Phenylacetylene and Carbon Dioxide Activation by an Organometallic Samarium Complex

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
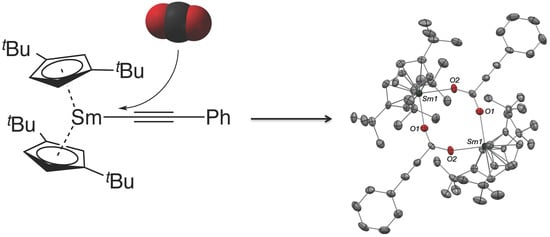
2. Results and Discussion
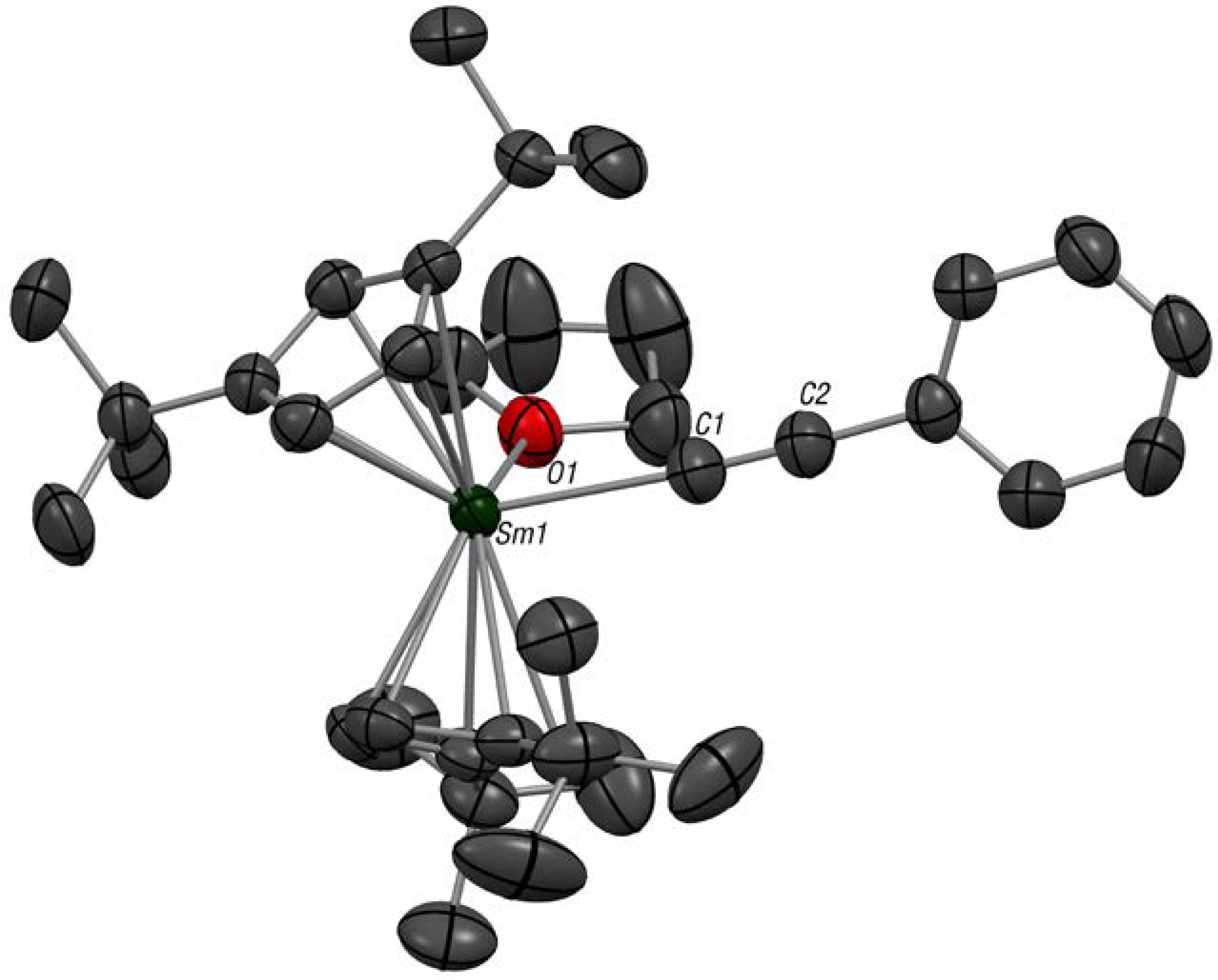
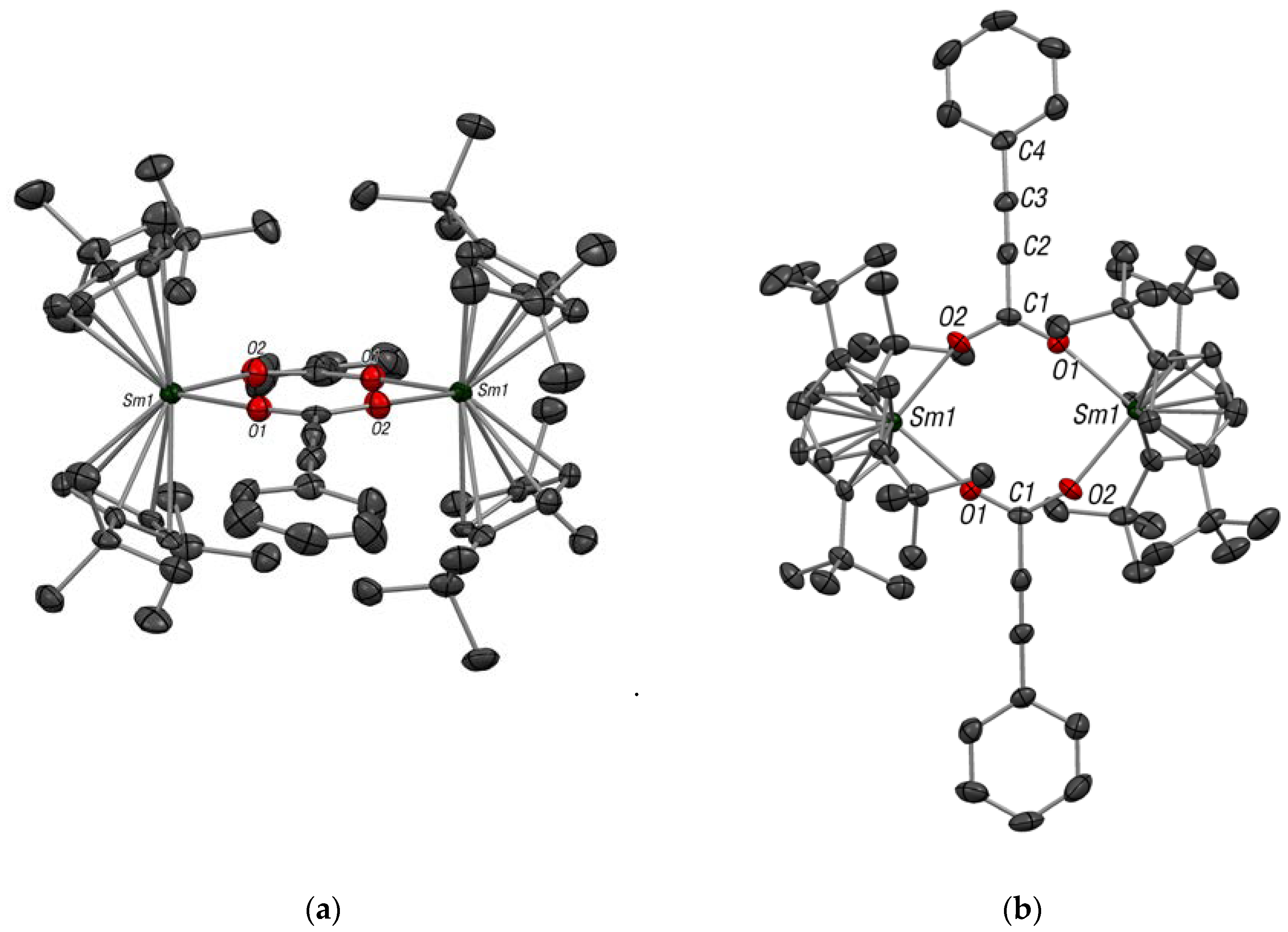
2.1. Synthesis, Solid-State and Solution Structures
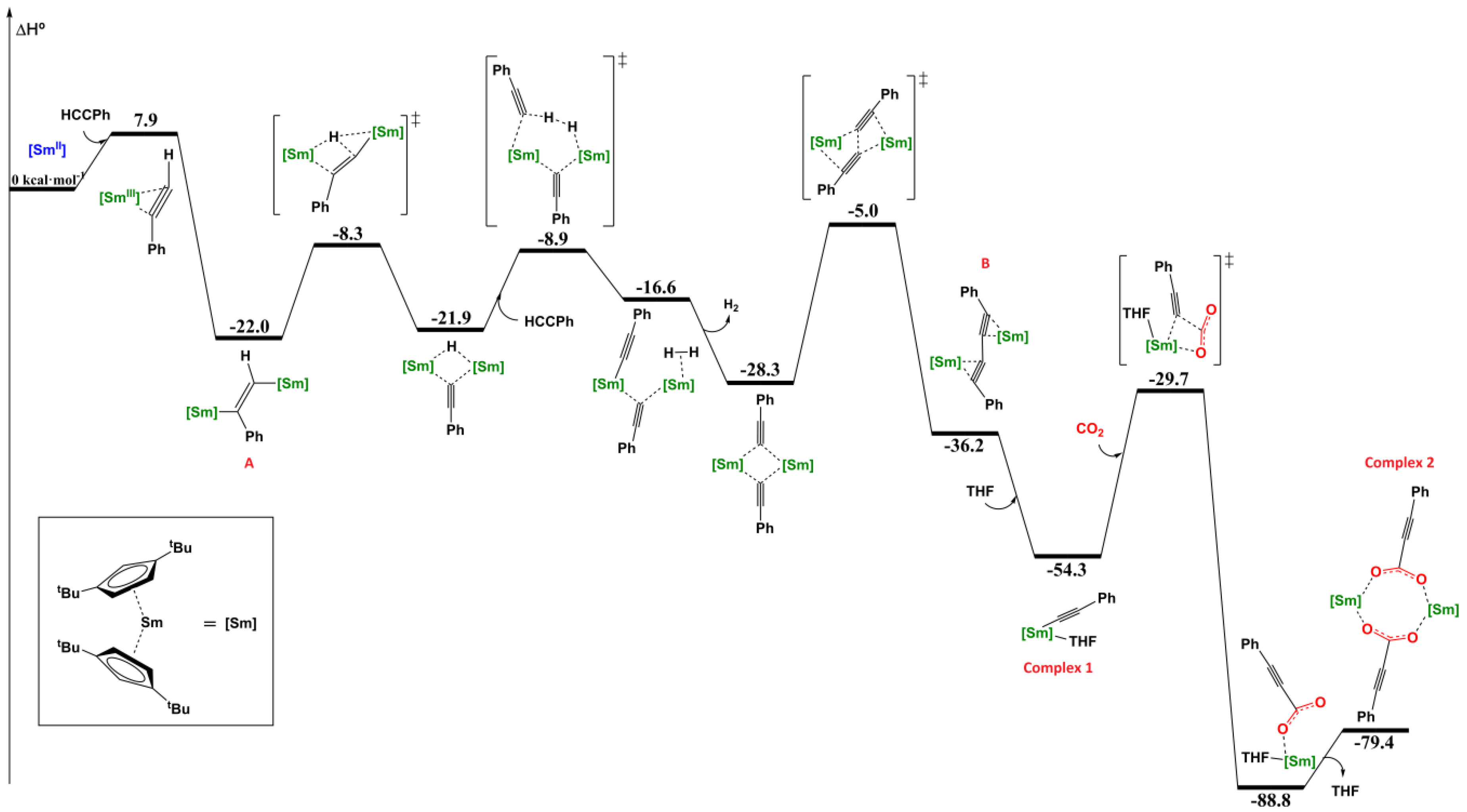
2.2. Theoretical Studies
3. Experimental Section
3.1. General Considerations
3.2. Synthesis of Cptt2Sm(C≡C–Ph)(thf) (1)
3.3. Synthesis of [Cptt2Sm(O2C–C≡C–Ph)]2 (2)
3.4. X-Ray Diffraction
3.5. Theoretical Computations Diffraction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crutchley, R.J. Preface. Coord. Chem. Rev. 2017, 334, 1. [Google Scholar] [CrossRef]
- Milani, B.; Licini, G.; Clot, E.; Albrecht, M. Small Molecule Activation. Dalton Trans. 2016, 45, 14419–14420. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-H.; Chuang, H.-J.; Lin, P.-H.; Ko, B.-T. Copolymerization of carbon dioxide with cyclohexene oxide catalyzed by bimetallic dysprosium complexes containing hydrazine-functionalized Schiff-base derivatives. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 321–328. [Google Scholar] [CrossRef]
- Tolman, W.B. Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights. Angew. Chem. Int. Ed. 2010, 49, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef] [PubMed]
