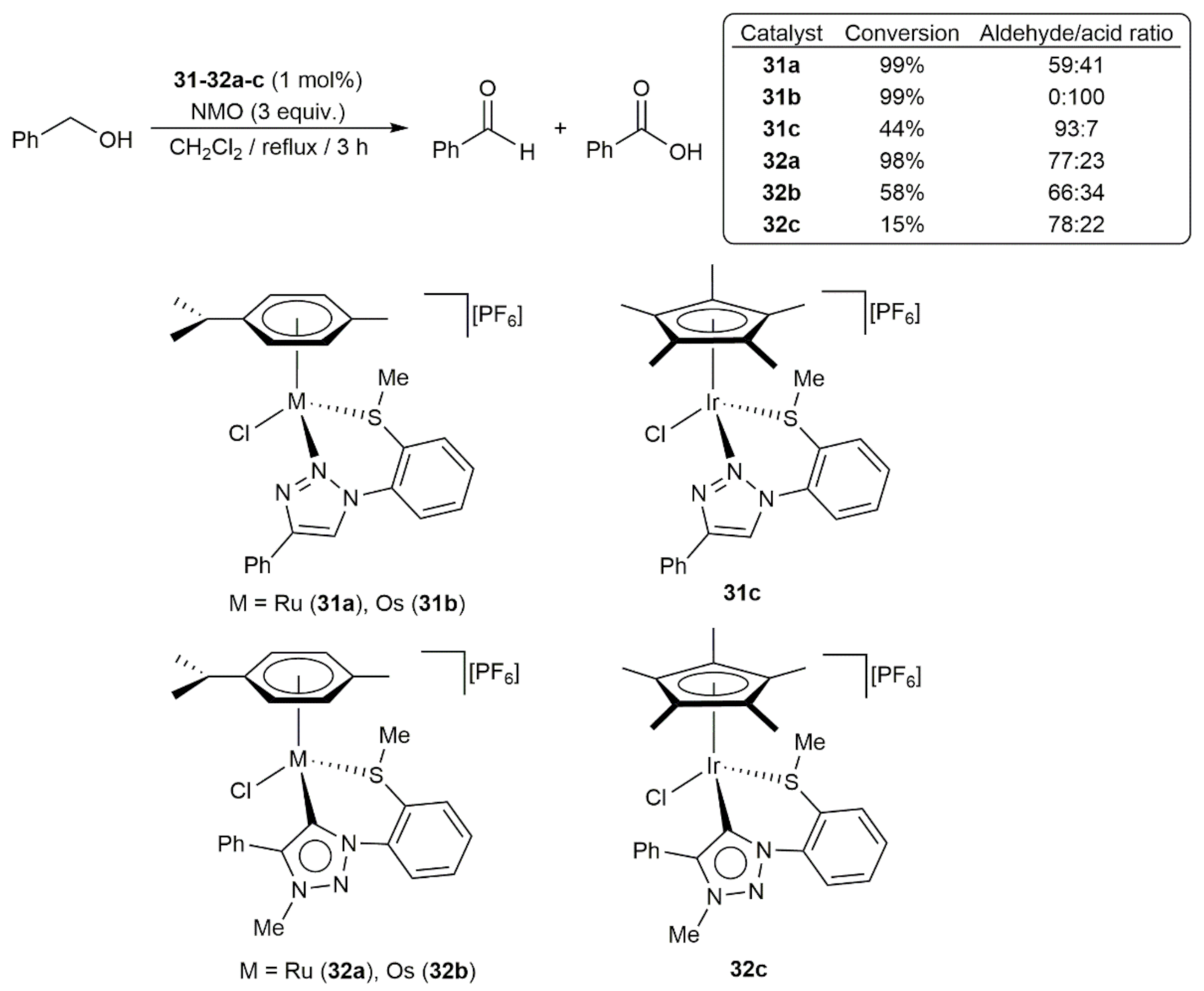
- Hohloch, S.; Hettmanczyk, L.; Sarkar, B. Introducing Potential Hemilability into “Click” Triazoles and Triazolylidenes: Synthesis and Characterization of d6-Metal Complexes and Oxidation Catalysis. Eur. J. Inorg. Chem. 2014, 2014, 3164–3171. [Google Scholar] [CrossRef]
- Gichumbi, J.; Omondi, B.; Friedrich, H.B. Oxidation of olefins catalyzed by half-sandwich osmium(II) arene complexes. J. Organomet. Chem. 2018, 856, 56–62. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Nesterov, D.S.; Shul´pina, L.S.; Pombeiro, A.J.L.; Shul´pin, G.B. Oxidation of hydrocarbons with H2O2/O2 catalyzed by osmium complexes containing p-cymene ligands in acetonitrile. Catal. Sci. Technol. 2014, 4, 3214–3226. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Shul´pina, L.S.; Kozlov, Y.N.; Kudinov, A.R.; Ikonnikov, N.S.; Shul´pin, G.B. Oxidation of hydrocarbons and alcohols with peroxides catalyzed by new π-cymene osmium complexes. J. Organomet. Chem. 2015, 784, 52–61. [Google Scholar] [CrossRef]
- Demonceau, A.; Lemoine, C.; Noels, A. Osmium-catalysed cyclopropanation of olefins. Tetrahedron Lett. 1996, 37, 1025–1026. [Google Scholar] [CrossRef]
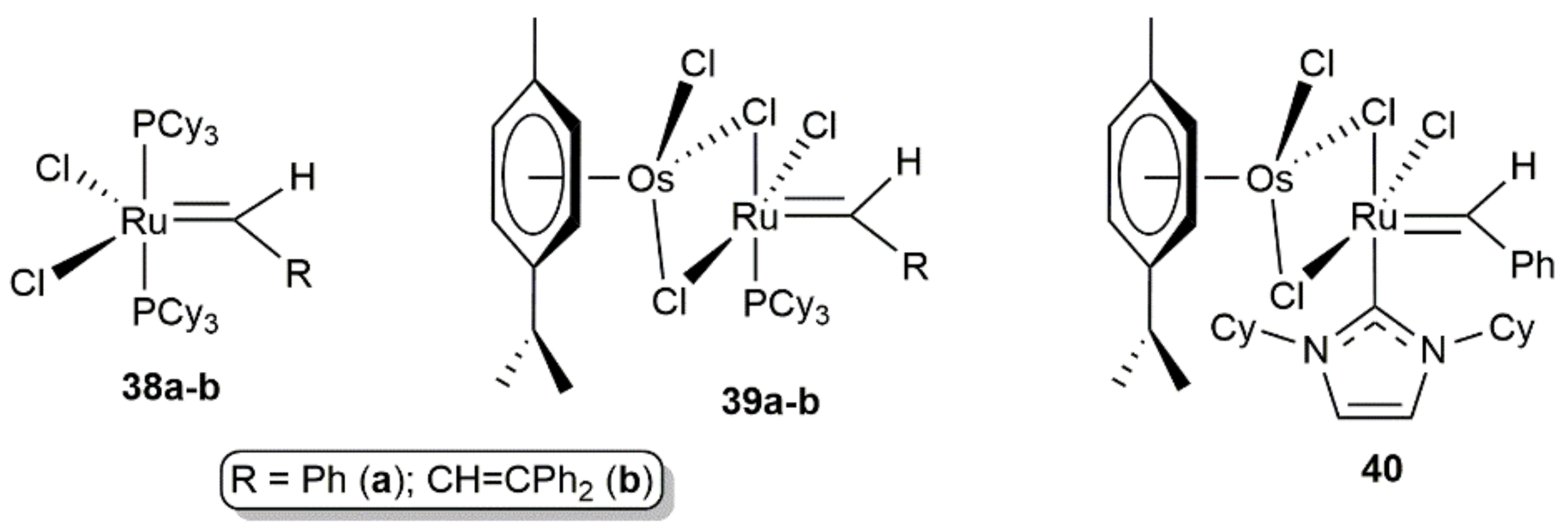
- Cui, M.; Guo, X.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Halide Effects on the Stability of Osmium Indenylidene Complexes: Isolation, Characterization, and Reactivities. Organometallics 2020, 39, 2142–2151. [Google Scholar] [CrossRef]
