Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments


Abstract
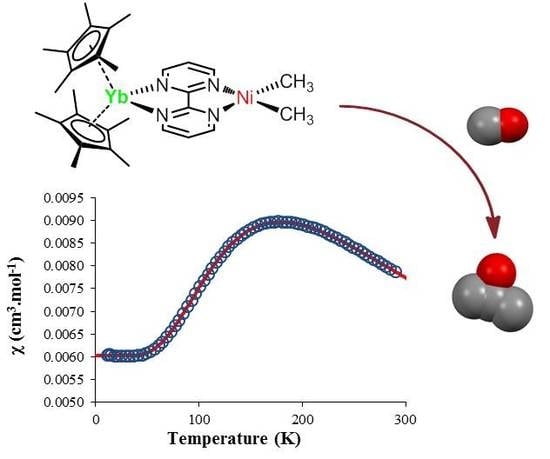
{kind=link}
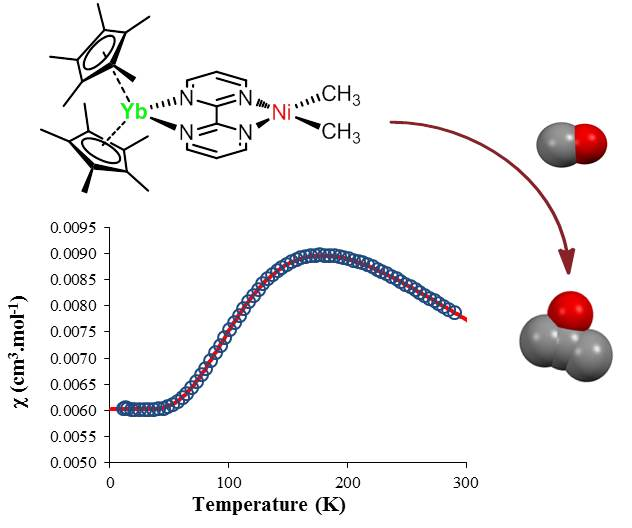
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
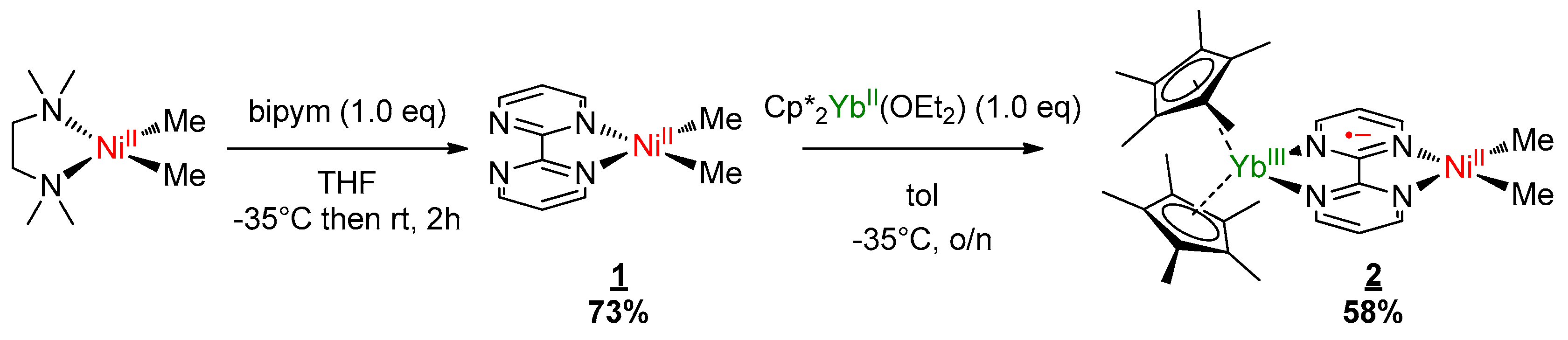{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
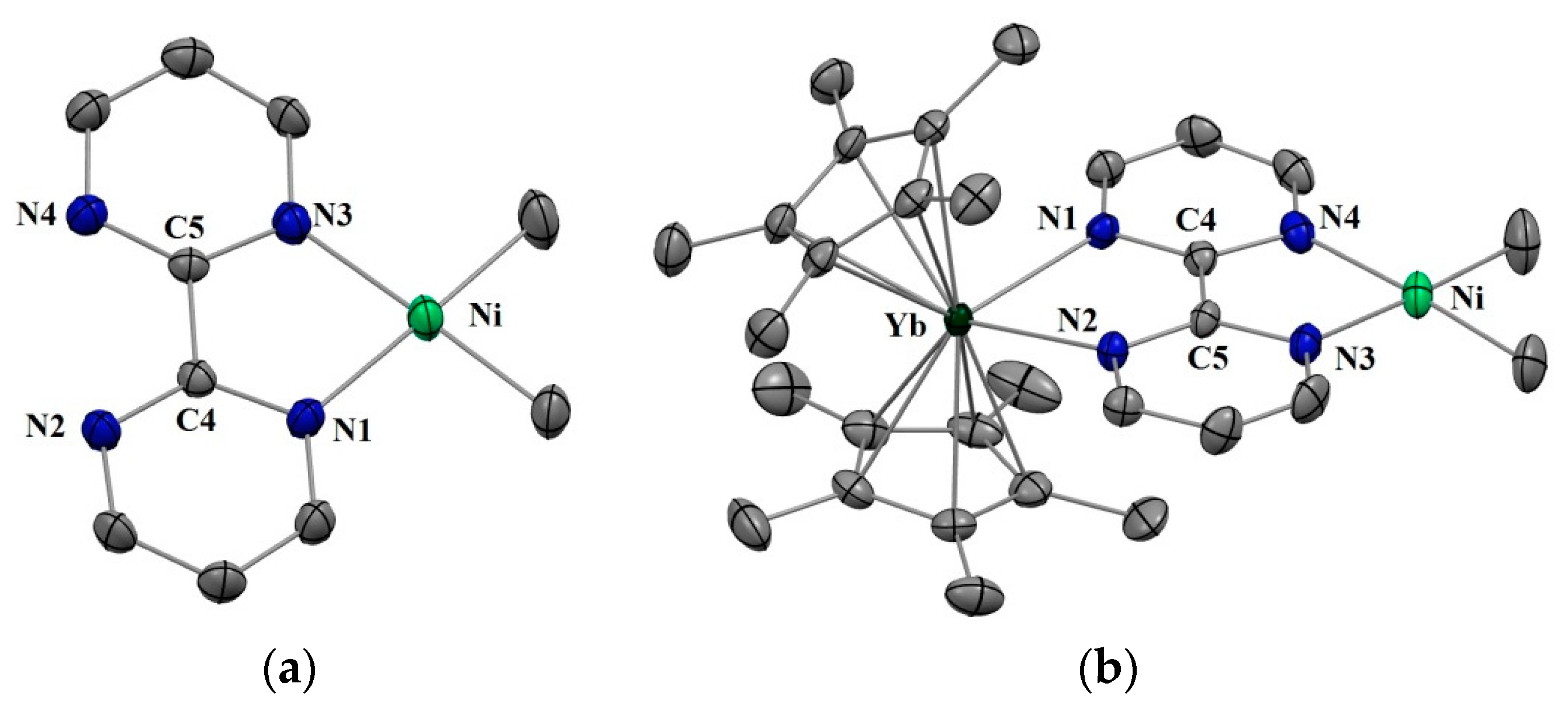
2.1. Synthesis and X-ray Diffraction
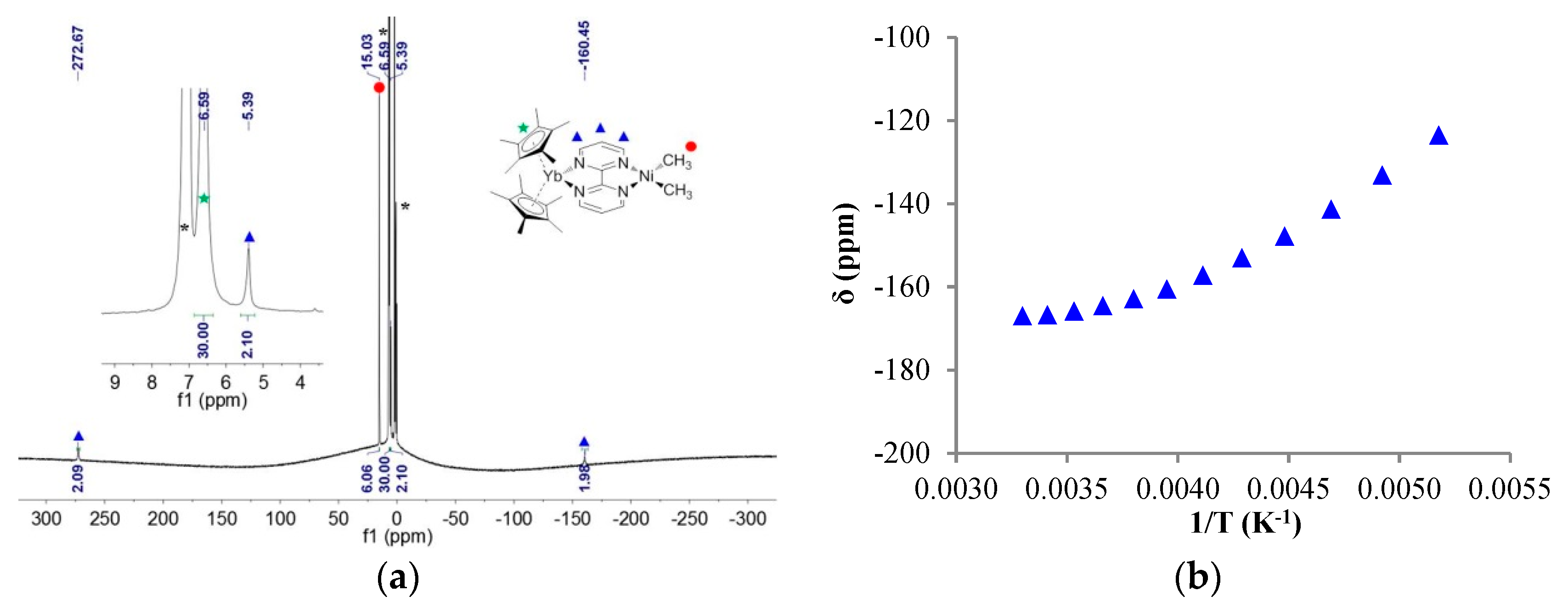
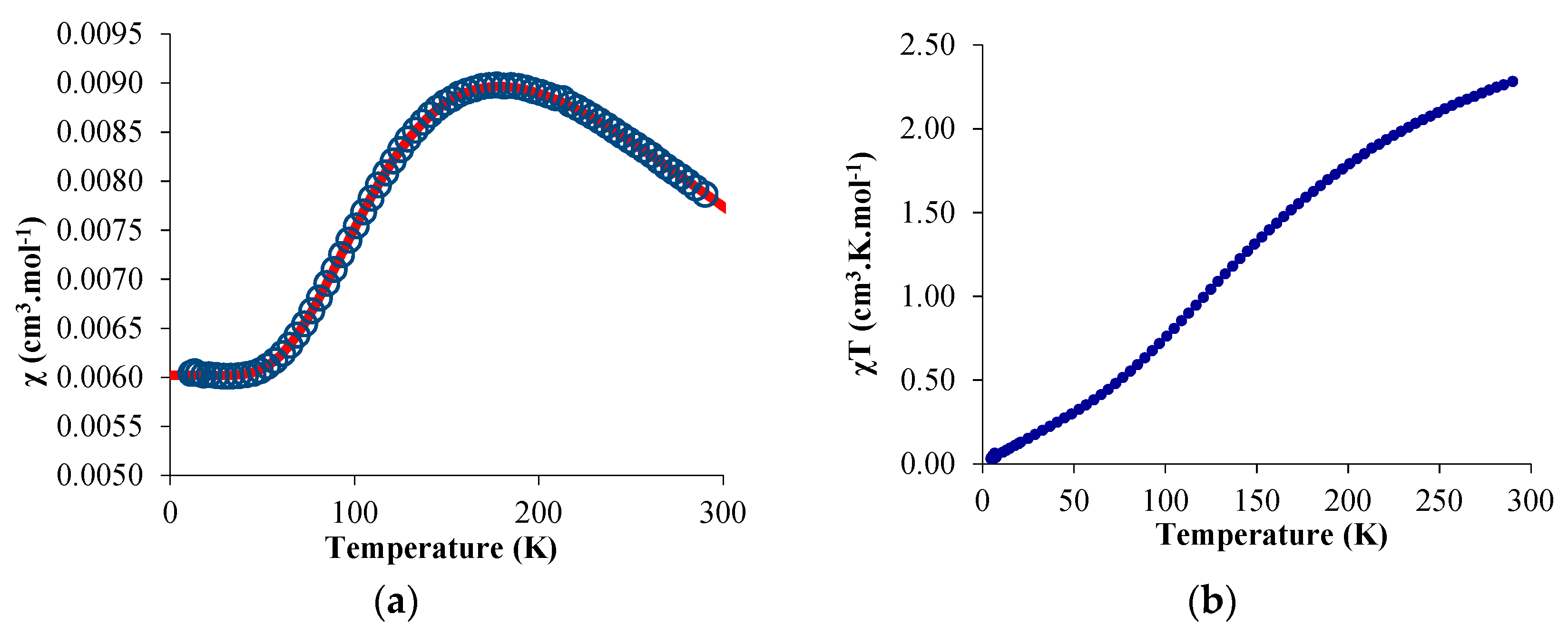
2.2. Solution NMR and Solid-State Magnetism
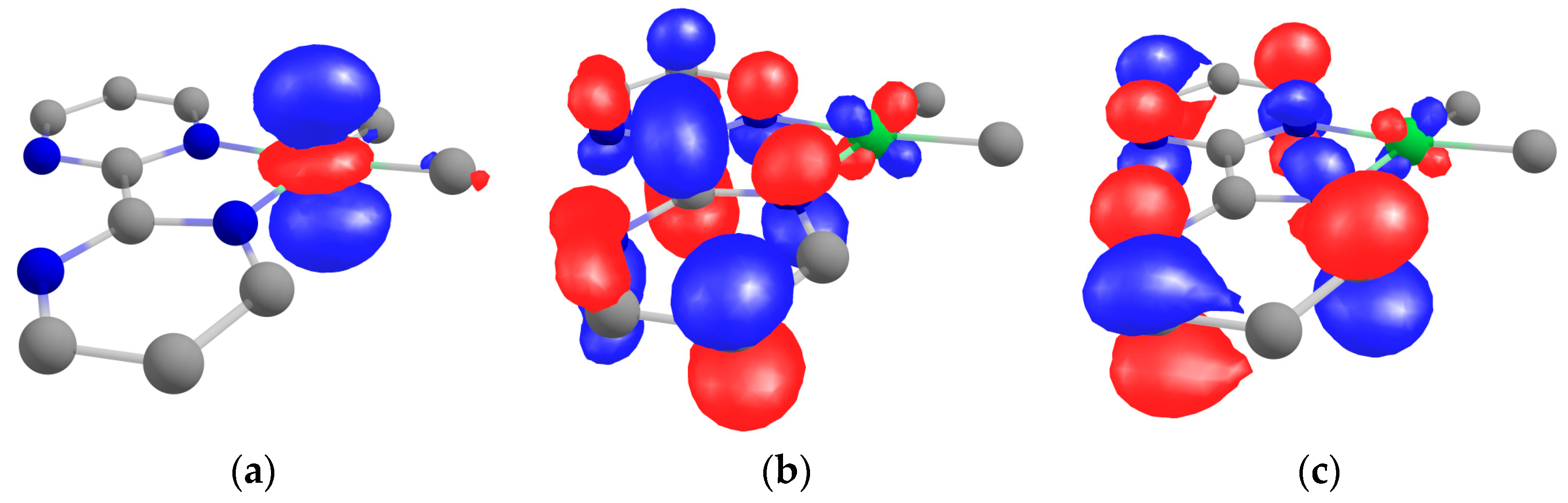
2.3. Theoretical Computations on 1
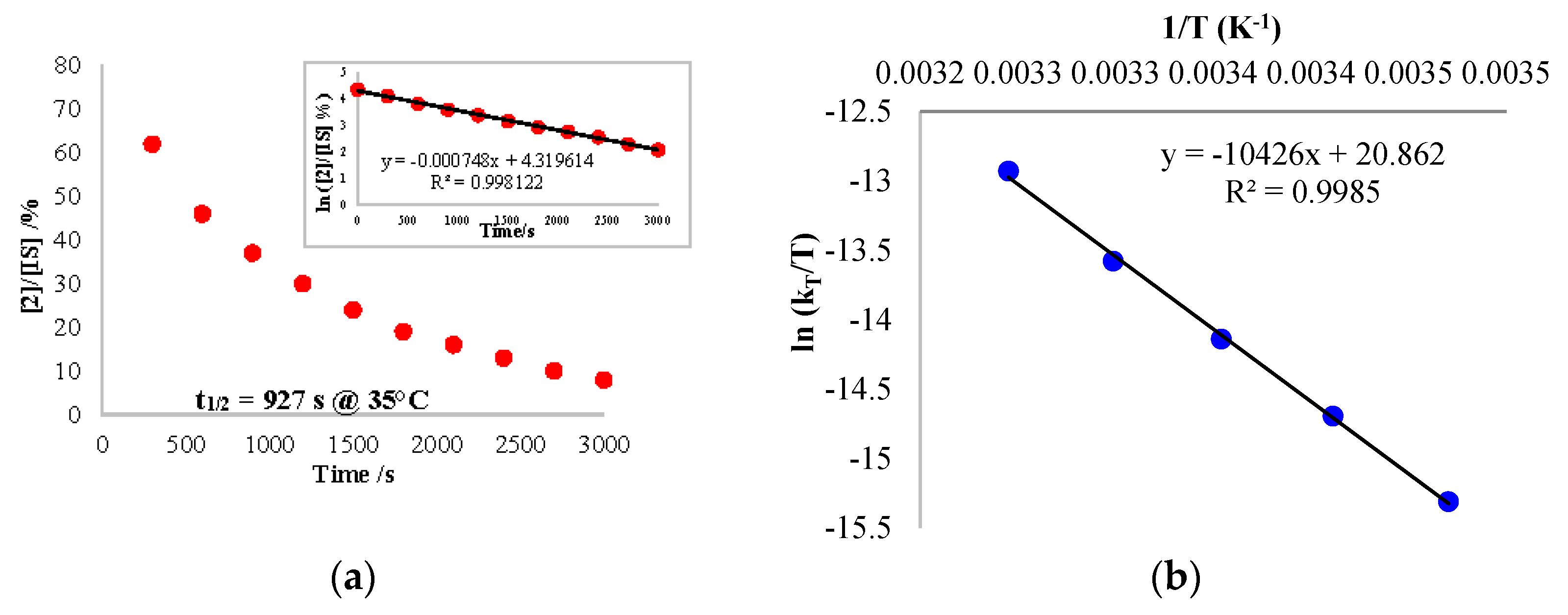
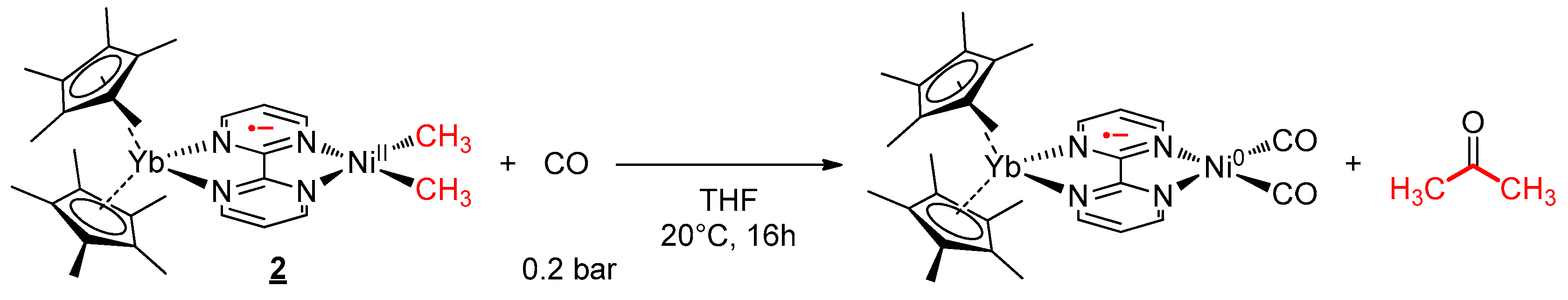
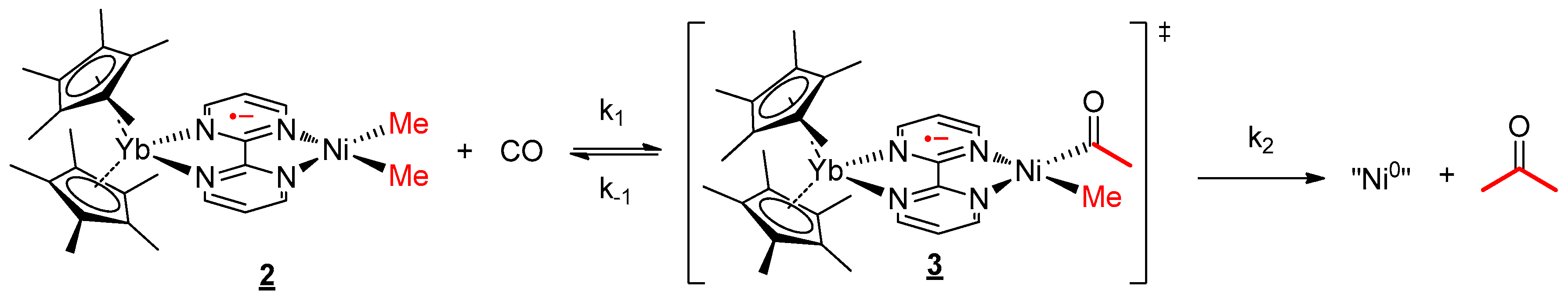
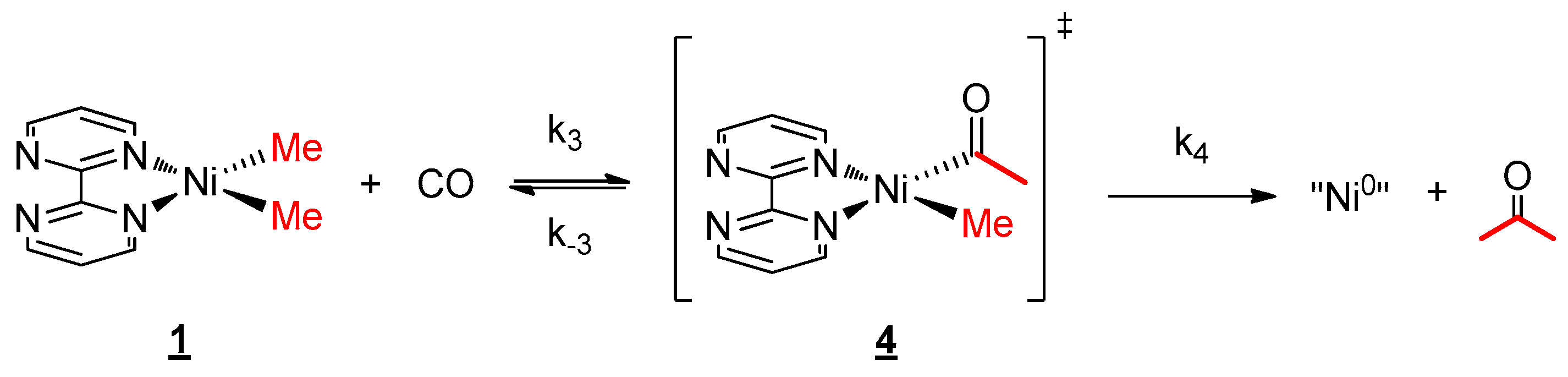
2.4. Reactivity with Carbon Monoxide
3. Materials and Methods
3.1. Synthesis of (bipym)Ni(Me)2 (1)
3.2. Synthesis of (Cp*)2Yb(bipym)Ni(Me)2 (2)
3.3. Theoretical Calculations
3.4. Crystal Structure Determinations
3.5. CO Migratory Insertion Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Buchwalter, P.; Rosé, J.; Braunstein, P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef] [PubMed]
- Sil, A.; Ghosh, U.; Mishra, V.K.; Mishra, S.; Patra, S.K. Synthesis, structure, electrochemical, and spectroscopic properties of hetero-bimetallic Ru(II)/Fe(II)-alkynyl organometallic complexes. Inorg. Chem. 2019, 58, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Saito, K.; Kikuchi, S.; Akita, M.; Inagaki, A. Visible-light-controlled homo-and copolymerization of styrenes by a bichromophoric Ir–Pd catalyst. Chem. Commun. 2015, 51, 5717–5720. [Google Scholar] [CrossRef]
