Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K) §


Abstract
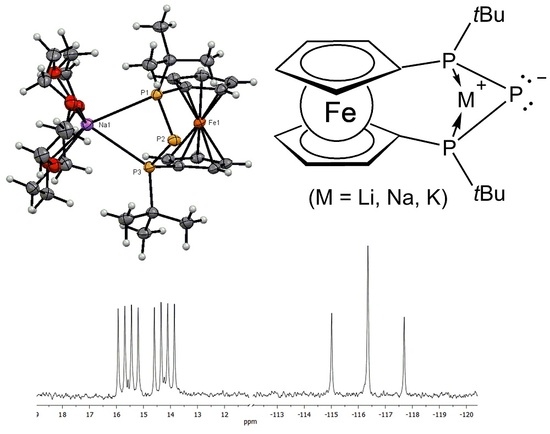
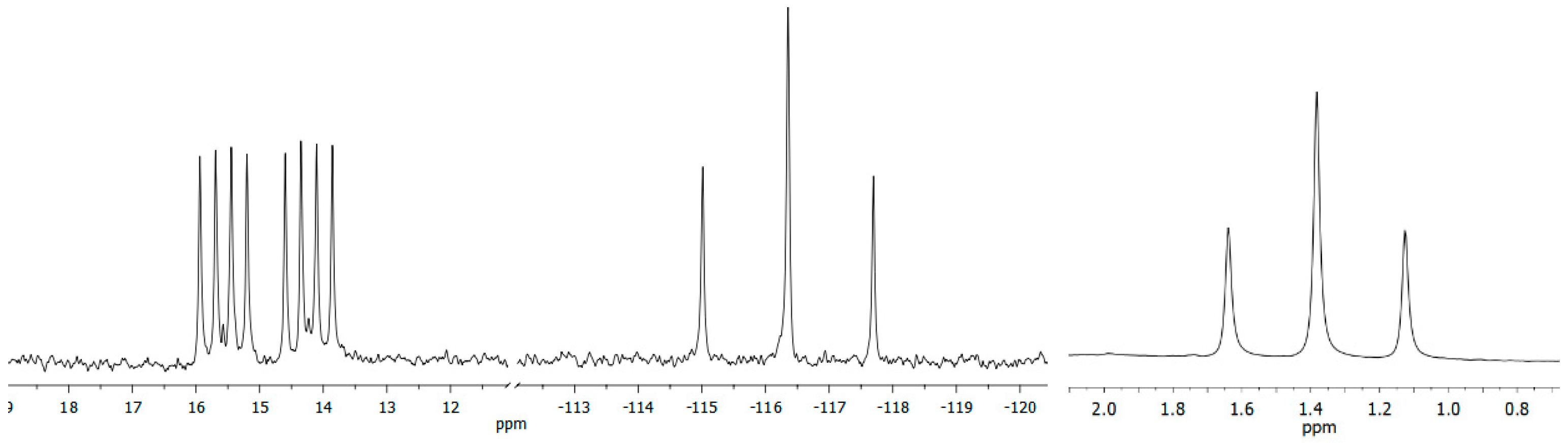
:
1. Introduction
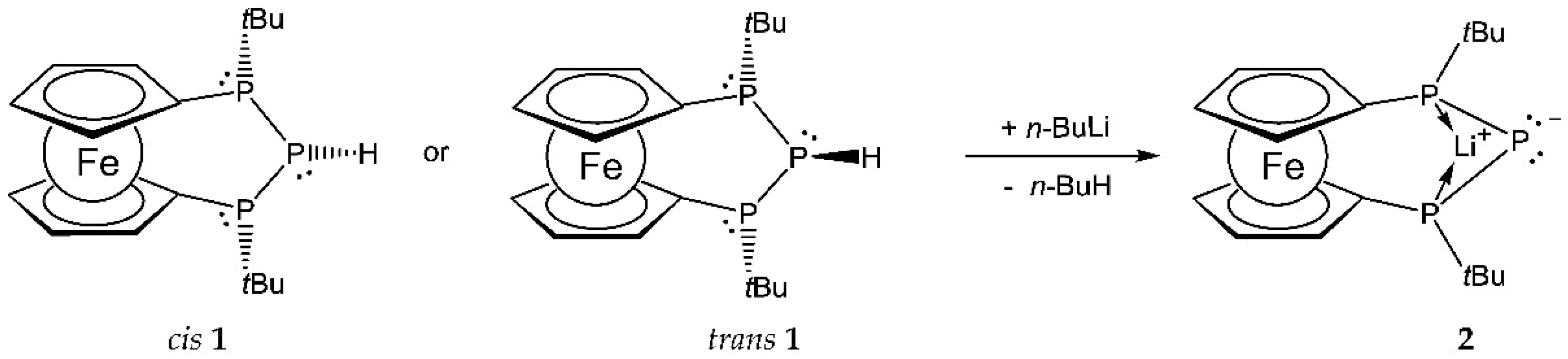
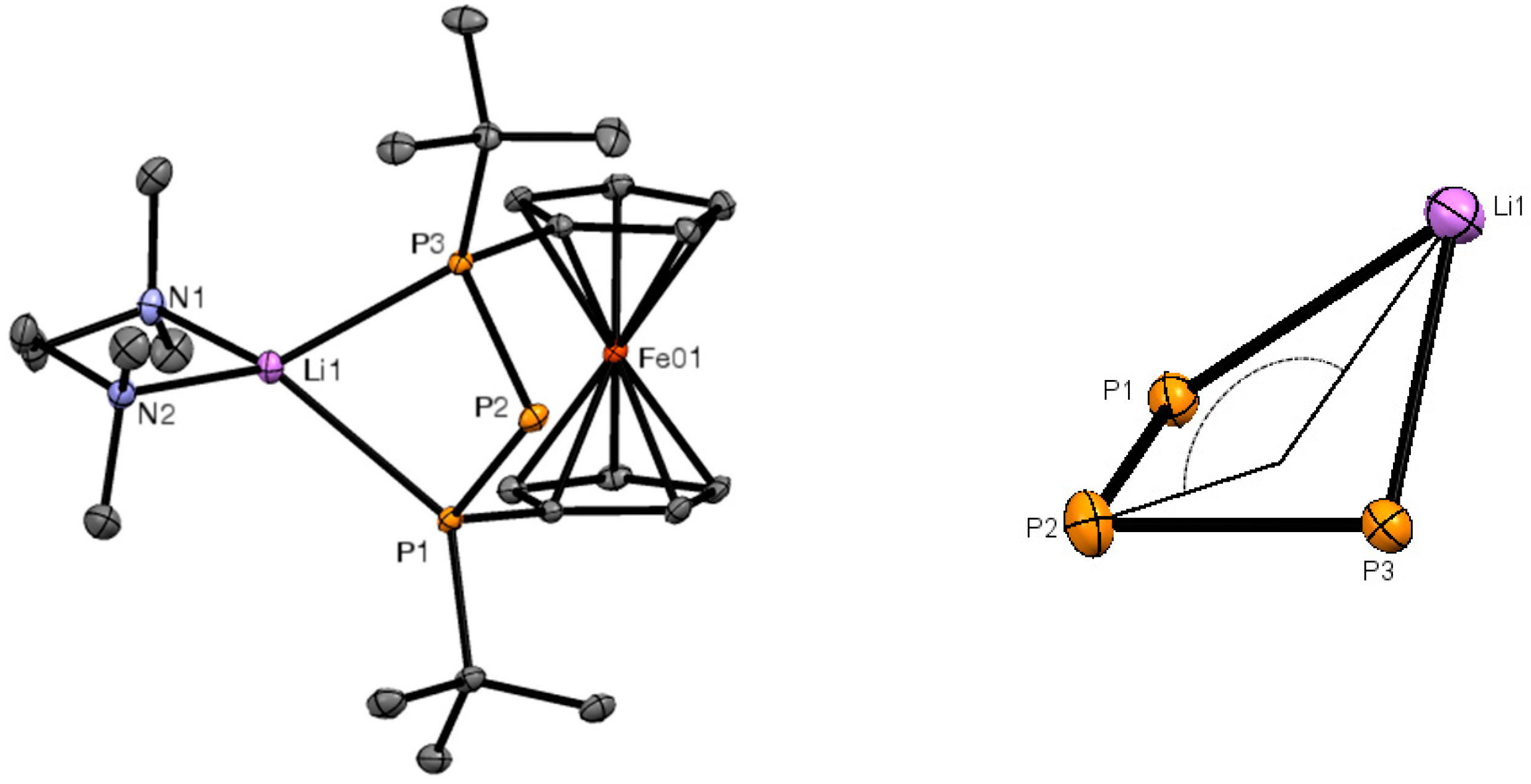
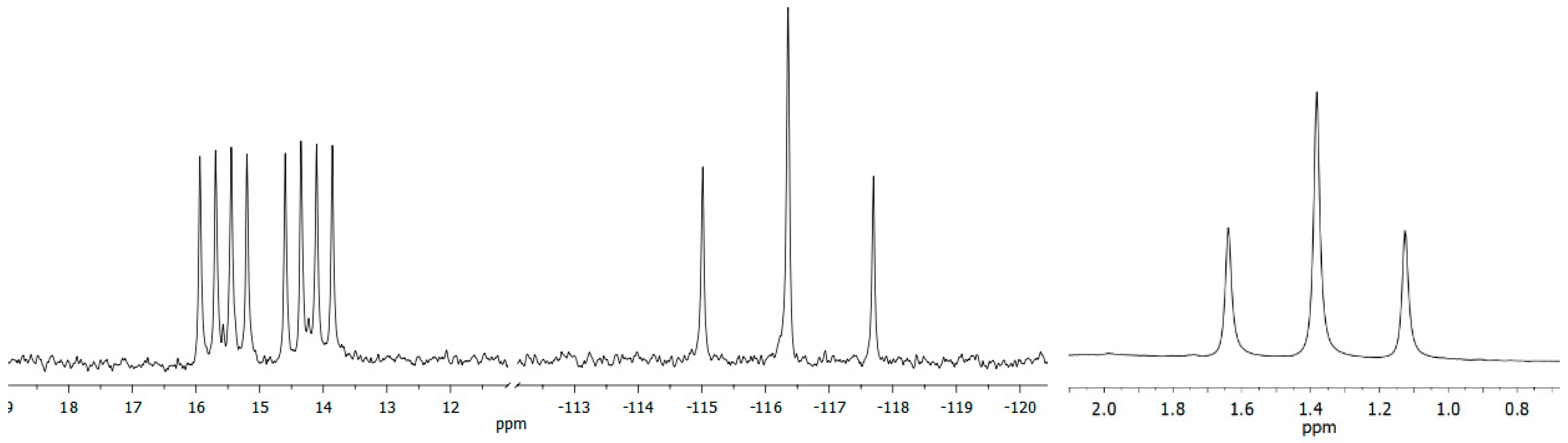
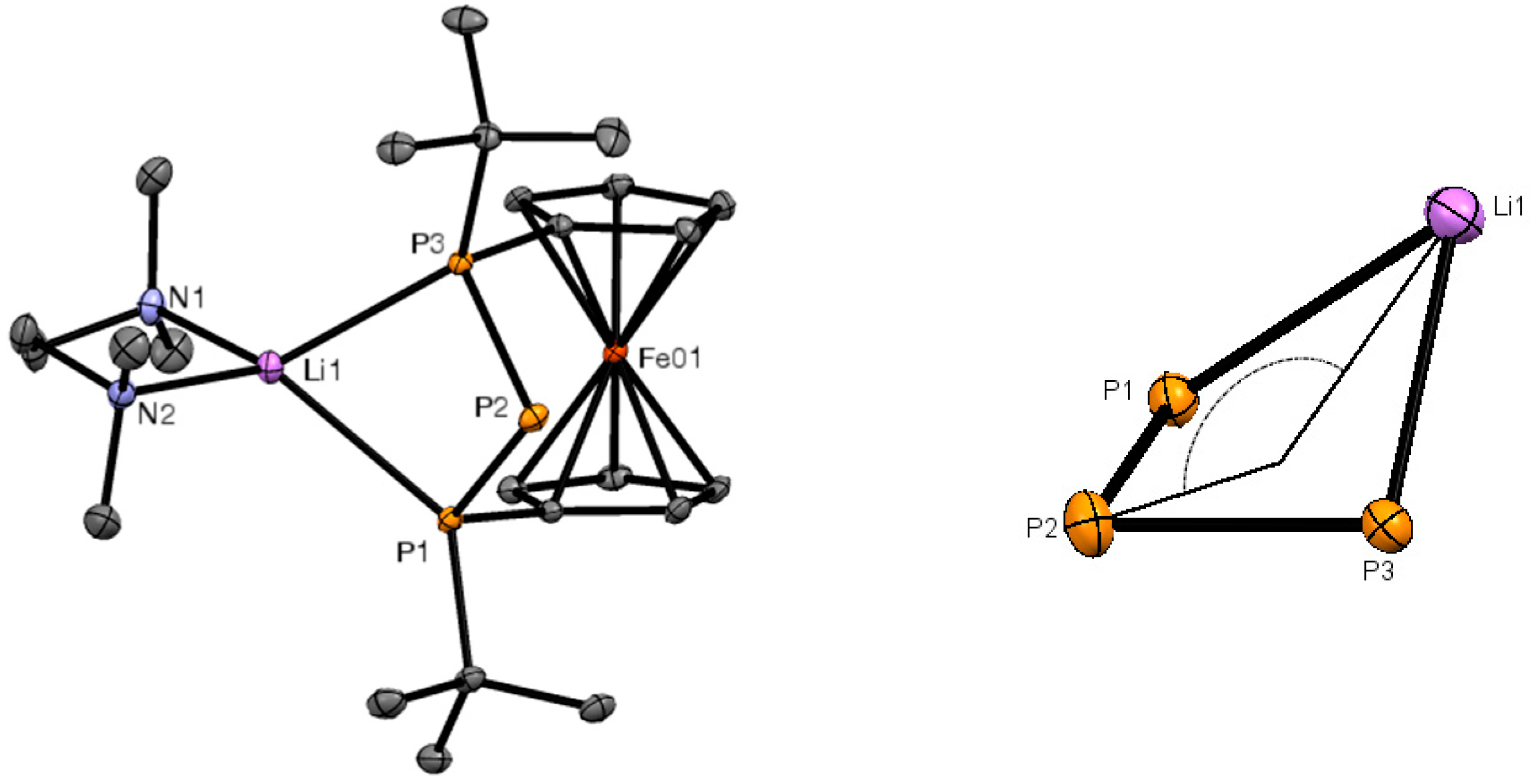
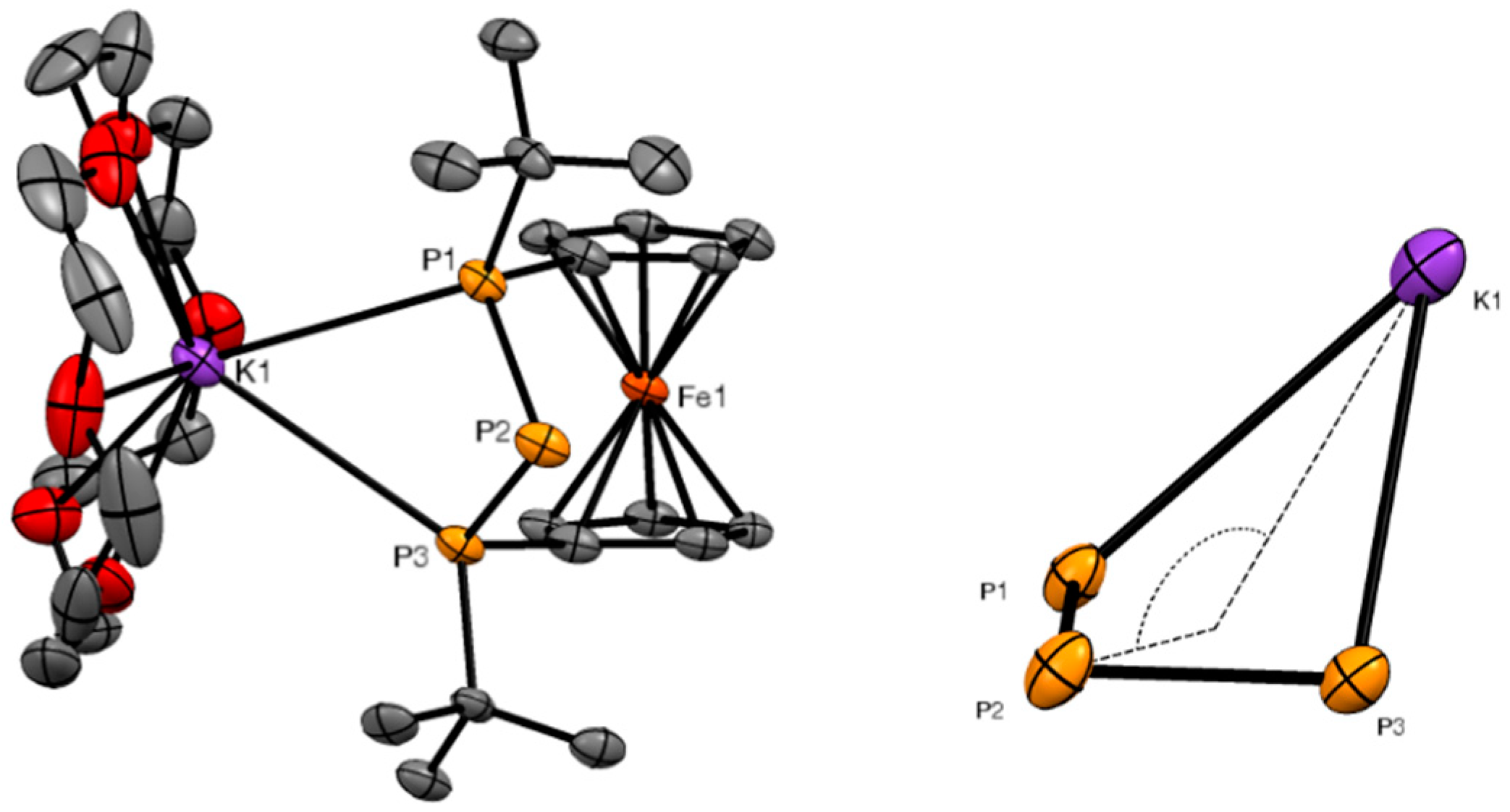
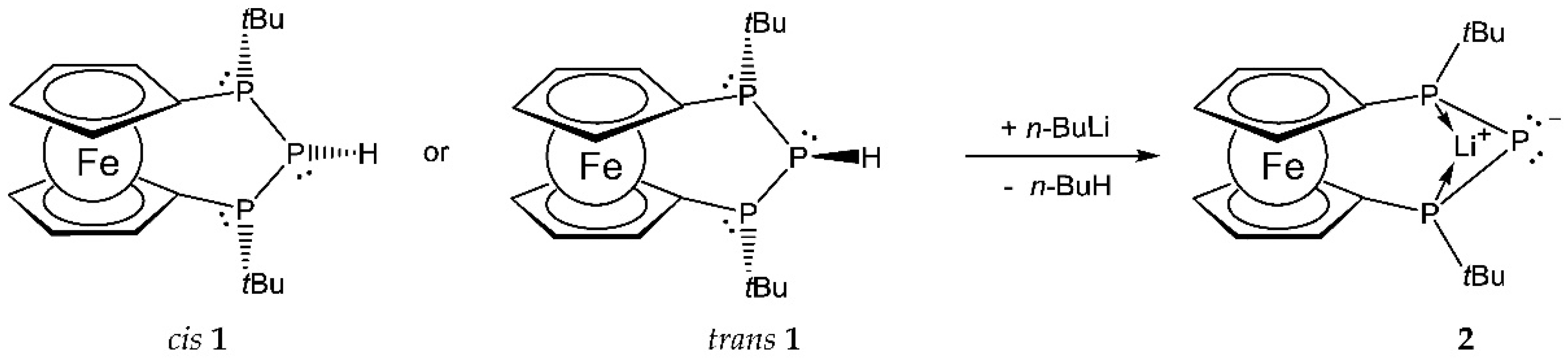
2. Results and Discussion
3. Experimental Details
3.1. Synthesis of Compound 2
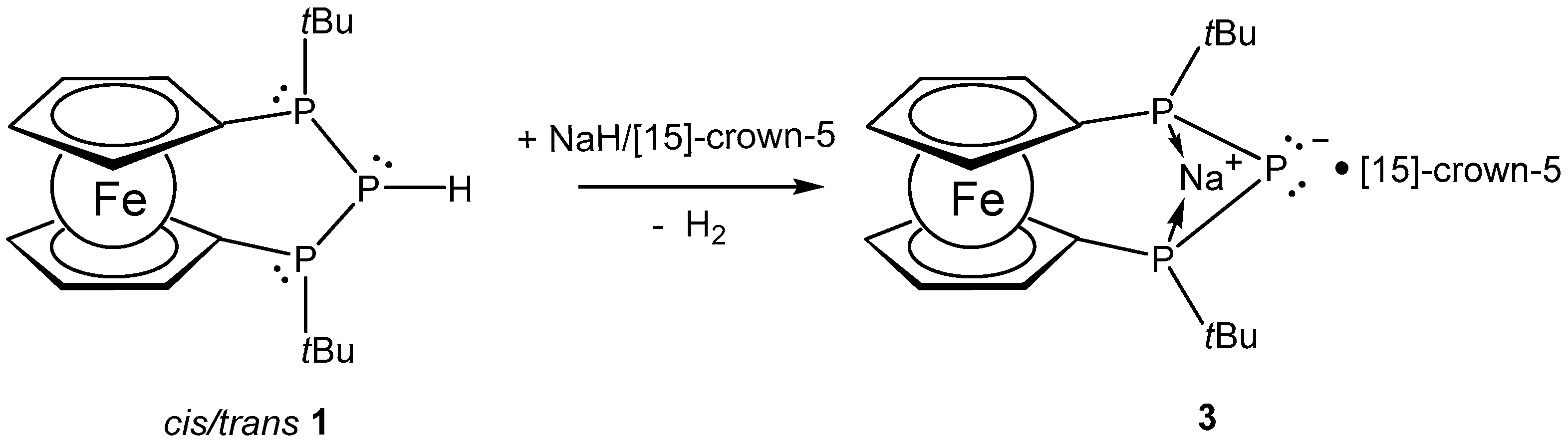
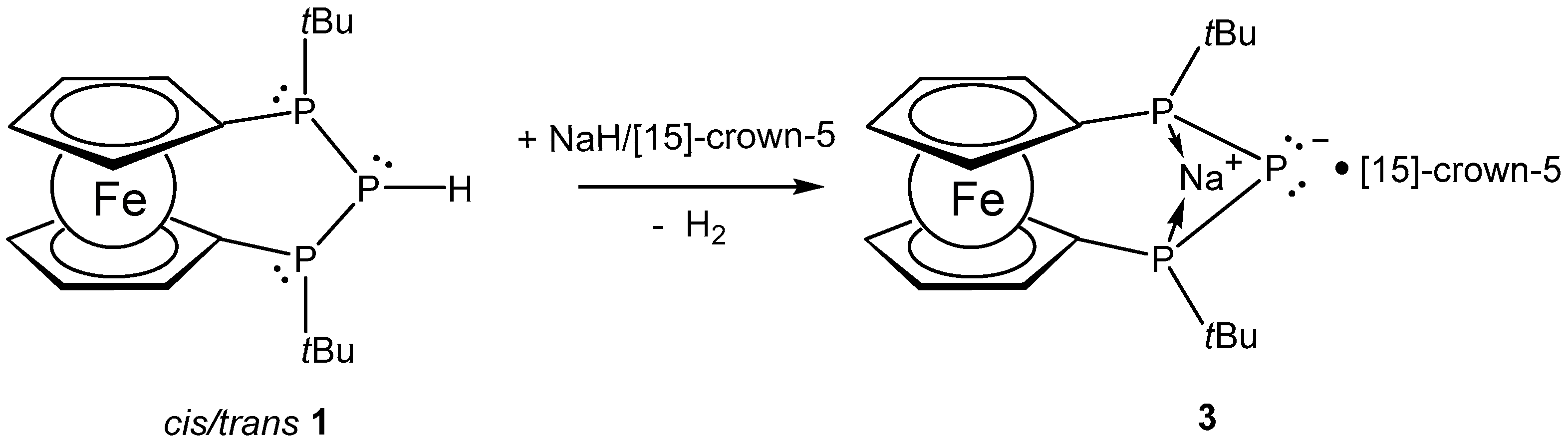
3.2. Synthesis of Compound 3
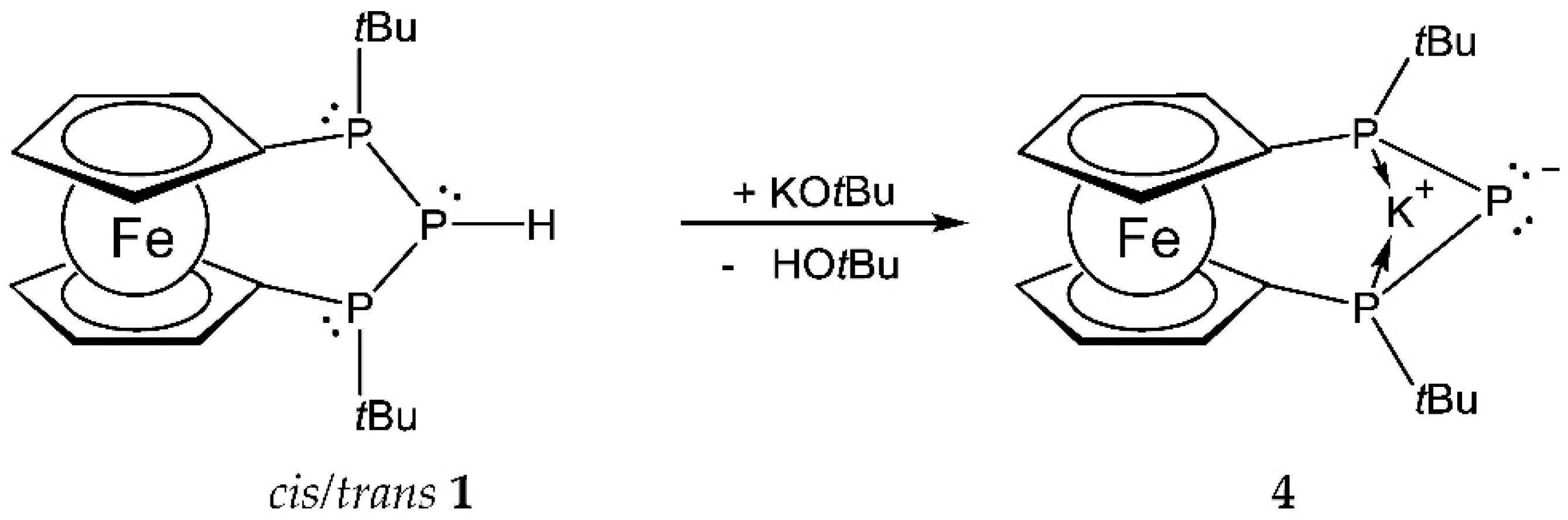
3.3. Synthesis of Compound 4
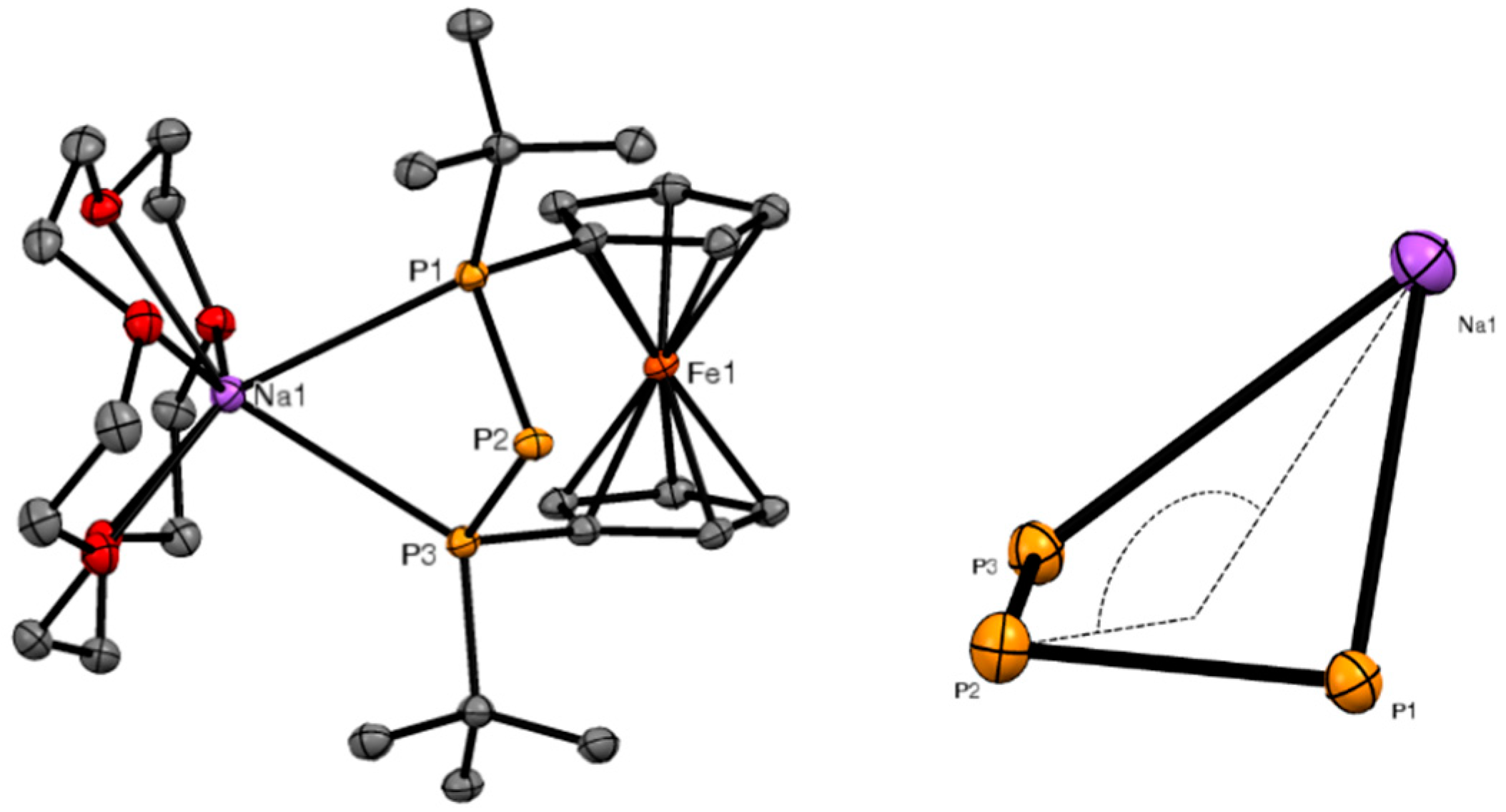
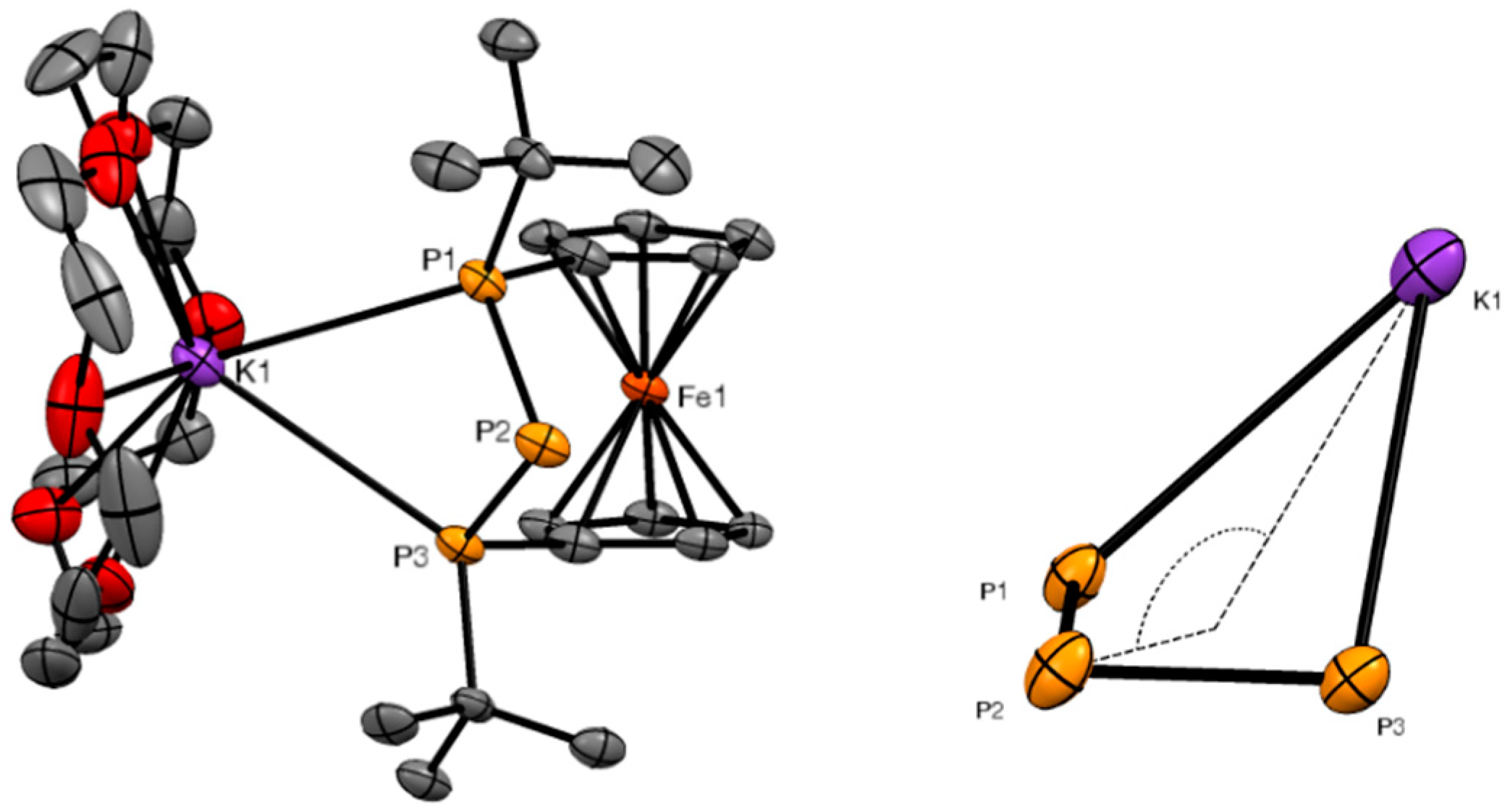
3.4. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Izod, K. Group I complexes of P- and as-donor ligands. Adv. Inorg. Chem. 2000, 50, 33–107. [Google Scholar]
- Fritz, G.; Scheer, P. Silylphosphanes: Developments in Phosphorus Chemistry. Chem. Rev. 2000, 100, 3341–3402. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L. Metal complexes of planar PR2 ligands: Examining the carbene analogy. Coord. Chem. Rev. 2012, 256, 606–626. [Google Scholar] [CrossRef]