- Faller, J.; Parr, J. Diastereoselectivity in Chiral Osmium Complexes of a Bidentate Bisphosphine Monoxide Ligand. Organometallics 2000, 19, 3556–3561. [Google Scholar] [CrossRef]
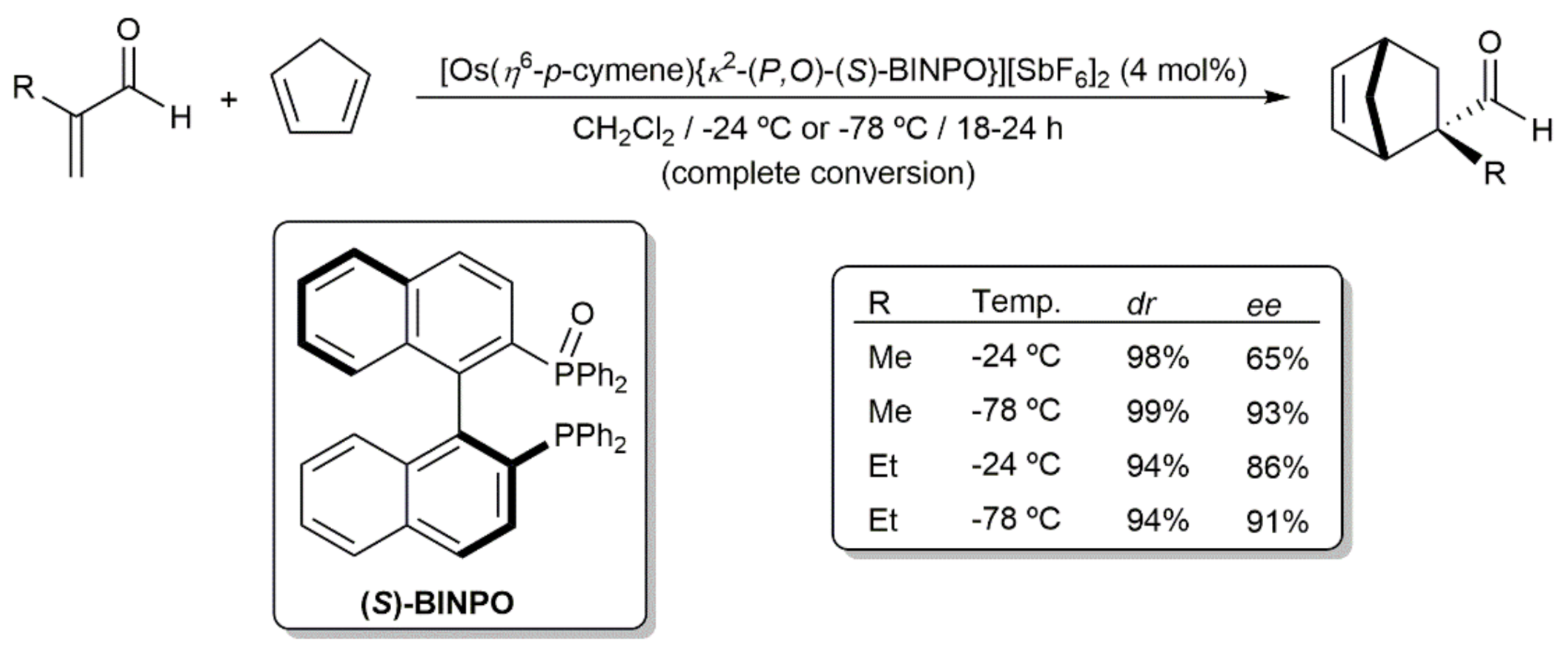
- Faller, J.W.; Parr, J. Utility of Osmium(II) in the Catalysis of Asymmetric Diels−Alder Reactions. Organometallics 2001, 20, 697–699. [Google Scholar] [CrossRef]
- Faller, J.W.; Parr, J. Synthesis, Characterization, Diastereoselectivity, and Catalytic Activity of Complexes of Ruthenium with BINAP Monoxide. Organometallics 2000, 19, 1829–1832. [Google Scholar] [CrossRef]
- Carmona, D.; Vega, C.; García, N.; Lahoz, F.J.; Elipe, S.; Oro, L.A.; Lamata, M.P.; Viguri, F.; Borao, R. Chiral phosphinooxa-zoline-ruthenium(II) and –osmium(II) complexes as catalysts in Diels-Alder reactions. Organometallics 2006, 23, 1592–1606. [Google Scholar] [CrossRef]
- Suzuki, T.; Torii, T. Catalytic asymmetric Michael reactions using a chiral rhodium complex. Tetrahedron Asymmetry 2001, 12, 1077–1081. [Google Scholar] [CrossRef]
- Hafner, A.; Mühlebach, A.; van der Schaaf, P.A. One-component catalysts for thermal and photoinduced ring opening metathesis polymerization. Angew. Chem. Int. Ed. Engl. 1997, 36, 2121–2124. [Google Scholar] [CrossRef]
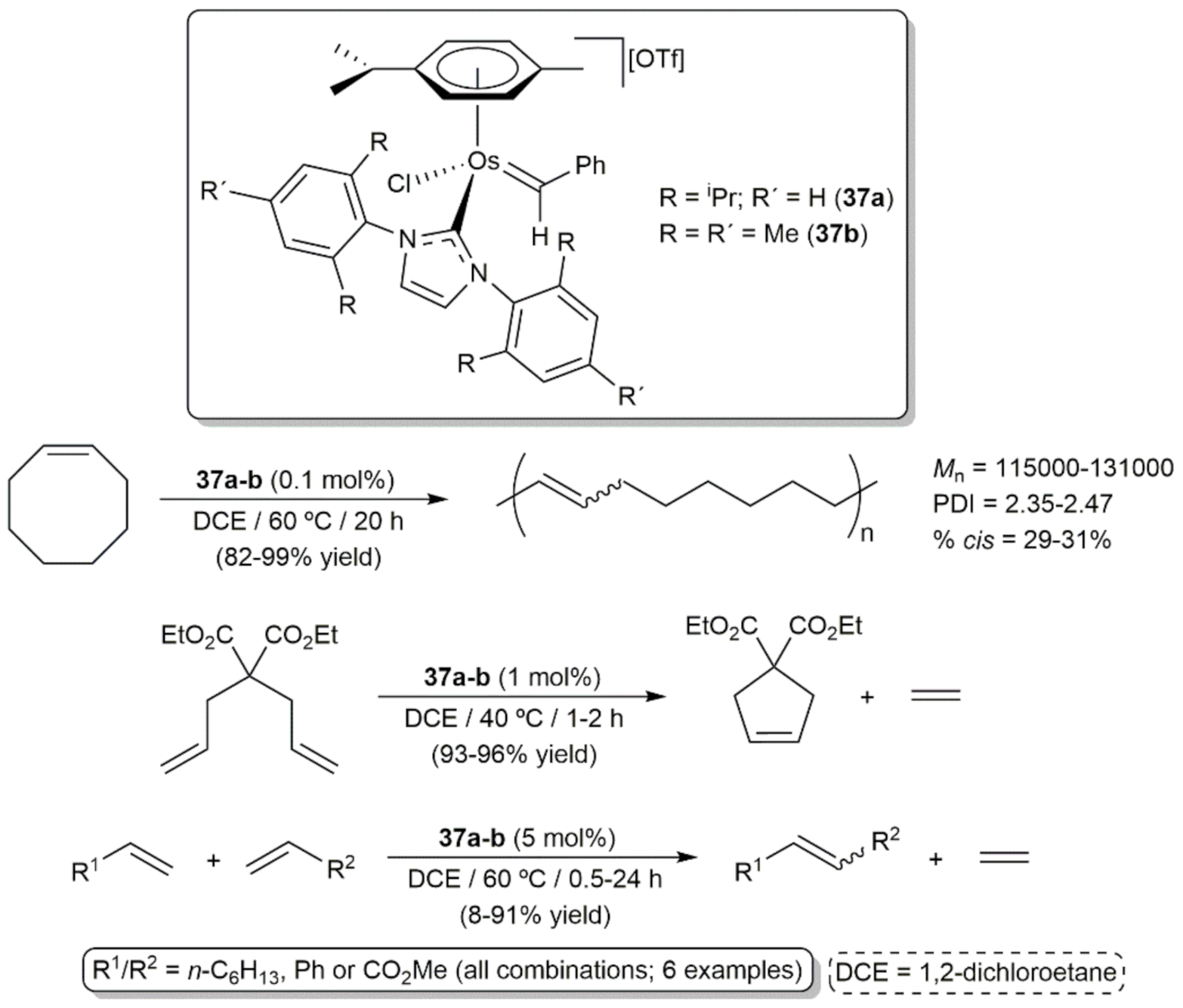
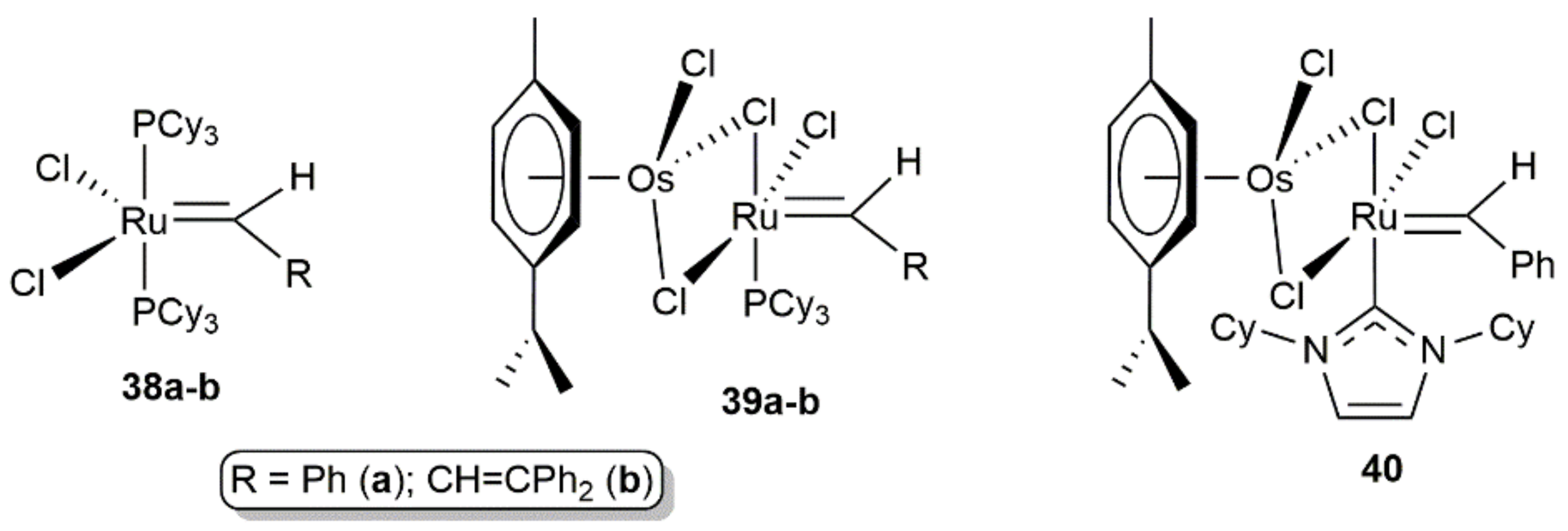
- Castarlenas, R.; Esteruelas, M.A.; Oñate, E. N-Heterocyclic Carbene−Osmium Complexes for Olefin Metathesis Reactions. Organometallics 2005, 24, 4343–4346. [Google Scholar] [CrossRef]
