Heterobimetallic Chromium Manganese Carbonyl Nitrosyls: Comparison with Isoelectronic Homometallic Binuclear Chromium Carbonyl Nitrosyls and Manganese Carbonyls

Abstract
1. Introduction
2. Theoretical Methods
3. Results and Discussion
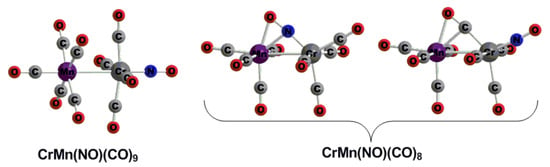
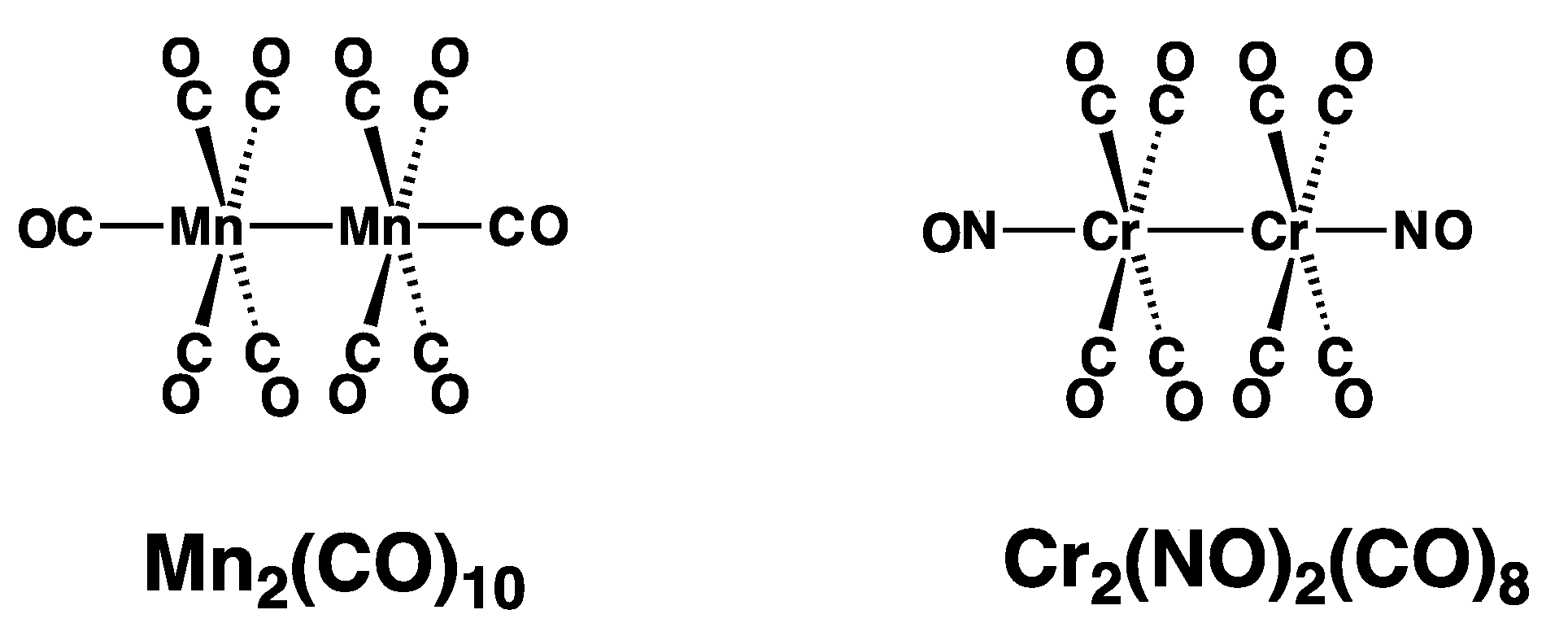
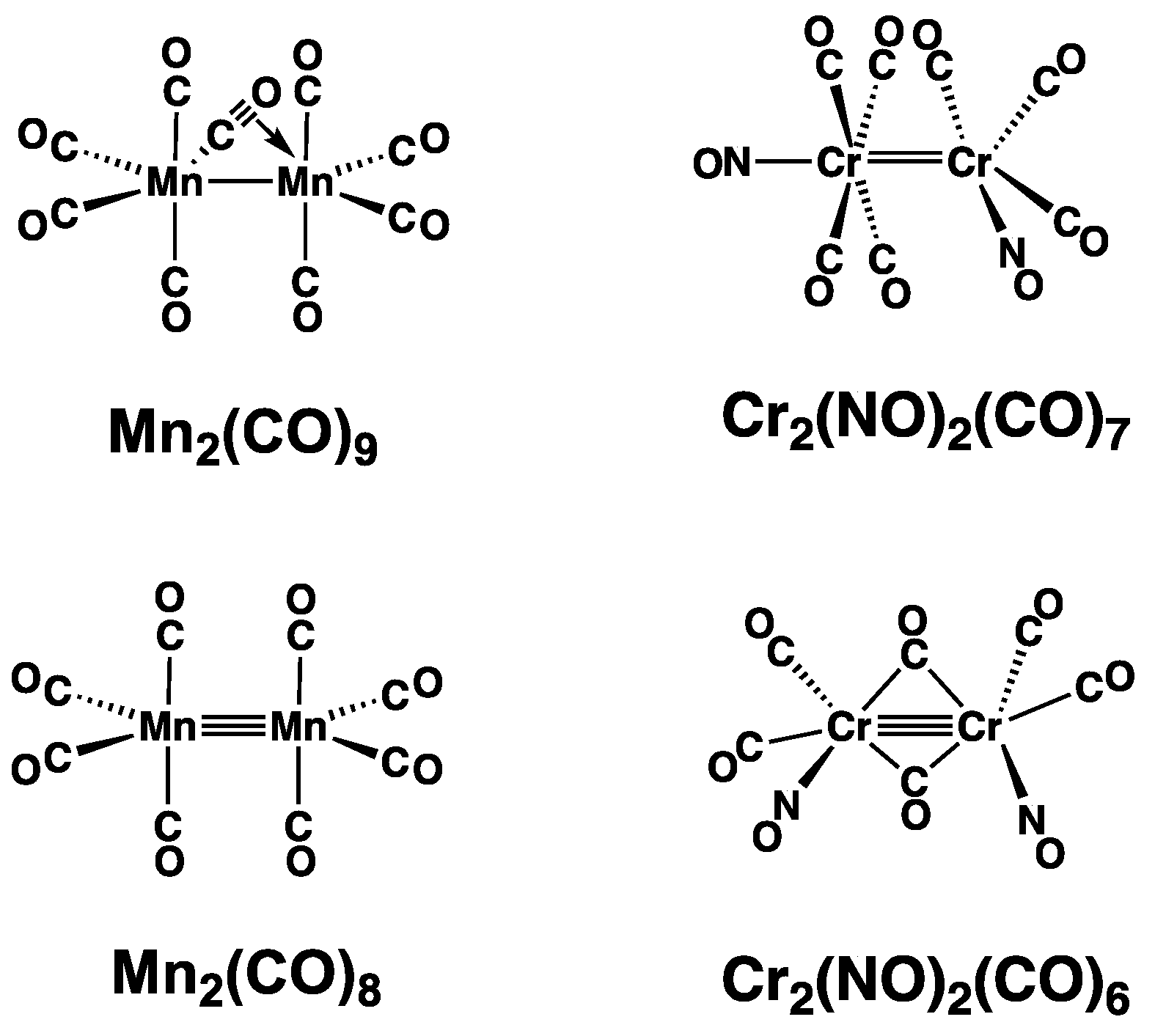
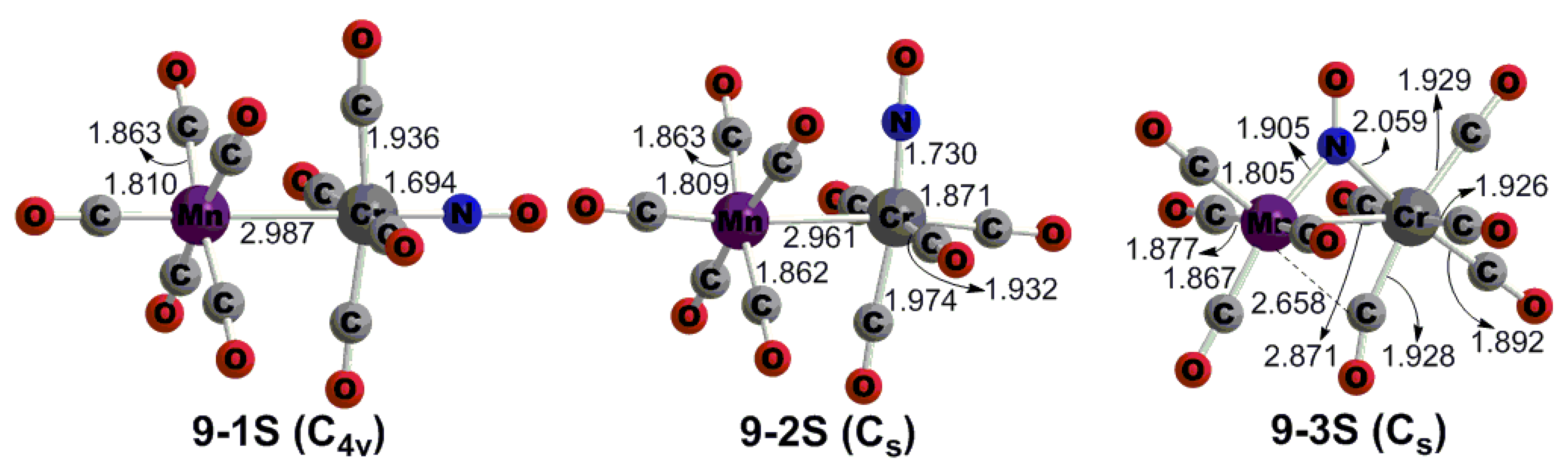
3.1. CrMn(NO)(CO)9
- (1)
- The staggered (OC)5Mn-Mn(CO)5 structure with D4d symmetry, which is the global minimum;
- (2)
- The eclipsed (OC)5Mn-Mn(CO)5 structure with D4h symmetry, which is a transition state with a small imaginary vibrational frequency and somewhat higher energy;
- (3)
- The doubly bridged (OC)4Mn(μ-CO)2Mn(CO)4 structure with D2h symmetry, like the global minimum of Cr2(CO)10 [45], which is also a transition state and has much higher energy.
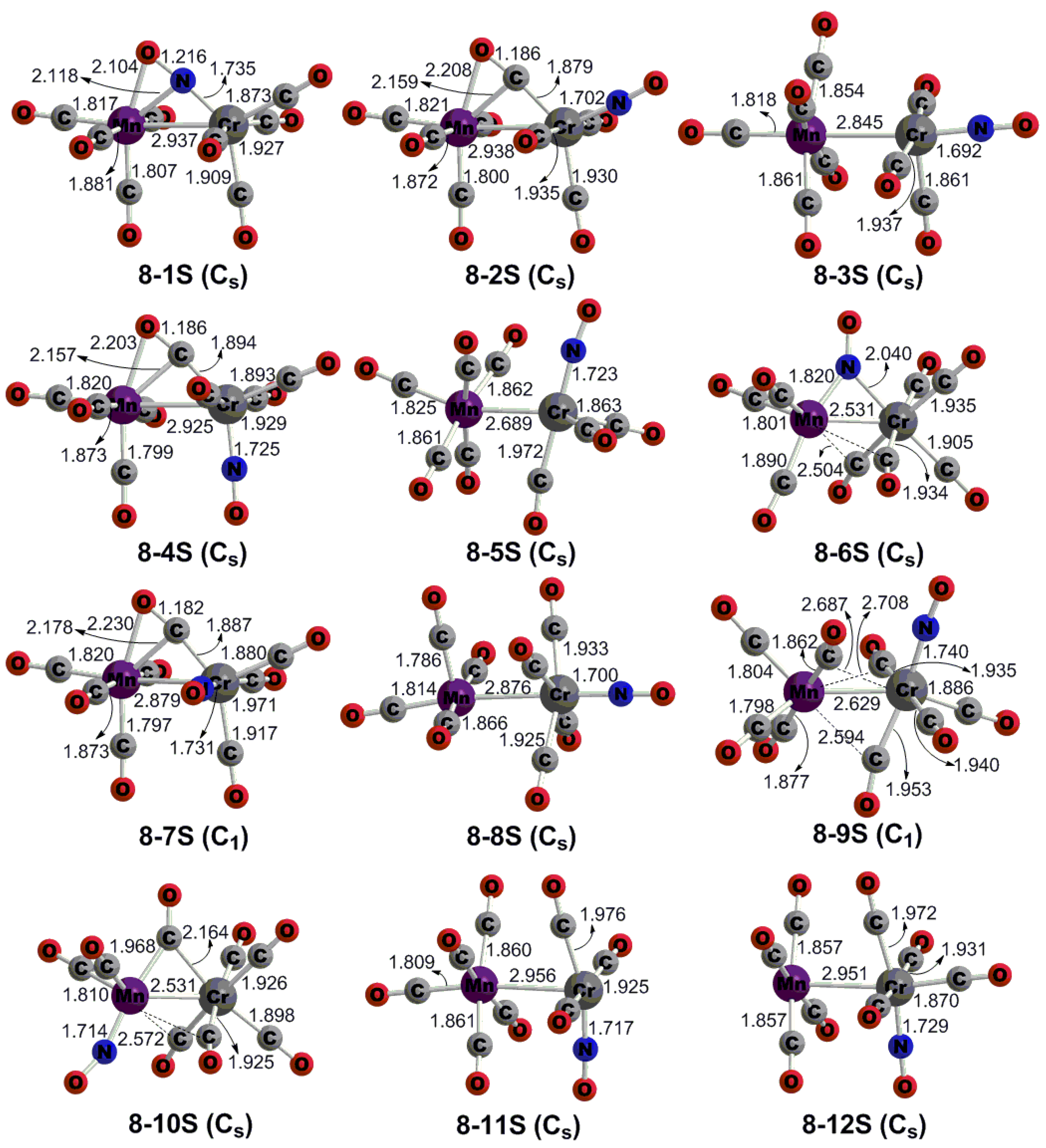
3.2. CrMn(NO)(CO)8
3.3. The Cr–Mn Wiberg Bond Indices Given by Natural Bond Orbital Analysis
3.4. Vibrational Frequencies
3.5. Carbonyl Dissociation Energies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brimm, E.O.; Lynch, M.A.; Sesny, W.J. Preparation and properties of manganese carbonyl. J. Am. Chem. Soc. 1954, 76, 3831–3835. [Google Scholar] [CrossRef]
- Closson, R.D.; Buzbee, L.R.; Ecke, G.C. A new metal carbonyl synthesis. J. Am. Chem. Soc. 1958, 80, 6167–6170. [Google Scholar] [CrossRef]
- Podall, H.E.; Dunn, J.H.; Shapiro, H. Reductive carbonylation synthesis of metal carbonyls. II. Synthesis of manganese carbonyl and group VI-B metal carbonyls by the alkylaluminum method. J. Am. Chem. Soc. 1960, 82, 1325–1330. [Google Scholar] [CrossRef]