- Dobbek, H.; Gremer, L.; Kiefersauer, R.; Huber, R.; Meyer, O. Catalysis at a dinuclear [CuSMo(O)OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 15971–15976. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, T.S.; Hollingsworth, R.L.; Lord, R.L.; Groysman, S. Cooperative bimetallic reactivity of a heterodinuclear molybdenum–copper model of Mo–Cu CODH. Dalton Trans. 2018, 47, 10017–10024. [Google Scholar] [CrossRef] [PubMed]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase enzymes and their synthetic models: The role of metal hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, S.; Bruschi, M.; De Gioia, L.; Le Roy, C.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. FeMo heterobimetallic dithiolate complexes: Investigation of their electron transfer chemistry and reactivity toward acids, a density functional theory rationalization. Inorg. Chem. 2019, 58, 679–694. [Google Scholar] [CrossRef]
- Ogo, S.; Mori, Y.; Ando, T.; Matsumoto, T.; Yatabe, T.; Yoon, K.-S.; Hayashi, H.; Asano, M. One model, two enzymes: Activation of hydrogen and carbon monoxide. Angew. Chem. Int. Ed. 2017, 56, 9723–9726. [Google Scholar] [CrossRef]
- Mankad, N.P. Selectivity effects in bimetallic catalysis. Chem. Eur. J. 2016, 22, 5822–5829. [Google Scholar] [CrossRef]
- Pye, D.R.; Mankad, N.P. Bimetallic catalysis for C–C and C–X coupling reactions. Chem. Sci. 2017, 8, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Ajamian, A.; Gleason, J.L. Two birds with one metallic stone: Single-pot catalysis of fundamentally different transformations. Angew. Chem. Int. Ed. 2004, 43, 3754–3760. [Google Scholar] [CrossRef]