- Summerscales, O.T.; Cloke, F.G.N.; Hitchcock, P.B.; Green, J.C.; Hazari, N. Reductive Cyclotrimerization of Carbon Monoxide to the Deltate Dianion by an Organometallic Uranium Complex. Science 2006, 311, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.; Selent, D.; Borner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Hugues, L.A.; Hanusa, T.P. Synthesis and crystallographic characterization of an unsolvated, monomeric samarium bis(pentamethylcyclopentadienyl) organolanthanide complex, (C5Me5)2Sm. J. Am. Chem. Soc. 1984, 106, 4270–4272. [Google Scholar] [CrossRef]
- Evans, W.J.; Grate, J.W.; Bloom, I.; Hunter, W.E.; Atwood, J.L. Synthesis and X-ray crystallographic characterization of an oxo-bridged bimetallic organosamarium complex, [(C5Me)2Sm]2(µ-O). J. Am. Chem. Soc. 1985, 107, 405–409. [Google Scholar] [CrossRef]
- Evans, W.J.; Seibel, C.A.; Ziller, J.W. Organosamarium-mediated transformations of CO2 and COS: Monoinsertion and disproportionation reactions and the reductive coupling of CO2 to [O2CCO2]2−. Inorg. Chem. 1998, 37, 770–776. [Google Scholar] [CrossRef]
- Davies, N.W.; Frey, A.S.P.; Gardiner, M.G.; Wang, J. Reductive disproportionation of carbon dioxide by a Sm(II) complex: Unprecedented f-block element reactivity giving a carbonate complex. Chem. Commun. 2006, 4853–4855. [Google Scholar] [CrossRef]
- Castro, L.; Labouille, S.; Kindra, D.R.; Ziller, J.W.; Nief, F.; Evans, W.J.; Maron, L. Insights into the mechanism of reaction of [(C5Me)2SmII(thf)2] with CO2 and COS by DFT studies. Chem. Eur. J. 2012, 18, 7886–7895. [Google Scholar] [CrossRef] [PubMed]