- King, R.B.; Stokes, J.C.; Korenowski, T.F. A convenient synthesis of dimanganese decarbonyl from inexpensive starting materials at atmospheric pressure. J. Organometal. Chem. 1968, 11, 641–643. [Google Scholar] [CrossRef]
- Masters, A.P.; Parvez, M.; Sorensen, T. Preparation of Cr2(CO)8(NO)2 and X-ray crystal structures of Cr2(CO)8(NO)2 and Cr2(CO)9NO−. Stereochemical comparisons to Cr2(CO)102−. Can. J. Chem. 1991, 69, 2136–2141. [Google Scholar] [CrossRef]
- Dahl, L.F.; Ishishi, E.; Rundle, R.E. Polynuclear metal carbonyls. I. Structures of Mn2(CO)10 and Re2(CO)10. J. Chem. Phys. 1957, 26, 1750–1751. [Google Scholar] [CrossRef]
- Almenningen, A.; Jacobsen, G.G.; Seip, H.M. On the molecular structure of dimanganese decacarbonyl Mn2(CO)10. Acta Chem. Scand. 1969, 23, 685–686. [Google Scholar] [CrossRef]
- Churchill, M.R.; Amoh, K.N.; Wasserman, H.J. Redetermination of the crystal structure of dimanganese decacarbonyl and determination of the crystal structure of dirhenium decacarbonyl. Revised values for the manganese–manganese and rhenium–rhenium bond lengths in dimanganese decacarbonyl and dirhenium decacarbonyl. Inorg. Chem. 1981, 20, 1609–1611. [Google Scholar]
- Hepp, A.F.; Wrighton, M.S. Relative importance of metal–metal bond scission and loss of carbon monoxide from photoexcited dimanganese decacarbonyl: Spectroscopic detection of a coordinatively unsaturated, carbonyl-bridged dinuclear species in low-temperature alkane matrices. J. Am. Chem. Soc. 1983, 105, 5934–5935. [Google Scholar] [CrossRef]
- Kvietok, F.A.; Bursten, B.E. Matrix photochemistry of Mn2(CO)10: Reversible formation of Mn2(CO)8 from Mn2(CO)8(μ-η1:η2-CO). Organometallics 1995, 14, 2395–2399. [Google Scholar] [CrossRef]
- Dunkin, I.R.; Härter, P.; Shields, C.J. Recognition of a semibridging carbonyl group in dimanganese nonacarbonyl from plane-polarized photolysis of dimanganese decacarbonyl in argon matrixes at 12 K. J. Am. Chem. Soc. 1984, 106, 7248–7249. [Google Scholar] [CrossRef]