- Mata, J.A.; Hahn, F.E.; Peris, E. Heterometallic complexes, tandem catalysis and catalytic cooperativity. Chem. Sci. 2014, 5, 1723–1732. [Google Scholar] [CrossRef]
- Lorber, C.; Vendier, L. Imido–titanium/molybdenum heterobimetallic systems. Switching from η6-arene to fischer-type aminocarbene complexes by tuning reactivity conditions. Organometallics 2010, 29, 1127–1136. [Google Scholar] [CrossRef]
- Stark, H.S.; Altmann, P.J.; Sproules, S.; Hess, C.R. Structural characterization and photochemical properties of mono- and bimetallic Cu–Mabiq complexes. Inorg. Chem. 2018, 57, 6401–6409. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Kawashima, M.; Yamashita, H. Visible-light-enhanced suzuki–miyaura coupling reaction by cooperative photocatalysis with an Ru–Pd bimetallic complex. Chem. Commun. 2014, 50, 14501–14503. [Google Scholar] [CrossRef]
- Goudy, V.; Jaoul, A.; Cordier, M.; Clavaguéra, C.; Nocton, G. Tuning the Stability of Pd(IV) Intermediates using a redox non-innocent ligand combined with an organolanthanide fragment. J. Am. Chem. Soc. 2017, 139, 10633–10636. [Google Scholar] [CrossRef]
- Goudy, V.; Xémard, M.; Karleskind, S.; Cordier, M.; Alvarez Lamsfus, C.; Maron, L.; Nocton, G. Phenylacetylene and carbon dioxide activation by an organometallic samarium complex. Inorganics 2018, 6, 82. [Google Scholar] [CrossRef]
- Xémard, M.; Cordier, M.; Louyriac, E.; Maron, L.; Clavaguéra, C.; Nocton, G. Small molecule activation with divalent samarium triflate: A synergistic effort to cleave O2. Dalton Trans. 2018, 47, 9226–9230. [Google Scholar] [CrossRef]