- Andrez, J.; Pécaut, J.; Bayle, P.; Mazzanti, M. Tuning Lanthanide Reactivity towards Small Molecules with Electron-Rich Siloxide Ligands. Angew. Chem. Int. Ed. 2014, 53, 10448–10452. [Google Scholar] [CrossRef] [PubMed]
- Tsoureas, N.; Castro, L.; Kilpatrick, A.F.R.; Cloke, F.G.N.; Maron, L. Controlling selectivity in the reductive activation of CO2 by mixed sandwich uranium(III) complexes. Chem. Sci. 2014, 5, 3777–3788. [Google Scholar] [CrossRef]
- Xemard, M.; Goudy, V.; Braun, A.; Tricoire, M.; Cordier, M.; Ricard, L.; Castro, L.; Louyriac, E.; Kefalidis, C.E.; Clavaguéra, C.; et al. Reductive disproportionation of CO2 with bulky divalent samarium complexes. Organometallics 2017, 36, 4660–4668. [Google Scholar] [CrossRef]
- Evans, W.J.; Keyer, R.A.; Ziller, J.W. Carbon–carbon bond formation by coupling of two phenylethynyl ligand in an organolanthanide system. Organometallics 1990, 9, 2628–2631. [Google Scholar] [CrossRef]
- Kefalidis, C.E.; Maron, L. On the dehydrocoupling of alkenylacetylenes mediated by various samarocene complexes: A charming story of metal cooperativity revealing a novel dual metal σ-bond metathesis type of mechanism (DM|σ-BM). Inorganics 2015, 3, 573–588. [Google Scholar] [CrossRef]
- Bel’sky, V.K.; Gunko, Y.K.; Bulychev, B.M.; Sizov, A.I.; Soloveichik, G.L. Dicyclopentadienylsamarium complexes with t-butyl substituents in the ring. Crystal and molecular structures of the solvate (η5-C5H3Bu2t)2Sm·OC4H8 and homoleptic ate complex [NaSm(η5:η2-C5H4But)3·OC4H8]n. J. Organomet. Chem. 1990, 390, 35–44. [Google Scholar] [CrossRef]