- Church, S.P.; Hermann, H.; Grevels, F.W. The primary photoproducts of Mn2(CO)10: Direct I.R. observation and decay kinetics of Mn(CO)5 and Mn2(CO)9 in hydrocarbon solution at room temperature. Chem. Commun. 1984, 12, 785–786. [Google Scholar] [CrossRef]
- Xie, Y.; Jang, J.H.; King, R.B.; Schaefer, H.F. Binuclear homoleptic manganese carbonyls: Mn2(CO)x (x = 10, 9, 8, 7). Inorg. Chem. 2003, 42, 5219–5230. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xie, Y.; Zhang, J.D.; King, R.B.; Schaefer, H.F. Chromium carbonyl nitrosyls: Comparison with isoelectronic manganese carbonyl derivatives. Inorg. Chem. 2007, 46, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Anders, U.; Graham, W.A.G. Organometallic compounds with metal–metal bonds. VI. Preparation and infrared spectra of the carbonylmetalate ions [(OC)5M-M′(CO)5]− (M = Mn, Re; M′ = Cr, Mo, W). J. Am. Chem. Soc. 1967, 89, 539–541. [Google Scholar] [CrossRef]
- Ruff, J.K. The chemistry of the dinuelear carbonyl anions. II. Mixed-metal derivatives. Inorg. Chem. 1968, 7, 1818–1821. [Google Scholar] [CrossRef]
- Park, Y.K.; Kim, S.J.; Kim, J.H.; Han, I.S.; Lee, C.H. Kinetic studies of the cleavage of M+[M′M″(CO)9L]− (M+ = Na+, PPN+; M′ = Cr, W.; M″ = Mn, Re; L = CO, PR3). J. Organomet. Chem. 1991, 408, 193–202. [Google Scholar] [CrossRef]
- Park, Y.K.; Han, I.S.; Kim, J.S.; Lee, C.J.; Baek, Y.O.; Song, G.O. Kinetic studies on the reactions of the heterobimetallic anion (OC)5CrMn(CO)5−M+ (M+ = Na+, PPN+) with CH3I. Bull. Korean Chem. Soc. 1994, 15, 537–541. [Google Scholar]