- Dias, E.L.; Grubbs, R.H. Synthesis and investigation of homo- and heterobimetallic ruthenium olefin metathesis catalysts exhibiting increased activities. Organometallics 1998, 17, 2758–2767. [Google Scholar] [CrossRef]
- Weskamp, T.; Kohl, F.J.; Herrmann, A.W. N-heterocyclic carbenes: Novel ruthenium–alkylidene complexes. J. Organomet. Chem. 1999, 582, 362–365. [Google Scholar] [CrossRef]
- Frenzel, U.; Weskamp, T.; Kohl, F.J.; Schattenmann, W.C.; Nuyken, O.; Herrmann, A.W. N-Heterocyclic carbenes: Application of ruthenium–alkylidene complexes in ring-opening metathesis polymerization. J. Organomet. Chem. 1999, 586, 263–265. [Google Scholar] [CrossRef]
- Guillena, G.; Ramón, D.J.; Yus, M. Alcohols as electrophiles in C-C bond-forming reactions: The hydrogen autotransfer process. Angew. Chem. Int. Ed. 2007, 46, 2358–2364. [Google Scholar] [CrossRef]
- Nixon, T.D.; Whittlesey, M.K.; Williams, J.M.J. Transition metal catalysed reactions of alcohols using borrowing hydrogen methodology. Dalton Trans. 2009, 40, 753–762. [Google Scholar] [CrossRef]
- Obora, Y. Recent Advances in α-Alkylation Reactions using Alcohols with Hydrogen Borrowing Methodologies. ACS Catal. 2014, 4, 3972–3981. [Google Scholar] [CrossRef]
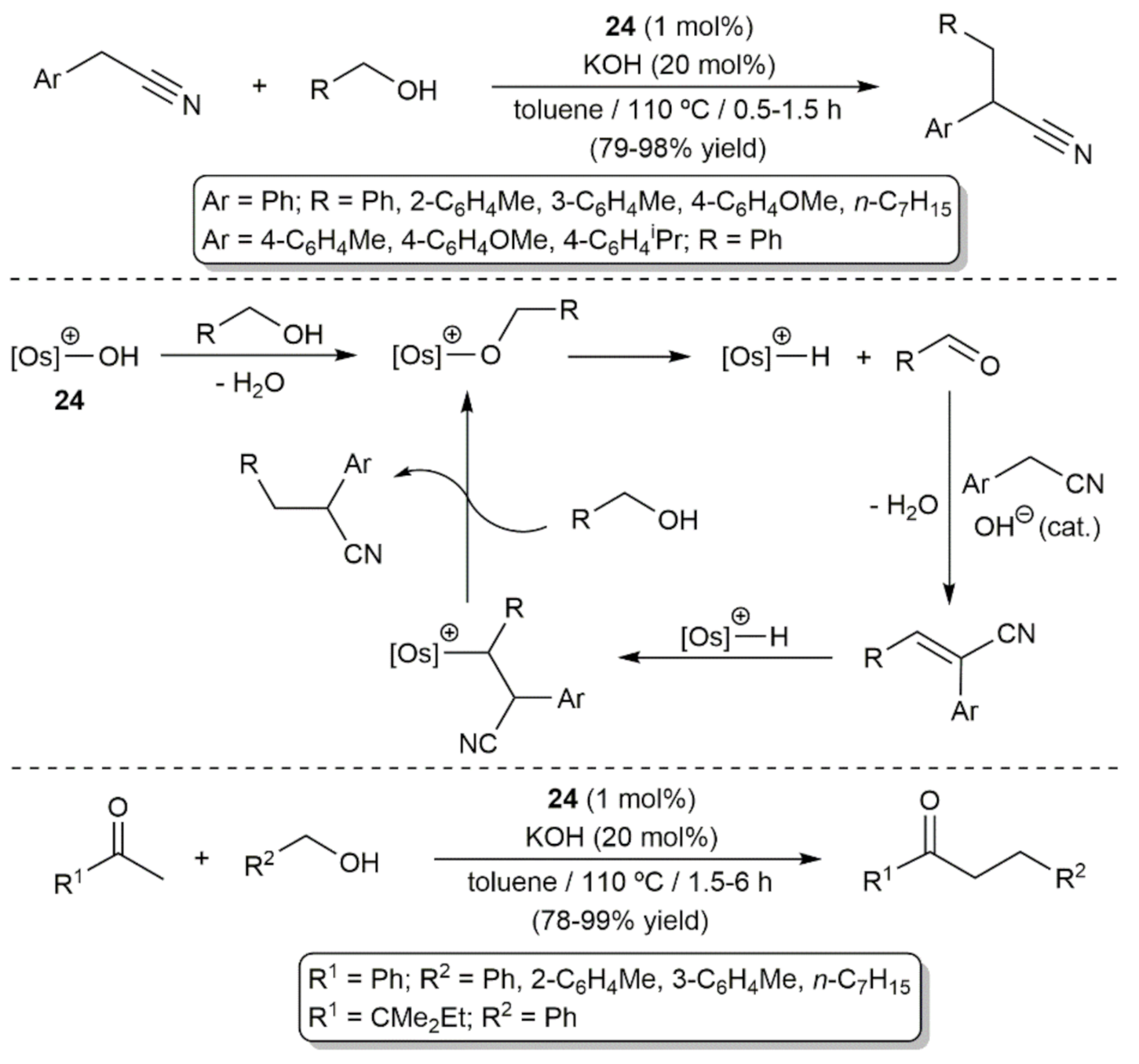
- Buil, M.L.; Esteruelas, M.A.; Herrero, J.; Izquierdo, S.; Pastor, I.M.; Yus, M. Osmium Catalyst for the Borrowing Hydrogen Methodology: α-Alkylation of Arylacetonitriles and Methyl Ketones. ACS Catal. 2013, 3, 2072–2075. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; García-Yebra, C.; Oliván, M.; Oñate, E.; Valencia, M. Osmium-Catalyzed Allylic Alkylation. Organometallics 2008, 27, 4892–4902. [Google Scholar] [CrossRef]