- Evans, W.J.; Keyer, R.A.; Ziller, J.W. Investigation of organolanthanide-based carbon-carbon bond formation: Synthesis, structure, and coupling reactivity of organolanthanide alkynide complexes, including the unusual structures of the trienediyl complex [(C5Me5)2Sm]2[µ-η2:η2-Ph(CH2)2C=C=C=C–(CH2)2Ph] and the unsolvated alkynide [(C5Me5)2Sm(C≡CCMe3)]2. Organometallics 1993, 12, 2618–2633. [Google Scholar] [CrossRef]
- Evans, W.J.; Ulibarri, T.A. Reactivity of (C5Me5)2Sm with cyclopentadiene and cyclopentadienide: Isolation of the mixed-valence complex (C5Me5)2Sm(III)(µ-C5H5)Sm(II)(C5Me5)2. J. Am. Chem. Soc. 1987, 109, 4292–4297. [Google Scholar] [CrossRef]
- Evans, W.J.; Chamberlain, L.R.; Ulibarri, T.A.; Ziller, J.W. Reactivity of trimethylaluminum with (C5Me5)2Sm(THF)2: Synthesis, structure, and reactivity of the samarium methyl complexes (C5Me5)2Sm[(µ-Me)AlMe2(µ-Me)]2Sm(C5Me5)2 and (C5Me5)2SmMe(THF). J. Am. Chem. Soc. 1988, 110, 6423–6432. [Google Scholar] [CrossRef]
- Evans, W.J.; Perotti, J.M.; Ziller, J.W. Synthetic Utility of [(C5Me5)2Ln][(μ-Ph)2BPh2] in Accessing [(C5Me5)2LnR]x Unsolvated Alkyl Lanthanide Metallocenes, Complexes with High C−H Activation Reactivity. J. Am. Chem. Soc. 2005, 127, 3894–3909. [Google Scholar] [CrossRef] [PubMed]