- Evans, W.J.; Ulibarri, T.A.; Ziller, J.W. Isolation and x-ray crystal structure of the first dinitrogen complex of an f-element metal, [(C5Me5)2Sm]2N2. J. Am. Chem. Soc. 1988, 110, 6877–6879. [Google Scholar] [CrossRef]
- Jaroschik, F.; Momin, A.; Nief, F.; Le Goff, X.F.; Deacon, G.B.; Junk, P.C. Dinitrogen reduction and C–H activation by the divalent organoneodymium complex [(C5H2tBu3)2Nd(µ-I)K([18]crown-6)]. Angew. Chem. Int. Ed. 2009, 48, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.H.; Walter, M.D.; Kazhdan, D.; Hu, Y.-J.; Lukens, W.W.; Bauer, E.D.; Maron, L.; Eisenstein, O.; Andersen, R.A. Decamethylytterbocene complexes of bipyridines and diazabutadienes: Multiconfigurational ground states and open-shell singlet formation. J. Am. Chem. Soc. 2009, 131, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.H.; Kazhdan, D.; Werkema, E.L.; Walter, M.D.; Lukens, W.W.; Bauer, E.D.; Hu, Y.-J.; Maron, L.; Eisenstein, O.; Head-Gordon, M.; et al. Intermediate-valence tautomerism in decamethylytterbocene complexes of methyl-substituted bipyridines. J. Am. Chem. Soc. 2010, 132, 17537–17549. [Google Scholar] [CrossRef] [PubMed]
- Nocton, G.; Lukens, W.L.; Booth, C.H.; Rozenel, S.S.; Melding, S.A.; Maron, L.; Andersen, R.A. Reversible sigma C–C bond formation between phenanthroline ligands activated by (C5Me5)2Yb. J. Am. Chem. Soc. 2014, 136, 8626–8641. [Google Scholar] [CrossRef]
- Nocton, G.; Booth, C.H.; Maron, L.; Andersen, R.A. Thermal dihydrogen elimination from Cp*2Yb(4,5-diazafluorene). Organometallics 2013, 32, 1150–1158. [Google Scholar] [CrossRef]
- Nocton, G.; Booth, C.H.; Maron, L.; Andersen, R.A. Influence of the torsion angle in 3,3′-dimethyl-2,2′-bipyridine on the intermediate valence of Yb in (C5Me5)2Yb(3,3′-Me2-bipy). Organometallics 2013, 32, 5305–5312. [Google Scholar] [CrossRef]