- Driess, M.; Nöth, H. Molecular Clusters of the Main Group Elements; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Wolf, R.; Schisler, A.; Lönnecke, P.; Jones, C.; Hey-Hawkins, E. Syntheses and Molecular Structures of Novel Alkali Metal Tetraorganylcyclopentaphosphanides and Tetraorganyltetraphosphane-1, 4-diides. Eur. J. Inorg. Chem. 2004, 16, 3277–3286. [Google Scholar] [CrossRef]
- Wolf, R.; Gomez-Ruiz, S.; Reinhold, J.; Boehlmann, W.; Hey-Hawkins, E. The (P4HMes4)− anion: Lability, fluxionality, and structural ambiguity (Mes = 2,4,6-Me3C6H2). Inorg. Chem. 2006, 45, 9107–9113. [Google Scholar] [CrossRef] [PubMed]
- Dielmann, F.; Heindl, C.; Hastreiter, F.; Peresypkina, E.V.; Virovets, A.V.; Gschwind, R.M.; Scheer, M. A nano-sized Supramolecule beyond the Fullerene Topology. Angew. Chem. Int. Ed. 2014, 53, 13605–13608. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Schindler, A.; Merkle, R.; Johnson, B.P.; Linseis, M.; Winter, R.; Anson, C.E.; Virovets, A.V. Fullerene C60 as an Endohedral Molecule within an Inorganic Supramolecule. J. Am. Chem. Soc. 2007, 129, 13386–13387. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Virovets, A.V.; Scheer, M. Synthesis of Inorganic Fullerene-Like Molecules. Science 2003, 300, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Borucki, S.; Kelemen, Z.; Maurer, M.; Bruhn, C.; Nyulászi, L.; Pietschnig, R. Stereochemical alignment in triphospha-[3]ferrocenophanes. Chem. Eur. J. 2017, 23, 10438–10450. [Google Scholar] [CrossRef] [PubMed]
- Kargin, D.; Kelemen, Z.; Krekić, K.; Maurer, M.; Bruhn, C.; Nyulászi, L.; Pietschnig, R. [3]Ferrocenophanes with bisphosphanotetryl bridge: Inorganic rings on the way to tetrylenes. Dalton Trans. 2016, 45, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Belaj, F.; Pietschnig, R. Phosphorus-Rich Ferrocenophanes. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 837–844. [Google Scholar] [CrossRef]
- Moser, C.; Belaj, F.; Pietschnig, R. Diphospha[2]ferrocenophane alias (1,4-dihydrotetraphosphaneoxide): Stereoselective formation via hydrolytic P-P bond formation. Chem. Eur. J. 2009, 15, 12589–12591. [Google Scholar] [CrossRef] [PubMed]
- Lange, D.; Klein, E.; Bender, H.; Niecke, E.; Nieger, M.; Pietschnig, R.; Schoeller, W.W.; Ranaivonjatovo, H. 1,3-Diphospha-2-silaallylic Lithium Complexes and Anions: Synthesis, Crystal Structures, Reactivity, and Bonding Properties. Organometallics 1998, 17, 2425–2432. [Google Scholar] [CrossRef]
- Hitzel, S.; Färber, C.; Bruhn, C.; Siemeling, U. Phosphido complexes derived from 1,1’-ferrocenediyl-bridged secondary diphosphines. Dalton Trans. 2017, 46, 6333–6348. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; Naseri, V.; Wright, D.S. Formation of an organometallic phosphanediide via main-group dehydrocoupling. Organometallics 2009, 28, 1995–1997. [Google Scholar] [CrossRef]
- Reich, H.J.; Dykstra, R.R. Solution Structure of Lithium Benzeneselenolate and Lithium Diphenylphosphide: NMR Identification of Cyclic Dimers and Mixed Dimers. Organometallics 1994, 13, 4578–4585. [Google Scholar] [CrossRef]
- Poonia, N.S.; Bajaj, A.V. Coordination chemistry of alkali and alkaline earth cations. Chem. Rev. 1979, 79, 389–445. [Google Scholar] [CrossRef]
- Bajaj, A.V.; Poonia, N.S. Comprehensive coordination chemistry of alkali and alkaline earth cations with macrocyclic multidentates: Latest position. Coord. Chem. Rev. 1988, 87, 55–213. [Google Scholar] [CrossRef]