- Ackermann, L. Carboxylate-Assisted Ruthenium-Catalyzed Alkyne Annulations by C–H/Het–H Bond Functionalizations. Acc. Chem. Res. 2013, 47, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.S. Recent Advances in C–H Bond Functionalization with Ruthenium-Based Catalysts. Catalysts 2019, 9, 173. [Google Scholar] [CrossRef] [Green Version]
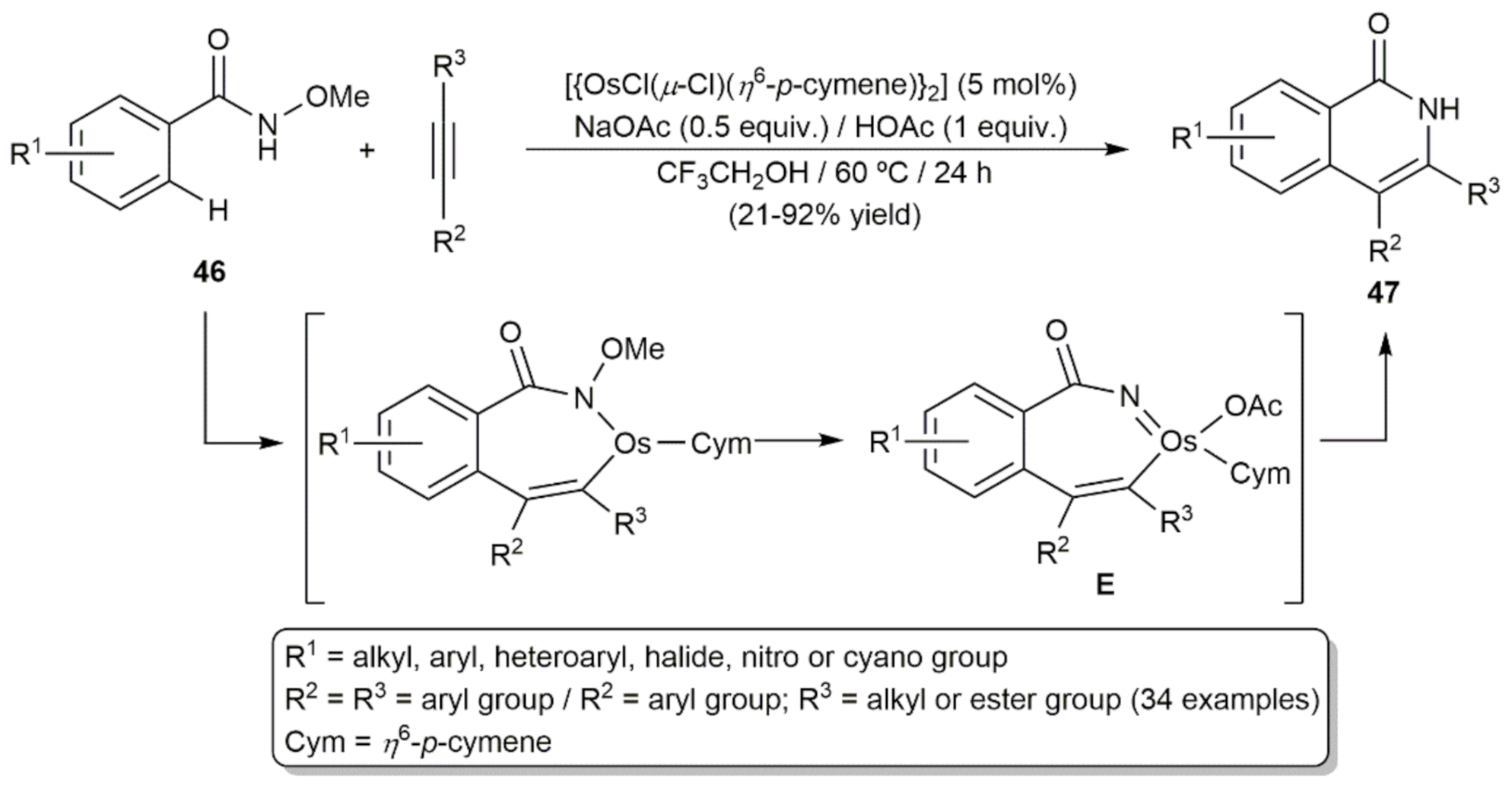
- Yang, J.; Wu, L.; Xu, H.; Gao, H.; Zhou, Z.; Yi, W. Redox-Neutral [4 + 2] Annulation ofN-Methoxybenzamides with Alkynes Enabled by an Osmium(II)/HOAc Catalytic System. Org. Lett. 2019, 21, 9904–9908. [Google Scholar] [CrossRef]
- Li, B.; Feng, H.; Xu, S.; Wang, B. Ruthenium-Catalyzed Isoquinolone Synthesis through C-H Activation Using an Oxidizing Directing Group. Chem. A Eur. J. 2011, 17, 12573–12577. [Google Scholar] [CrossRef]
- Ackermann, L.; Fenner, S. Ruthenium-Catalyzed C–H/N–O Bond Functionalization: Green Isoquinolone Syntheses in Water. Org. Lett. 2011, 13, 6548–6551. [Google Scholar] [CrossRef]
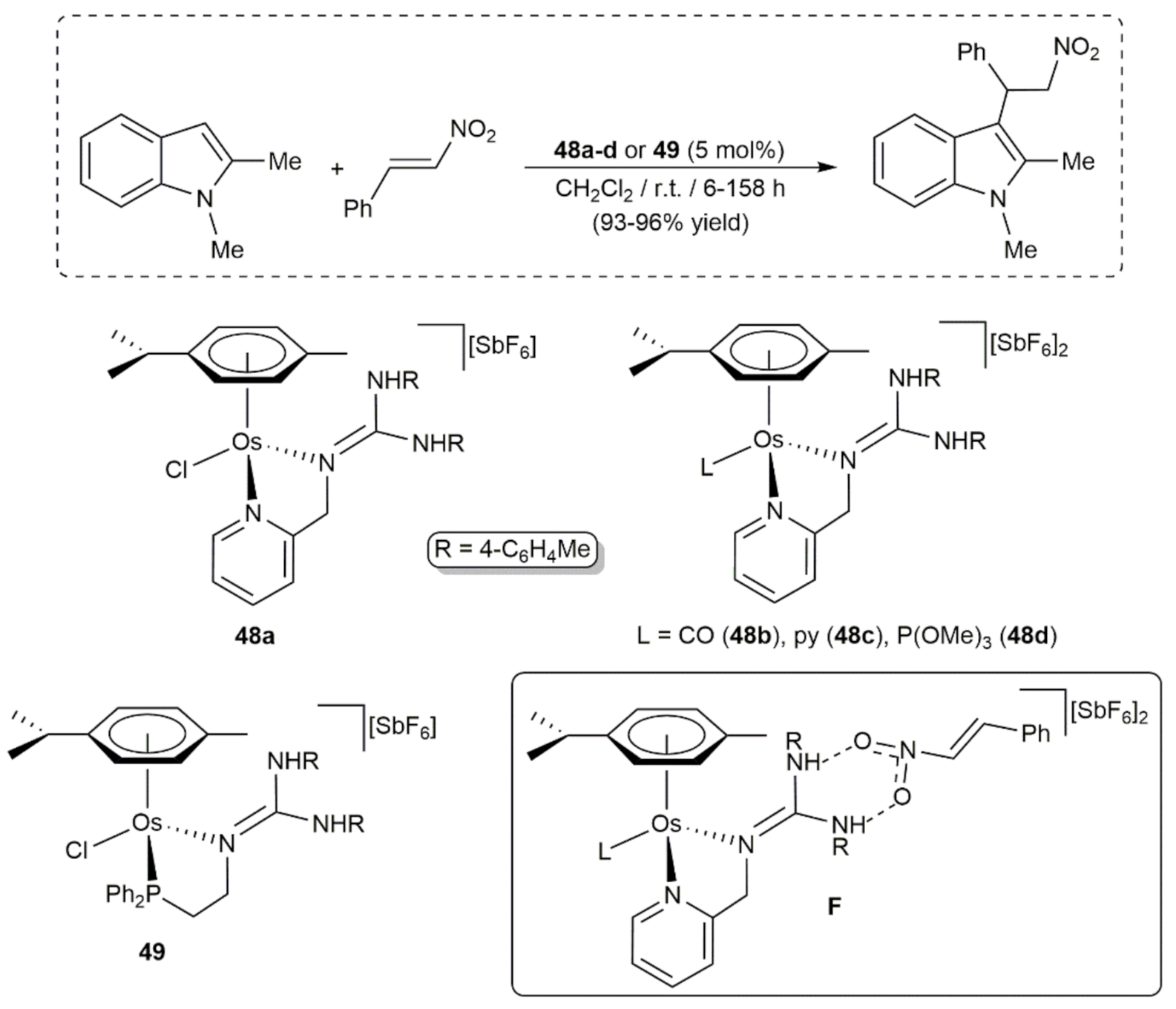
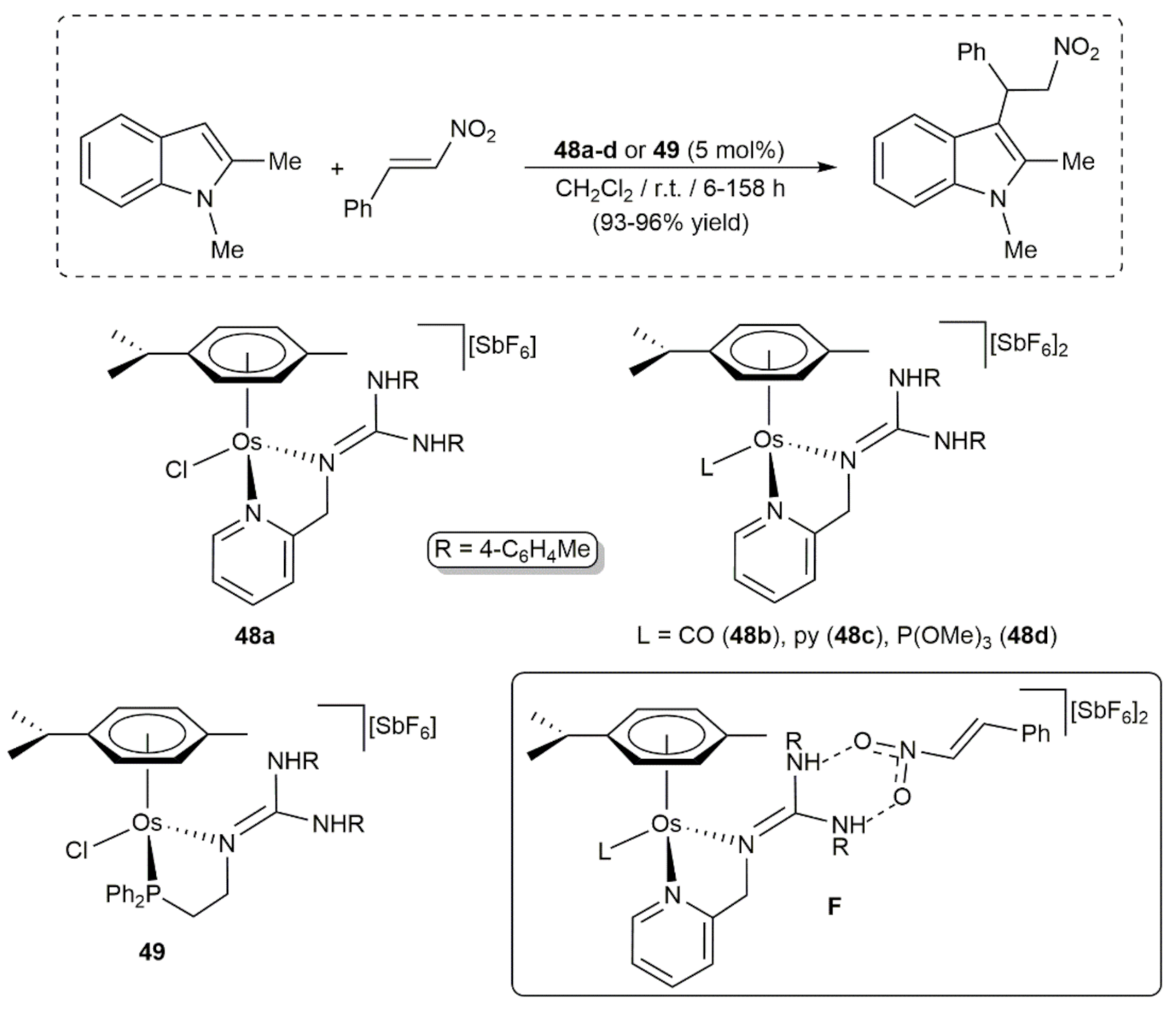
- Parker, A.; Lamata, P.; Viguri, F.; Rodríguez, R.; López, J.A.; Lahoz, F.J.; García-Orduña, P.; Carmona, D. Half-sandwich complexes of osmium containing guanidine-derived ligands. Dalton Trans. 2020, 49, 13601–13617. [Google Scholar] [CrossRef]
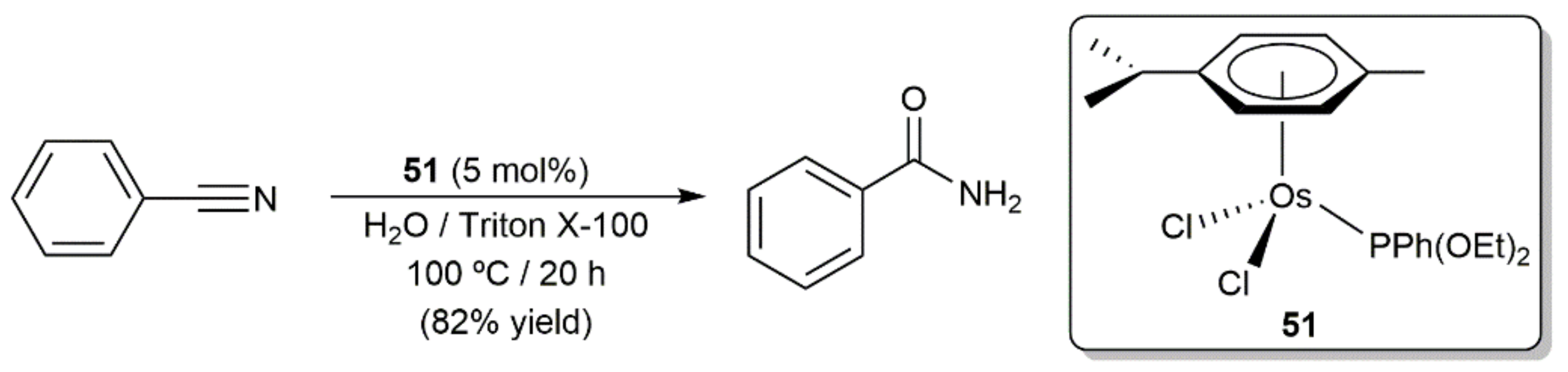
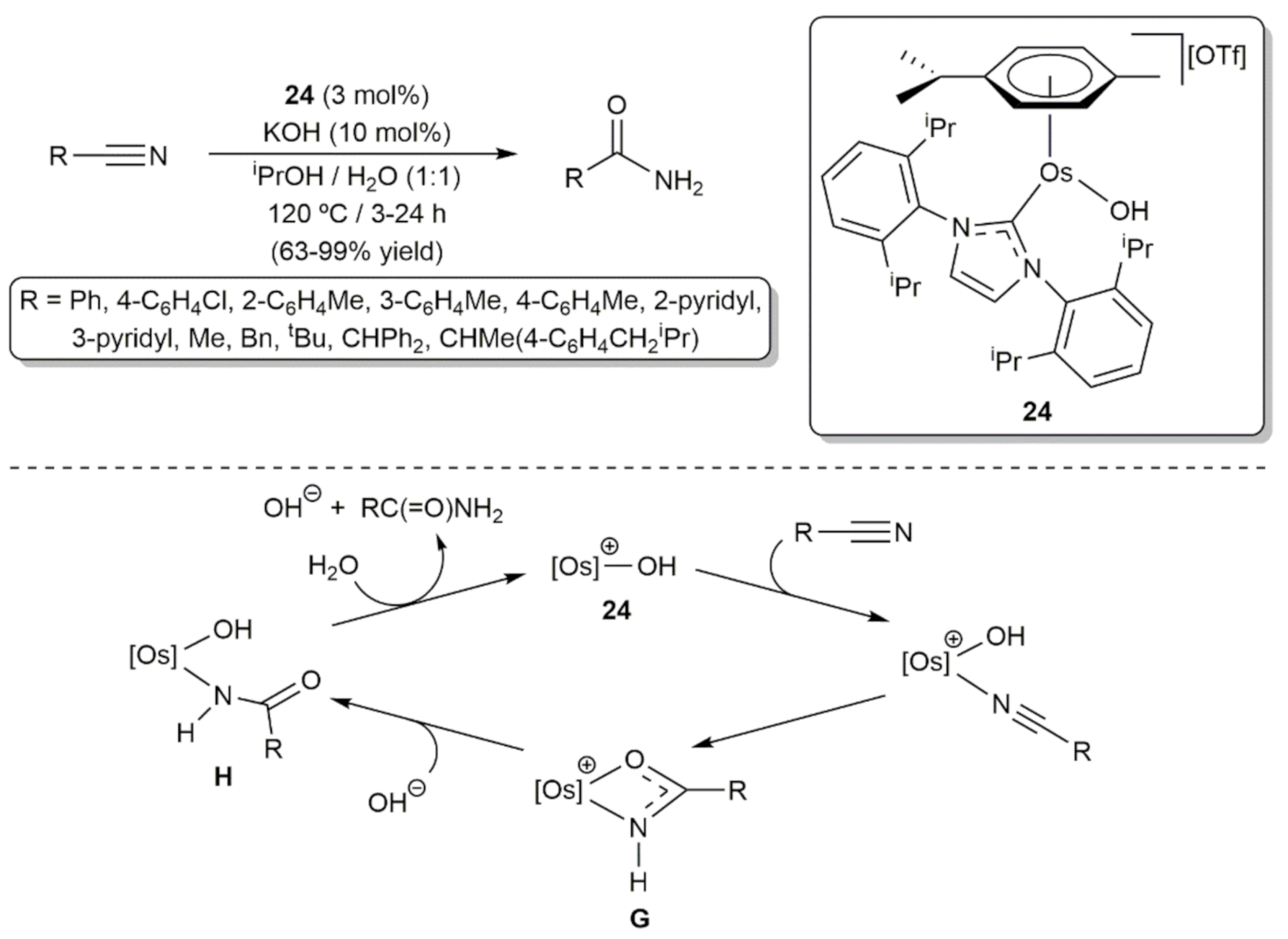
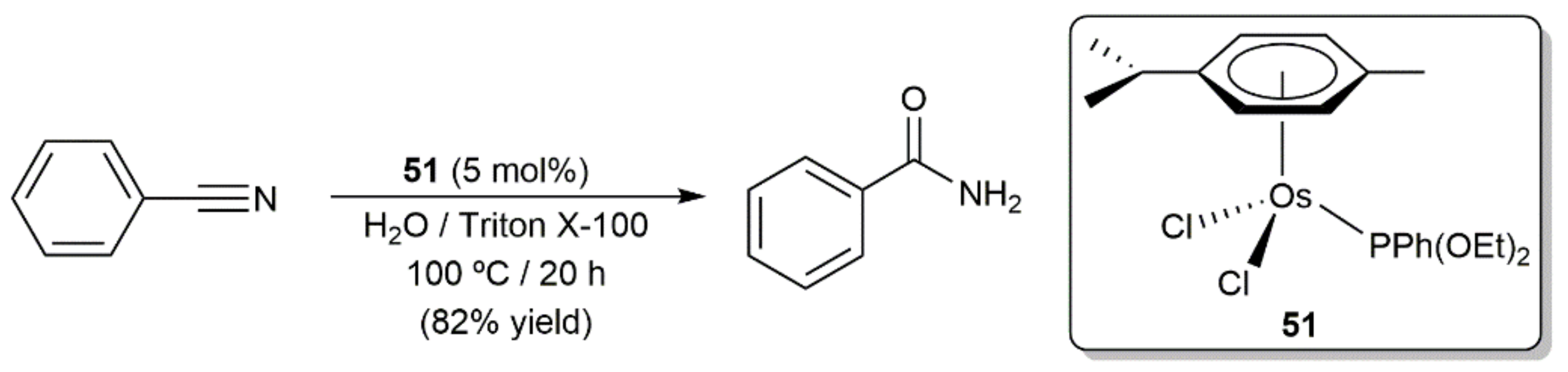