- Jacquot, L.; Xémard, M.; Clavaguéra, C.; Nocton, G. Multiple one-electron transfers in bipyridine complexes of Bis(phospholyl) thulium. Organometallics 2014, 33, 4100–4106. [Google Scholar] [CrossRef]
- Nocton, G.; Booth, C.H.; Maron, L.; Ricard, L.; Andersen, R.A. Carbon–hydrogen bond breaking and making in the open-shell singlet molecule, Cp*2Yb(4,7-Me2phen). Organometallics 2014, 44, 6819–6829. [Google Scholar] [CrossRef]
- Nocton, G.; Ricard, L. Reversible C–C coupling in phenanthroline complexes of divalent samarium and thulium. Chem. Commun. 2015, 51, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Jaoul, A.; Clavaguera, C.; Nocton, G. Electron transfer in tetramethylbiphosphinine complexes of Cp*2Yb and Cp*2Sm. New J. Chem. 2016, 40, 6643–6649. [Google Scholar] [CrossRef]
- Dick, A.R.; Kampf, J.W.; Sanford, M.S. Unusually stable palladium(IV) Complexes: Detailed mechanistic investigation of C–O bond-forming reductive elimination. J. Am. Chem. Soc. 2005, 127, 12790–12791. [Google Scholar] [CrossRef] [PubMed]
- Shabashov, D.; Daugulis, O. Auxiliary-assisted palladium-catalyzed arylation and Alkylation of sp2 and sp3 carbon–hydrogen bonds. J. Am. Chem. Soc. 2010, 132, 3965–3972. [Google Scholar] [CrossRef] [PubMed]
- Rovira, M.; Roldán-Gómez, S.; Martin-Diaconescu, V.; Whiteoak, C.J.; Company, A.; Luis, J.M.; Ribas, X. Trifluoromethylation of a well-defined square-planar aryl–NiII complex involving NiIII/CF3· and NiIV–CF3 intermediate species. Chem. Eur. J. 2017, 23, 11662–11668. [Google Scholar] [CrossRef] [PubMed]