- Berry, A.; Green, M.L.H.; Bandy, J.A.; Prout, K. Transition Metal-Hydrogen-Alkali Metal Bonds: Synthesis and Crystal Structures of [K(18-crown-6)][W(PMe3)3H5], [Na(15-crown-5)][W(PMe3)3H5] and [{W(PMe3)3H5Li}4] and Related Studies. J. Chem. Soc. Dalton Trans. 1991, 2185–2206. [Google Scholar] [CrossRef]
- Gjikaj, M.; Adam, A. Complexation of Alkali Triflates by Crown Ethers: Synthesis and Crystal Structure of [Na(12-crown-4)2][SO3CF3], [Na(15-crown-5)][SO3CF3], [Rb(18-crown-6)][SO3CF3], and [Cs(18-crown-6)][SO3CF3]. Z. Anorg. Allg. Chem. 2006, 632, 2475–2480. [Google Scholar] [CrossRef]
- Nöth, H.; Warchhold, M. Sodium Hydro(isothiocyanato)borates: Synthesis and Structures. Eur. J. Inorg. Chem. 2004, 2004, 1115–1124. [Google Scholar] [CrossRef]
- Cole, M.L.; Jones, C.; Junk, P.C. Ether and crown ether adduct complexes of sodium and potassium cyclopentadienide and methylcyclopentadienide—Molecular structures of [Na(dme)Cp]∞, [K(dme)0.5Cp]∞, [Na(15-crown-5)Cp], [Na(18-crown-6)CpMe] and the “naked Cp–” complex [K(15-crown-5)2][Cp]. J. Chem. Soc. Dalton Trans. 2002, 896–905. [Google Scholar] [CrossRef]