- Park, Y.K.; Kim, G.S.; Song, G.O. Kinetic studies on the reactions of the heterobimetallic anion (OC)5CrMn(CO)5−M+ (M+ = Na+, PPN+) with allyl bromide. Bull. Korean Chem. Soc. 1995, 16, 310–315. [Google Scholar]
- Kruck, T.; Höfler, M.; Noack, M. Reaktionsweisen von Rhenium(I)-Kohlenoxidkomplexen und neue Anschauungen über den Mechanismus der Basenreaktion von Metallcarbonylen. Chem. Ber. 1966, 99, 1153–1167. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis functions for use in molecular calculations. I. Contraction of (9s5p) atomic basis sets for the first-row atoms. J. Chem. Phys. 1970, 53, 2823–2833. [Google Scholar] [CrossRef]
- Huzinaga, S. Gaussian-type functions for polyatomic systems. I. J. Chem. Phys. 1965, 42, 1293–1302. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian basis set for molecular wavefunctions containing third-row atoms. J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar] [CrossRef]
- Hood, D.M.; Pitzer, R.M.; Schaefer, H.F. Electronic structure of homoleptic transition metal hydrides: TiH4, VH4, CrH4, MnH4, FeH4, CoH4, and NiH4. J. Chem. Phys. 1979, 71, 705–712. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Balabanov, N.B.; Peterson, K.A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc–Zn. J. Chem. Phys 2005, 123, 064107. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision D 01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Hileman, J.C.; Huggins, D.K.; Kaesz, H.D. Derivatives of technetium carbonyl. Synthesis and properties of the carbonyl halides and the pentacarbonyl hydride. Inorg. Chem. 1962, 1, 933–938. [Google Scholar] [CrossRef]
- Flitcroft, N.; Huggins, D.K.; Kaesz, H.D. Infrared spectra of (CO)5Mn-Re(CO)5 and the carbonyls of manganese, technetium, and rhenium; assignment of C≡O and M=C stretching absorptions. Inorg. Chem. 1964, 3, 1123–1130. [Google Scholar] [CrossRef]