- Camasso, N.M.; Sanford, M.S. Design, synthesis, and carbon–heteroatom coupling reactions of organometallic nickel(IV) complexes. Science 2015, 347, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- D’Accriscio, F.; Borja, P.; Saffon-Merceron, N.; Fustier-Boutignon, M.; Mézailles, N.; Nebra, N. C–H bond trifluoromethylation of arenes enabled by a robust, high-valent nickel(IV) complex. Angew. Chem. Int. Ed. 2017, 56, 12898–12902. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d transition metals for C–H activation. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Han, F.-S. Transition-metal-catalyzed Suzuki–Miyaura cross-coupling reactions: A remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. [Google Scholar] [CrossRef]
- Peng, J.-B.; Wu, F.-P.; Wu, X.-F. First-row transition-metal-catalyzed carbonylative transformations of carbon electrophiles. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Maitlis, P.M.; Haynes, A.; Sunley, G.J.; Howard, M.J. Methanol carbonylation revisited: Thirty years on. J. Chem. Soc. Dalton Trans. 1996, 2187–2196. [Google Scholar] [CrossRef]
- Paulik, F.E.; Roth, J.F. Novel catalysts for the low-pressure carbonylation of methanol to acetic acid. Chem. Commun. 1968, 1578a. [Google Scholar] [CrossRef]
- Haynes, A.; Maitlis, P.M.; Morris, G.E.; Sunley, G.J.; Adams, H.; Badger, P.W.; Bowers, C.M.; Cook, D.B.; Elliott, P.I.P.; Ghaffar, T.; et al. Promotion of iridium-catalyzed methanol carbonylation: Mechanistic studies of the cativa process. J. Am. Chem. Soc. 2004, 126, 2847–2861. [Google Scholar] [CrossRef]