- Schmidpeter, A.; Burget, G. Die Reaktion Von Alkaliphosphiden Mit Weissem Phosphor, 11 Bildung Von 1,1,3,3-Tetraphenyl-Triphosphid, 2,3,4,5-Tetraphenyl-Cyclopentaphosphid und Phenyl-Tricycloheptaphosphid. Phosphorus Sulfur Silicon Relat. Elem. 1985, 22, 323–335. [Google Scholar] [CrossRef]
- Geier, J.; Ruegger, H.; Worle, M.; Grützmacher, H. Sodium oligophosphanediide ions in the PhPCl2/Na system: Syntheses and structural characterization. Angew. Chem. Int. Ed. 2003, 42, 3951–3954. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, N.; Wörner, A.; Lerner, H.W.; Karaghiosoff, K.; Fenske, D.; Baum, G.; Dransfeld, A.; von Ragué Schleyer, P. The Triphosphide (tBu3Si)2P3Na: Formation, X-ray and Ab initio Structure Analyses, Protonation and Oxidation to Triphosphane (tBu3Si)2P3H and Hexaphosphanes (tBu3Si)4P6. Eur. J. Inorg. Chem. 1998, 1998, 833–841. [Google Scholar] [CrossRef]
- Thiele, G.; Vondung, L.; Donsbach, C.; Pulz, S.; Dehnen, S. Organic Cation and Complex Cation-Stabilized (Poly-)Selenides, [Cation]x(Sey)z: Diversity in Structures and Properties. Z. Anorg. Allg. Chem. 2014, 640, 2684–2700. [Google Scholar] [CrossRef]