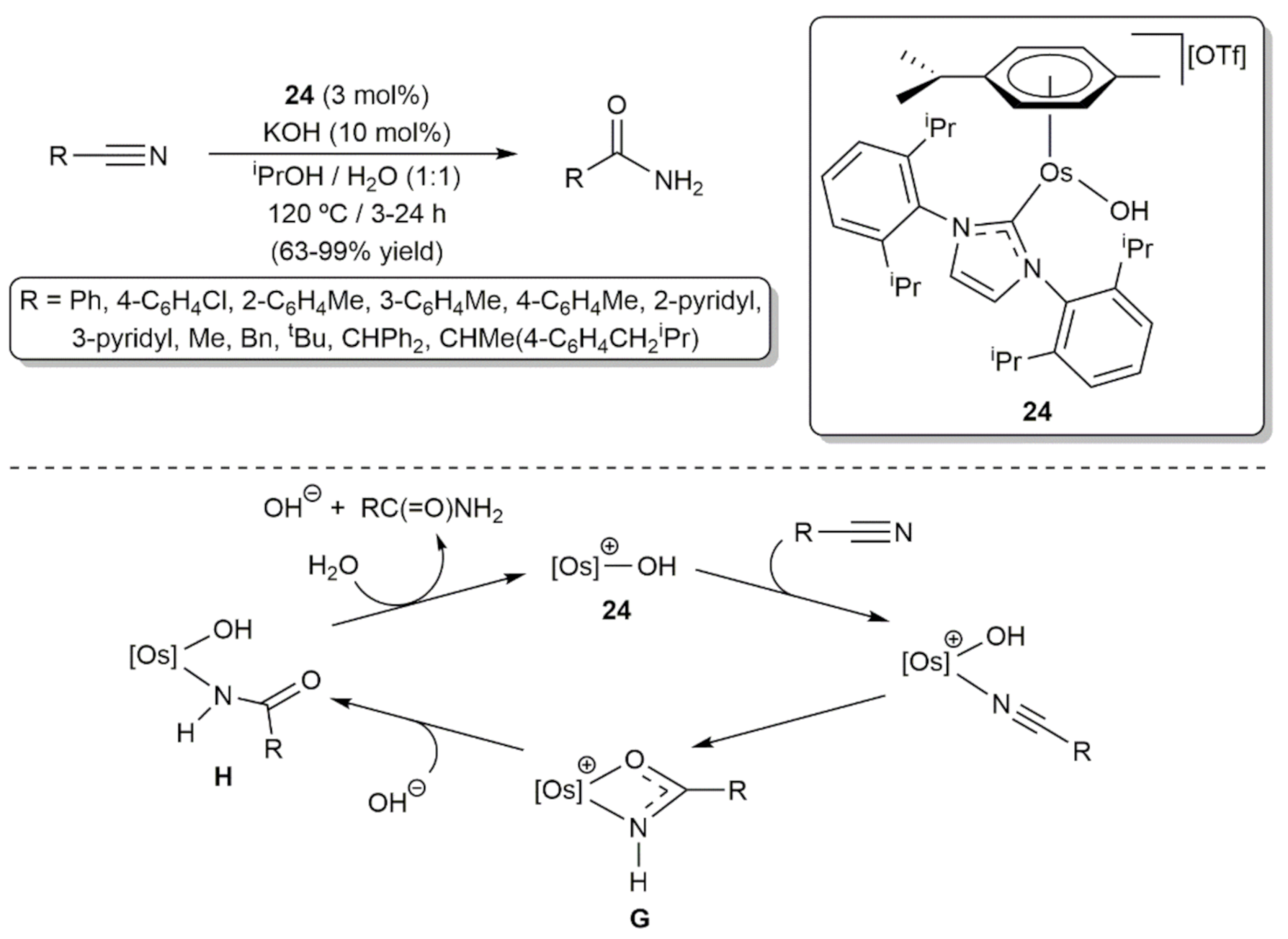
- Ahmed, T.J.; Knapp, S.M.; Tyler, D.R. Frontiers in catalytic nitrile hydration: Nitrile and cyanohydrin hydration catalyzed by homogeneous organometallic complexes. Coord. Chem. Rev. 2011, 255, 949–974. [Google Scholar] [CrossRef]
- García-Álvarez, R.; Crochet, P.; Cadierno, V.; Cadierno-Menéndez, V. Metal-catalyzed amide bond forming reactions in an environmentally friendly aqueous medium: Nitrile hydrations and beyond. Green Chem. 2012, 15, 46–66. [Google Scholar] [CrossRef]
- García-Álvarez, R.; Francos, J.; Tomás-Mendivil, E.; Crochet, P.; Cadierno, V. Metal-catalyzed nitrile hydration reactions: The specific contribution of ruthenium. J. Organomet. Chem. 2014, 771, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Sarbaina, A.; Bera, J.K. Bifunctional organometallic catalysts for selective hydration of nitriles to amides. J. Indian Chem. Soc. 2018, 95, 853–861. [Google Scholar]
- González-Fernández, R.; Crochet, P.; Cadierno, V. Arene-ruthenium(II) and osmium(II) complexes as catalysts for nitrile hydration and aldoxime rearrangement reactions. Inorg. Chim. Acta 2021, 517, 120180. [Google Scholar] [CrossRef]
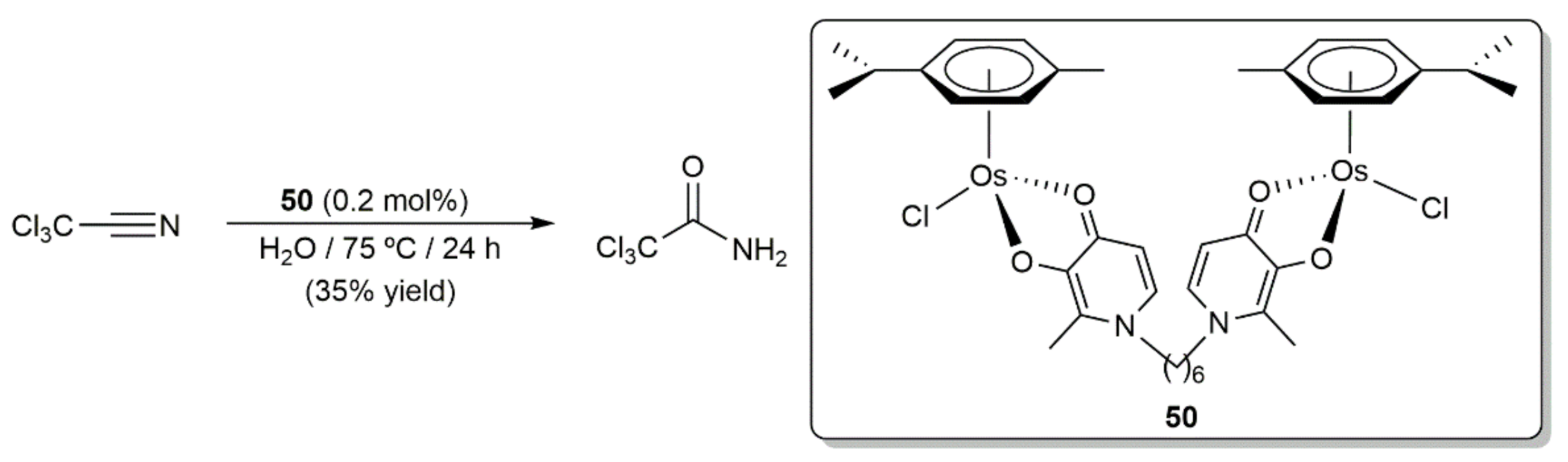
- Ashraf, S.M.; Kandioller, W.; Mendoza-Ferri, M.-G.; Nazarov, A.A.; Hartinger, C.G.; Keppler, B.K. The hydration of chloroacetonitriles catalyzed by mono- and dinuclear RuII- and OsII-arene complexes. Chem. Biodivers. 2008, 5, 2060–2066. [Google Scholar] [CrossRef]