- Kaschube, W.; Pörschke, K.R.; Wilke, G. tmeda-Nickel-Komplexe: III. (N,N,N′,N′-Tetramethylethylendiamin)-(dimethyl)nickel(II). J. Organomet. Chem. 1988, 355, 525–532. [Google Scholar] [CrossRef]
- Nocton, G.; Ricard, L. N-aromatic heterocycle adducts of bulky [1,2,4-(Me3C)3C5H2]2Sm: Synthesis, structure and solution analysis. Dalton Trans. 2014, 43, 4380–4387. [Google Scholar] [CrossRef] [PubMed]
- Van Vleck, J.H. The Theory of Electric and Magnetic Susceptibilities; Oxford University Press: London, UK, 1932. [Google Scholar]
- O’Connor, C.J. Magnetochemistry—advances in theory and experimentation. In Progress in Inorganic Chemistry; Lippard, S.J., Ed.; Wiley: Hoboken, NJ, USA, 1982; Chapter 4. [Google Scholar]
- Lukens, W.W.; Walter, M.D. Quantifying exchange coupling in f-ion pairs using the diamagnetic substitution method. Inorg. Chem. 2010, 49, 4458–4465. [Google Scholar] [CrossRef]
- Lukens, W.W.; Magnani, N.; Booth, C.H. Application of the hubbard model to Cp*2Yb(bipy), a model system for strong exchange coupling in lanthanide systems. Inorg. Chem. 2012, 51, 10105–10110. [Google Scholar] [CrossRef][Green Version]
- Scarborough, C.C.; Wieghardt, K. Electronic structure of 2,2′-bipyridine organotransition-metal complexes. Establishing the ligand oxidation level by density functional theoretical calculations. Inorg. Chem. 2011, 50, 9773–9793. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.J.; Boncella, J.M.; Andersen, R.A. Preparation of coordination compounds of Cp*2Yb with heterocyclic nitrogen bases: Examples of antiferromagnetic exchange coupling across bridging ligands. Organometallics 2002, 21, 4622–4631. [Google Scholar] [CrossRef]