- Kobrsi, I.; Zheng, W.; Knox, J.E.; Heeg, M.J.; Schlegel, H.B.; Winter, C.H. Experimental and theoretical study of the coordination of 1, 2, 4-triazolato, tetrazolato, and pentazolato ligands to the [K(18-crown-6)]+ fragment. Inorg. Chem. 2006, 45, 8700–8710. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J. Higher order 31P NMR Spectra of Polyphosphorus Compounds. In Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis; Verkade, J.G., Quin, L.D., Eds.; VCH: Weinheim, Germany, 1987; pp. 331–364. [Google Scholar]
- Albrand, J.P.; Faucher, H.; Gagnaire, D.; Robert, J.B. Calculation of the 1 J(PP) angular dependence in P2H4. Chem. Phys. Lett. 1976, 38, 521–523. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van der Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 | 3 | 4 | |
|---|---|---|---|
| Formula | C24H42FeLiN2P3,0.5(C6H14) | C28H46FeNaO5P3 | C30H50FeKO6P3 |
| Formular weight | 557.38 | 634.40 | 694.56 |
| Temperature (K) | 100 | 100 | 100 |
| Wavelength (Å) | 1.54186 | 0.71073 | 1.54186 |
| Crystal system | monoclinic | monoclinic | triclinic |
| Space group | C2/c | P21/c | P−1 |
| Unit-cell dimensions: | |||
| a (Å) | 10.2172(3) | 14.2758(6) | 10.791(1) |
| b (Å) | 17.3386(6) | 10.3266(3) | 10.7753(10) |
| c (Å) | 34.6955(9) | 21.4828(9) | 16.8369(16) |