- Cavarzan, A.; Scarso, A.; Strukul, G. Efficient nitrile hydration mediated by RuII catalysts in micellar media. Green Chem. 2010, 12, 790–794. [Google Scholar] [CrossRef]
- Buil, M.L.; Cadierno, V.; Esteruelas, M.A.; Gimeno, J.; Herrero, J.; Izquierdo, S.; Oñate, E.; Cadierno-Menéndez, V. Selective Hydration of Nitriles to Amides Promoted by an Os–NHC Catalyst: Formation and X-ray Characterization of κ2-Amidate Intermediates. Organometallics 2012, 31, 6861–6867. [Google Scholar] [CrossRef]
- Tomás-Mendivil, E.; Cadierno, V.; Menéndez, M.I.; López, R.; Cadierno-Menéndez, V. Unmasking the Action of Phosphinous Acid Ligands in Nitrile Hydration Reactions Catalyzed by Arene-Ruthenium(II) Complexes. Chem. A Eur. J. 2015, 21, 16874–16886. [Google Scholar] [CrossRef]
- González-Fernández, R.; Crochet, P.; Cadierno, V.; Menéndez, M.I.; López, R. Phosphinous Acid-Assisted Hydration of Nitriles: Understanding the Controversial Reactivity of Osmium and Ruthenium Catalysts. Chem. A Eur. J. 2017, 23, 15210–15221. [Google Scholar] [CrossRef]
- González-Fernández, R.; Álvarez, D.; Crochet, P.; Cadierno, V.; Menéndez, M.I.; López, R. Catalytic hydration of cyanamides with phosphinous acid-based ruthenium(ii) and osmium(ii) complexes: Scope and mechanistic insights. Catal. Sci. Technol. 2020, 10, 4084–4098. [Google Scholar] [CrossRef]
- Cadierno, V. Synthetic applications of the Parkins nitrile hydration catalyst [PtH{(PMe2O)2H}(PMe2OH)]: A review. Appl. Sci. 2015, 5, 380. [Google Scholar] [CrossRef] [Green Version]