- Tilley, T.D.; Boncella, J.M.; Berg, D.J.; Burns, C.J.; Andersen, R.A.; Lawless, G.A.; Edelman, M.A.; Lappert, M.F. Bis[Bis(Trimethylsilyl)Amido]Bis(Diethyl Ether)Ytterbium and (Diethyl Ether)Bis(η5-Pentamethylcyclopentadienyl)Ytterbium. In Inorganic Syntheses; Ginsberg, A.P., Ed.; Wiley: Hoboken, NJ, USA, 2007; Chapter 27; pp. 146–150. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-electron scalar relativistic basis sets for third-row transition metal atoms. J. Chem. Theo. Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-electron scalar relativistic basis sets for the lanthanides. J. Chem. Theo. Comput. 2009, 5, 2229–2238. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallgr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Spek, A. Single-crystal structure validation with the program PLATON. J. App. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Moutet, J.; Tricoire, M.; Cordier, M.; Nocton, G. Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments. Inorganics 2019, 7, 58. https://doi.org/10.3390/inorganics7050058
Wang D, Moutet J, Tricoire M, Cordier M, Nocton G. Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments. Inorganics. 2019; 7(5):58. https://doi.org/10.3390/inorganics7050058
Chicago/Turabian StyleWang, Ding, Jules Moutet, Maxime Tricoire, Marie Cordier, and Grégory Nocton. 2019. "Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments" Inorganics 7, no. 5: 58. https://doi.org/10.3390/inorganics7050058
APA StyleWang, D., Moutet, J., Tricoire, M., Cordier, M., & Nocton, G. (2019). Reactive Heterobimetallic Complex Combining Divalent Ytterbium and Dimethyl Nickel Fragments. Inorganics, 7(5), 58. https://doi.org/10.3390/inorganics7050058