S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions

 , and
, and 
Abstract
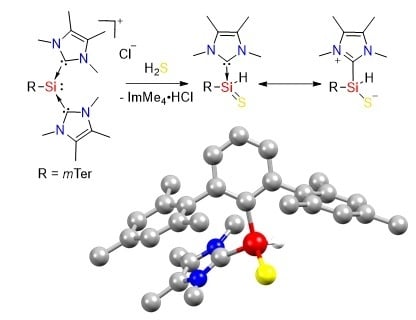
:
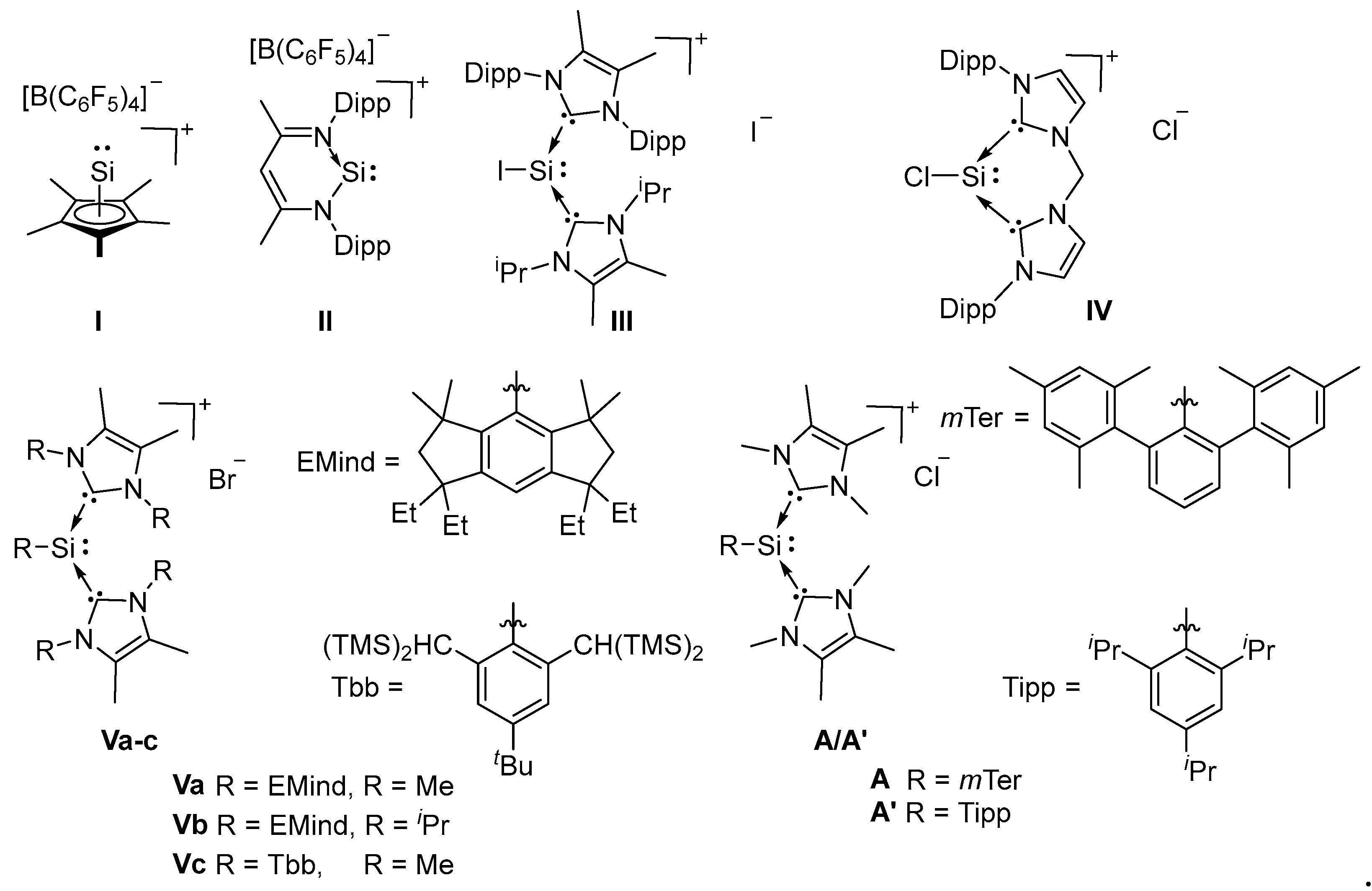