- Haas, H.; Sheline, R.K. Analysis of the infrared spectra of metal carbonyls. J. Chem. Phys. 1967, 47, 2996–3021. [Google Scholar] [CrossRef]
- Rosa, A.; Ricciarde, G.; Baerends, E.J.; Stufkens, D.J. Density functional study of ground and excited states of Mn2(CO)10. Inorg. Chem. 1995, 34, 3425–3432. [Google Scholar] [CrossRef]
- Rosa, A.; Ricciarde, G.; Baerends, E.J.; Stufkens, D.J. Density functional study of the photodissociation of Mn2(CO)10. Inorg. Chem. 1996, 35, 2886–2897. [Google Scholar] [CrossRef]
- Decker, S.A.; Donini, O.; Klobukowski, M. A contribution to the understanding of carbonyl migration in Mn2(CO)10 via the pairwise exchange mechanism. J. Phys. Chem. A 1997, 101, 8734–8740. [Google Scholar] [CrossRef]
- Liddell, M.J. Organometallic clusters: What is an appropriate DFT treatment? J. Organomet. Chem. 1998, 565, 271–277. [Google Scholar] [CrossRef]
- van Gisbergen, S.J.A.; Groeneveld, J.A.; Rosa, A.; Snijders, J.G.; Baerends, E.J. Excitation energies for transition metal compounds from time-dependent density functional theory. Applications to MnO4−, Ni(CO)4, and Mn2(CO)10. J. Phys. Chem. A 1999, 103, 6835–6844. [Google Scholar] [CrossRef]
- Barchholtz, T.A.; Bursten, B.E. Density functional calculations of dinuclear organometallic carbonyl complexes. Part I: Metal–metal and metal–CO bond energies. J. Organomet. Chem. 2000, 596, 212–220. [Google Scholar]
- Li, S.; Richardson, N.R.; Xie, Y.; King, R.B.; Schaefer, H.F. The rule breaking Cr2(CO)10. A 17 electron Cr system or a Cr=Cr double bond? Faraday Discuss. 2003, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Richardson, N.R.; Xie, Y.; King, R.B.; Schaefer, H.F. Flat potential energy surface of the saturated binuclear homoleptic chromium carbonyl Cr2(CO)11 with one, two, and three bridging carbonyls: Comparison with the well-known [HCr2(CO)10]− anion and the related [(μ-H)2Cr2(CO)9]2− and [(μ-H)2Cr2(CO)8]2− dianions. J. Phys. Chem. A 2001, 105, 11134–11143. [Google Scholar]