| α (°) | 90 | 90 | 73.904(7) |
| β (°) | 90.735(2) | 99.536(3) | 71.663(7) |
| γ (°) | 90 | 90 | 69.620(7) |
| Volume (Å3) | 6145.9(3) | 3123.2(2) | 1710.8(3) |
| Z | 8 | 4 | 2 |
| Calculated density (mg/m3) | 1.205 | 1.349 | 1.348 |
| μ (mm−1) | 5.526 | 0.685 | 6.265 |
| Θ-range for data collected (°) | 5.100–68.990 | 1.446–25.498 | 2.815–68.998 |
| Data/parameters | 5340/318 | 5814/349 | 6198/376 |
| Goodness-of-fit on F2 | 1.021 | 1.047 | 1.024 |
| R1 (observed data) | 0.0309 | 0.0531 | 0.1104 |
| wR2 (all data) | 0.0733 | 0.1559 | 0.3201 |
| Rint | 0.0240 | 0.0654 | 0.0772 |
| r.e.d. min/max (e·Å−3) | 0.539/0.834 | −0.944/0.776 | 0.451/0.887 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isenberg, S.; Frenzel, L.-M.; Bruhn, C.; Pietschnig, R.
Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K)
Isenberg S, Frenzel L-M, Bruhn C, Pietschnig R.
Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K)
Isenberg, Stefan, Lisa-Marie Frenzel, Clemens Bruhn, and Rudolf Pietschnig.
2018. "Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K)
Isenberg, S., Frenzel, L.-M., Bruhn, C., & Pietschnig, R.
(2018). Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K)