- González-Fernández, R.; Crochet, P.; Cadierno, V. Cymene-osmium(II) complexes with amino-phosphane ligands as precat-alysts for nitrile hydration reactions. ChemistrySelect 2018, 3, 4324–4329. [Google Scholar] [CrossRef]
- Francos, J.; González-Liste, P.J.; Menéndez-Rodríguez, L.; Crochet, P.; Cadierno, V.; Borge, J.; Antiñolo, A.; Fernández-Galán, R.; Carrillo-Hermosilla, F. Half-sandwich guanidinate-osmium(II) complexes: Synthesis and application in the selective de-hydration of aldoximes. Eur. J. Inorg. Chem. 2016, 2016, 393–402. [Google Scholar] [CrossRef]
- Crochet, P.; Cadierno, V. Catalytic synthesis of amides via aldoximes rearrangement. Chem. Commun. 2015, 51, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Debnath, P. Recent Advances in the Synthesis of Amides via Oxime Rearrangements and its Applications. Curr. Org. Synth. 2018, 15, 666–706. [Google Scholar] [CrossRef]
- Reddy, T.N.; Beatriz, A.; Rao, V.J.; de Lima, D.P. Carbonyl compounds´ journey to amide bond formation. Chem. Asian, J. 2019, 14, 344–388. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Rivera, S.A.; Francos, J.; Borge, J.; Cadierno, V. Mononuclear Ruthenium and Osmium Complexes with a Bicyclic Guanidinate Ligand: Synthesis and Catalytic Behavior in Olefin Isomerization Processes. Eur. J. Inorg. Chem. 2017, 2017, 4138–4146. [Google Scholar] [CrossRef]
- Lastra-Barreira, B.; Francos, J.; Crochet, P.; Cadierno, V. Ruthenium(IV) catalysts for the selective estragole to trans-anethole isomerization in environmentally friendly media. Green Chem. 2011, 13, 307–313. [Google Scholar] [CrossRef]