- Wang, H.; Xie, Y.; King, R.B.; Schaefer, H.F. Remarkable aspects of unsaturation in trinuclear metal carbonyl clusters: The triiron species Fe3(CO)n (n = 12, 11, 10, 9). J. Am. Chem. Soc. 2006, 128, 11376–11384. [Google Scholar] [CrossRef] [PubMed]
- Jonas, V.; Thiel, W. Theoretical study of the vibrational spectra of the transition metal carbonyls M(CO)6 [M = Cr, Mo, W], M(CO)5 [M = Fe, Ru, Os], and M(CO)4 [M = Ni, Pd, Pt]. J. Phys. Chem. 1995, 102, 8474–8484. [Google Scholar] [CrossRef]
- Silaghi-Dumitrescu, I.; Bitterwolf, T.E.; King, R.B. Butterfly diradical intermediates in photochemical reactions of Fe2(CO)6(μ-S2). J. Am. Chem. Soc. 2006, 128, 5342–5343. [Google Scholar] [CrossRef]
- Sunderlin, L.S.; Wang, D.; Squires, R.R. Bond strengths in first-row-metal carbonyl anions. J. Am. Chem. Soc. 1993, 115, 12060–12070. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | State (Sym.) | ΔE | Cr–Mn | Structure | State (Sym.) | ΔE | Cr–Mn |
|---|---|---|---|---|---|---|---|
| 9-1S | 1A1 (C4v) | 0.0 | 2.987 | 8-5S | 1A′ (Cs) | 6.4 | 2.689 |
| 9-2S | 1A′ (Cs) | 5.8 | 2.961 | 8-6S | 1A′ (Cs) | 6.5 | 2.531 |
| 9-3S | 1A′ (Cs) | 20.2 | 2.871 | 8-7S | 1A (C1) | 7.4 | 2.879 |
| 8-8S | 1A′ (Cs) | 8.2 | 2.876 | ||||
| 8-1S | 1A′ (Cs) | 0.0 | 2.937 | 8-9S | 1A (C1) | 11.4 | 2.629 |
| 8-2S | 1A′ (Cs) | 1.1 | 2.938 | 8-10S | 1A′ (Cs) | 11.5 | 2.531 |
| 8-3S | 1A′ (Cs) | 3.5 | 2.845 | 8-11S | 1A′ (Cs) | 17.8 | 2.956 |
| 8-4S | 1A′ (Cs) | 3.6 | 2.925 | 8-12S | 1A′ (Cs) | 21.6 | 2.951 |