- Manjolinho, F.; Arndt, M.; Gooßen, K.; Gooßen, L.J. Catalytic C–H Carboxylation of Terminal Alkynes with Carbon Dioxide. ACS Catal. 2012, 2, 2014–2021. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A Found Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Burke, K.; Perdew, J.P.; Wang, Y. Electronic Density Functional Theory: Recent Progress and New Directions; Dobson, J.F., Vignale, G., Das, M.P., Eds.; Plenum: New York, NY, USA, 1998. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Savin, A.; Preuss, H. Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goudy, V.; Xémard, M.; Karleskind, S.; Cordier, M.; Alvarez Lamsfus, C.; Maron, L.; Nocton, G. Phenylacetylene and Carbon Dioxide Activation by an Organometallic Samarium Complex. Inorganics 2018, 6, 82. https://doi.org/10.3390/inorganics6030082
Goudy V, Xémard M, Karleskind S, Cordier M, Alvarez Lamsfus C, Maron L, Nocton G. Phenylacetylene and Carbon Dioxide Activation by an Organometallic Samarium Complex. Inorganics. 2018; 6(3):82. https://doi.org/10.3390/inorganics6030082
Chicago/Turabian StyleGoudy, Violaine, Mathieu Xémard, Simon Karleskind, Marie Cordier, Carlos Alvarez Lamsfus, Laurent Maron, and Grégory Nocton. 2018. "Phenylacetylene and Carbon Dioxide Activation by an Organometallic Samarium Complex" Inorganics 6, no. 3: 82. https://doi.org/10.3390/inorganics6030082
APA StyleGoudy, V., Xémard, M., Karleskind, S., Cordier, M., Alvarez Lamsfus, C., Maron, L., & Nocton, G. (2018). Phenylacetylene and Carbon Dioxide Activation by an Organometallic Samarium Complex. Inorganics, 6(3), 82. https://doi.org/10.3390/inorganics6030082