- Hassam, M.; Taher, A.; Arnott, G.; Green, I.R.; Van Otterlo, W.A.L. Isomerization of Allylbenzenes. Chem. Rev. 2015, 115, 5462–5569. [Google Scholar] [CrossRef]
- Brunner, H.; Zwack, T.; Zabel, M.; Beck, W.; Böhm, A. Optically active transition metal complexes. Synthesis, crystal structures, and catalytic properties of chiral-at-metal (η6-arene)ruthenium(II) and (η6-arene)osmium(II) half-sandwich com-plexes. Crystallization of pure diastereoisomers versus diastereomer mixtures in a 1:1 ratio. Organometallics 2003, 22, 1741–1750. [Google Scholar]
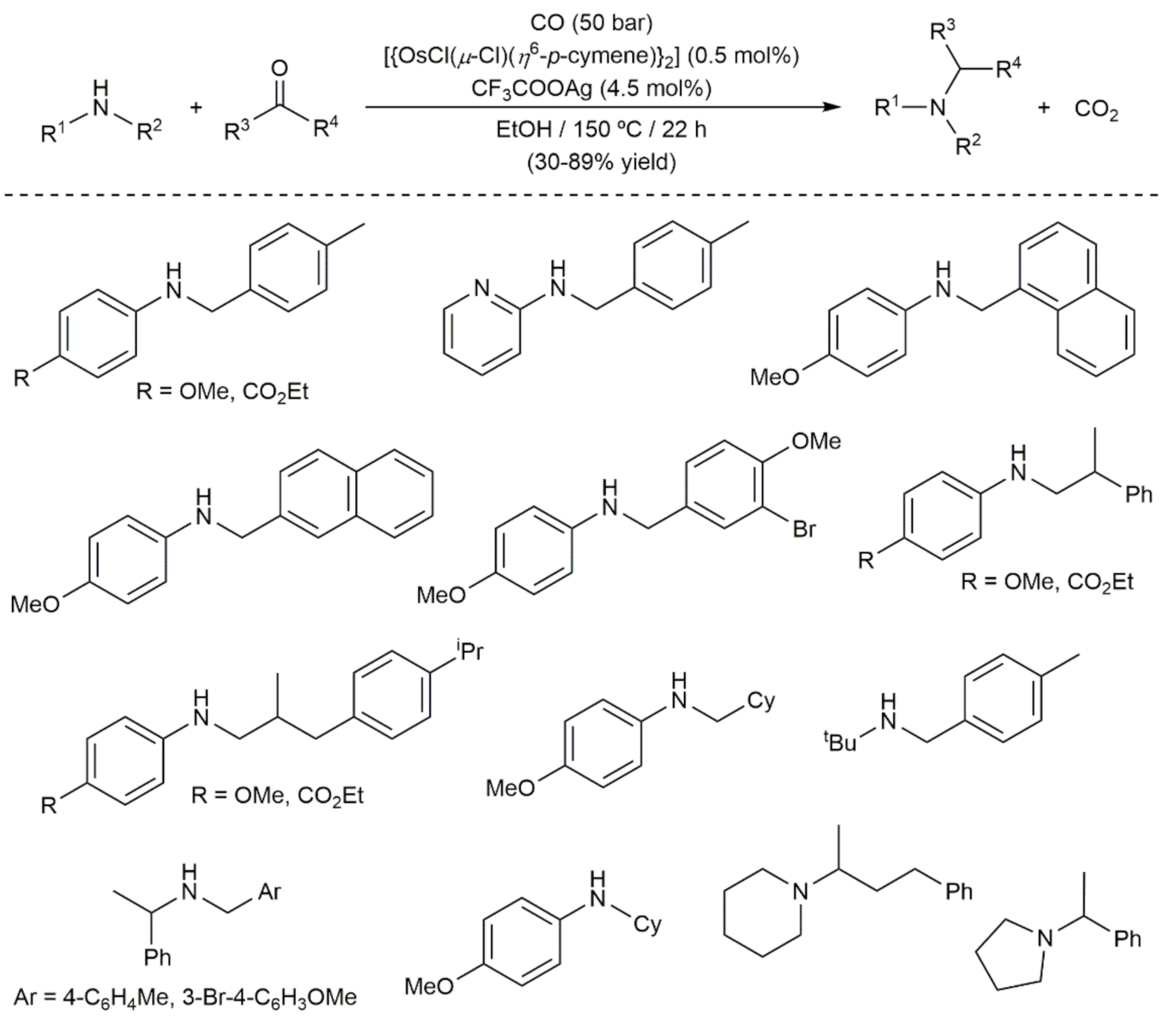
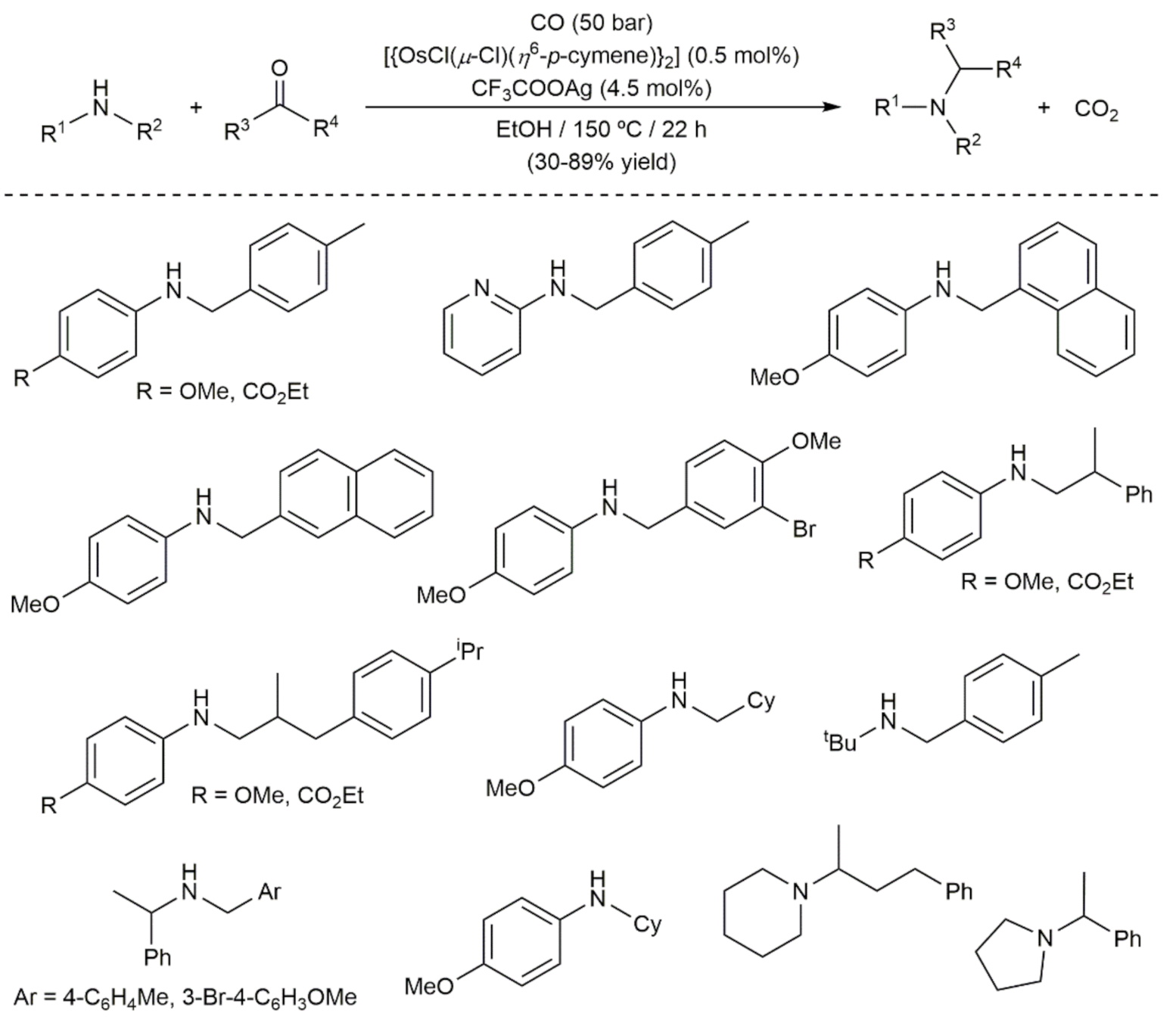
- Vinogradov, M.M.; Afanasyev, O.I.; Nelyubina, Y.V.; Denisov, G.L.; Loginov, D.A.; Chusov, D. Osmium catalysis in the re-ductive amination using carbon monoxide as a reducing agent. Mol. Catal. 2020, 498, 111260. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crochet, P.; Cadierno, V. Arene-Osmium(II) Complexes in Homogeneous Catalysis. Inorganics 2021, 9, 55. https://doi.org/10.3390/inorganics9070055
Crochet P, Cadierno V. Arene-Osmium(II) Complexes in Homogeneous Catalysis. Inorganics. 2021; 9(7):55. https://doi.org/10.3390/inorganics9070055
Chicago/Turabian StyleCrochet, Pascale, and Victorio Cadierno. 2021. "Arene-Osmium(II) Complexes in Homogeneous Catalysis" Inorganics 9, no. 7: 55. https://doi.org/10.3390/inorganics9070055
APA StyleCrochet, P., & Cadierno, V. (2021). Arene-Osmium(II) Complexes in Homogeneous Catalysis. Inorganics, 9(7), 55. https://doi.org/10.3390/inorganics9070055