| Structure | Cr–Mn (Å) | Formal Bond Order | WBI (Cr–Mn) | Structure | Cr–Mn (Å) | Formal Bond Order | WBI (Cr–Mn) |
|---|---|---|---|---|---|---|---|
| 9-1S | 2.987 | 1 | 0.11 | 8-5S | 2.689 | 2 | 0.18 |
| 9-2S | 2.961 | 1 | 0.11 | 8-6S | 2.531 | 2 | 0.22 |
| 9-3S | 2.871 | 1 | 0.13 | 8-7S | 2.879 | 1 | 0.14 |
| 8-8S | 2.876 | 1 | 0.15 | ||||
| 8-1S | 2.937 | 1 | 0.12 | 8-9S | 2.629 | 2 | 0.22 |
| 8-2S | 2.938 | 1 | 0.14 | 8-10S | 2.531 | 2 | 0.26 |
| 8-3S | 2.845 | 1 | 0.15 | 8-11S | 2.956 | 1 | 0.18 |
| 8-4S | 2.925 | 1 | 0.14 | 8-12S | 2.951 | 1 | 0.22 |
| Struct. | ν(CO) * | ν(NO) * |
|---|---|---|
| 9-1S | 1966 (0), 1966 (0), 1983 (298), 1994 (0), 1999 (2182), 1999 (2182), 2002 (0), 2020 (819), 2082 (86) | 1807 (984) |
| 9-2S | 1965 (5), 1970 (550), 1977 (601), 1983 (146), 1996 (2193), 2000 (1133), 2001 (37), 2021 (1285), 2078 (70) | 1804 (641) |
| 9-3S | 1923 (371), 1961 (184), 1981 (230), 1983 (988), 1996 (2151), 1997 (740), 2003 (145), 2031 (1642), 2076 (11) | 1595 (363) |
| 8-1S | 1954 (517), 1970 (86), 1970 (346), 1984 (869), 1995 (2232), 1996 (393), 2027 (1112), 2073 (93) | 1511 (397) |
| 8-2S | 1742 (406), 1964 (204), 1966 (39), 1987 (463), 1999 (2207), 2000 (609), 2024 (845), 2070 (189) | 1803 (991) |
| 8-3S | 1951 (20), 1965 (91), 1978 (1476), 1985 (320), 1988 (1127), 1993 (1058), 2011 (1039), 2068 (145) | 1792 (856) |
| 8-4S | 1739 (499), 1966 (2), 1971 (701), 1978 (700), 1990 (107), 1995 (2296), 2026 (1184), 2067 (44) | 1801 (430) |
| 8-5S | 1953 (250), 1962 (19), 1972 (1451), 1976 (992), 1993 (409), 1995 (309), 2004 (1818), 2065 (238) | 1785 (718) |
| 8-6S | 1942 (476), 1944 (228), 1966 (1020), 1975 (643), 1995 (1175), 2002 (566), 2013 (1844), 2063 (133) | 1618 (415) |
| 8-7S | 1756 (402), 1960 (128), 1974 (621), 1978 (1193), 1987 (651), 1999 (734), 2019 (1427), 2064 (180) | 1810 (673) |
| 8-8S | 1949 (11), 1960 (67), 1972 (260), 1977 (1830), 1981 (89), 1988 (2072), 2010 (785), 2063 (60) | 1805 (1093) |
| 8-9S | 1925 (208), 1941 (620), 1959 (97), 1970 (1165), 1978 (566), 1992 (1192), 2012 (1718), 2056 (51) | 1796 (753) |
| 8-10S | 1853 (230), 1950 (558), 1957 (879), 1977 (959), 1993 (545), 1995 (691), 2011 (1662), 2060 (273) | 1775 (897) |
| 8-11S | 1939 (557), 1955 (17), 1979 (384), 1987 (1996), 1987 (1448), 1999 (0), 2004 (827), 2071 (133) | 1779 (661) |
| 8-12S | 1941 (318), 1947 (933), 1969 (556), 1985 (2029), 1986 (106), 1989 (882), 2012 (712), 2065 (66) | 1800 (579) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Wen, L.; King, R.B. Heterobimetallic Chromium Manganese Carbonyl Nitrosyls: Comparison with Isoelectronic Homometallic Binuclear Chromium Carbonyl Nitrosyls and Manganese Carbonyls. Inorganics 2019, 7, 127. https://doi.org/10.3390/inorganics7100127
Li G, Wen L, King RB. Heterobimetallic Chromium Manganese Carbonyl Nitrosyls: Comparison with Isoelectronic Homometallic Binuclear Chromium Carbonyl Nitrosyls and Manganese Carbonyls. Inorganics. 2019; 7(10):127. https://doi.org/10.3390/inorganics7100127
Chicago/Turabian StyleLi, Guoliang, Limei Wen, and R. Bruce King. 2019. "Heterobimetallic Chromium Manganese Carbonyl Nitrosyls: Comparison with Isoelectronic Homometallic Binuclear Chromium Carbonyl Nitrosyls and Manganese Carbonyls" Inorganics 7, no. 10: 127. https://doi.org/10.3390/inorganics7100127
APA StyleLi, G., Wen, L., & King, R. B. (2019). Heterobimetallic Chromium Manganese Carbonyl Nitrosyls: Comparison with Isoelectronic Homometallic Binuclear Chromium Carbonyl Nitrosyls and Manganese Carbonyls. Inorganics, 7(10), 127. https://doi.org/10.3390/inorganics7100127