1. Introduction
2. Results and Discussion
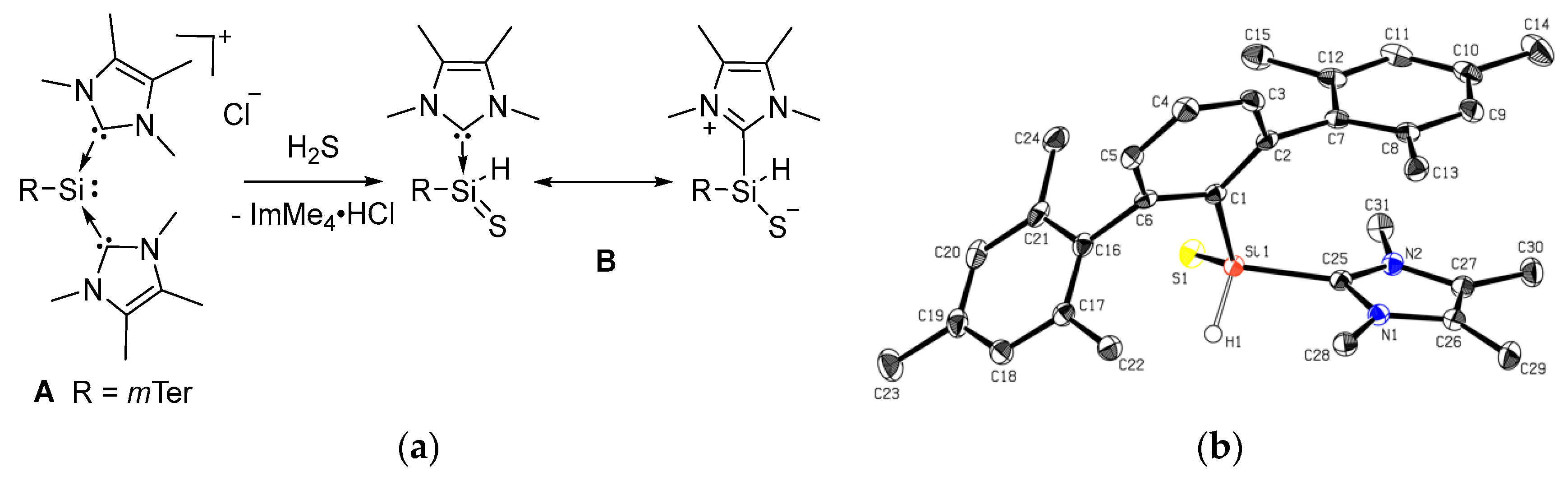
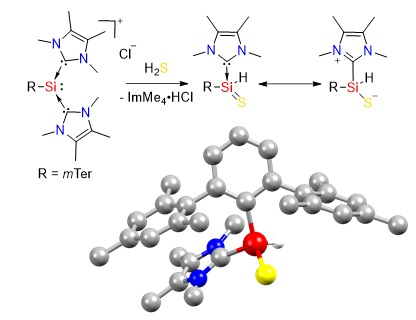
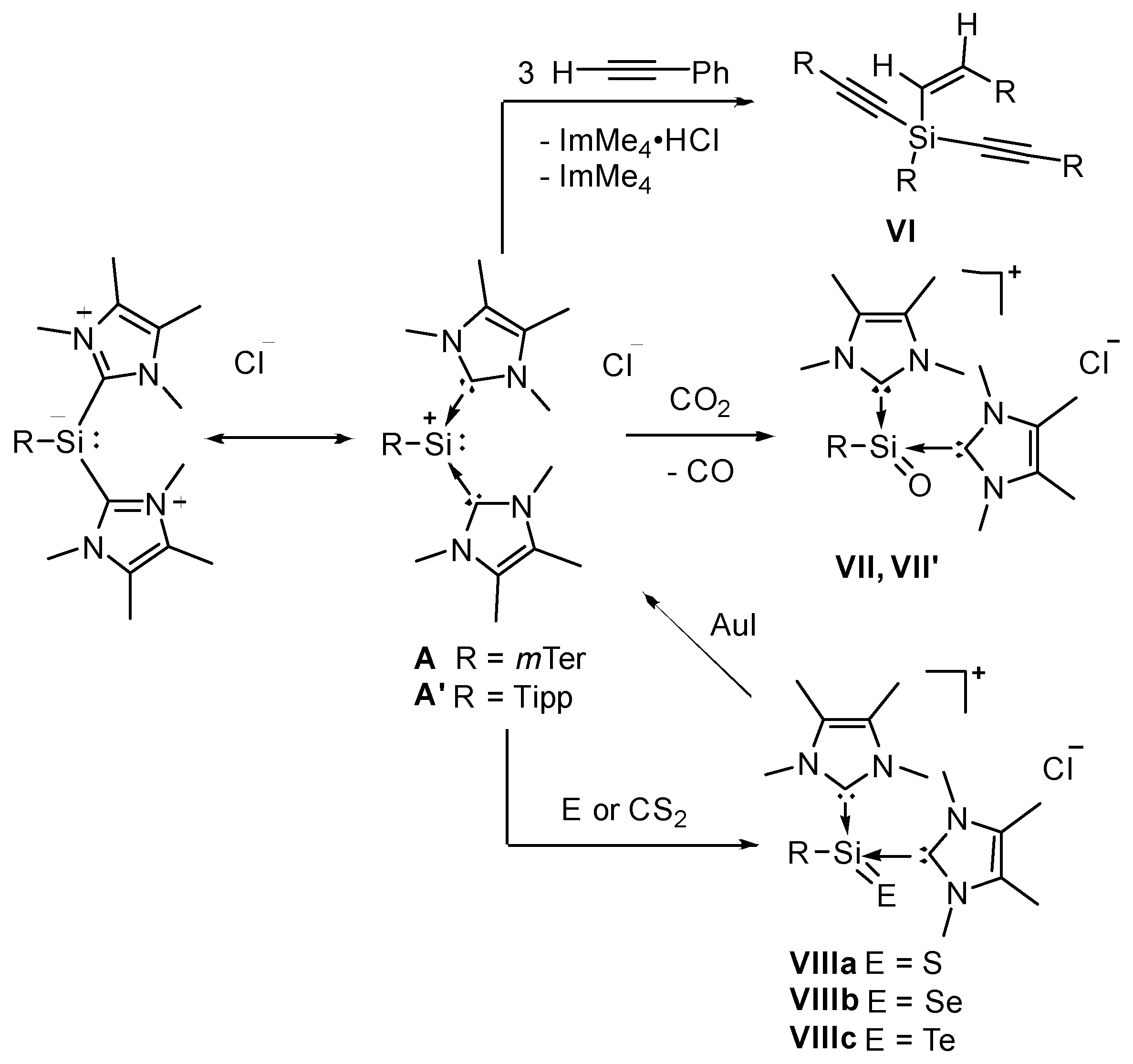
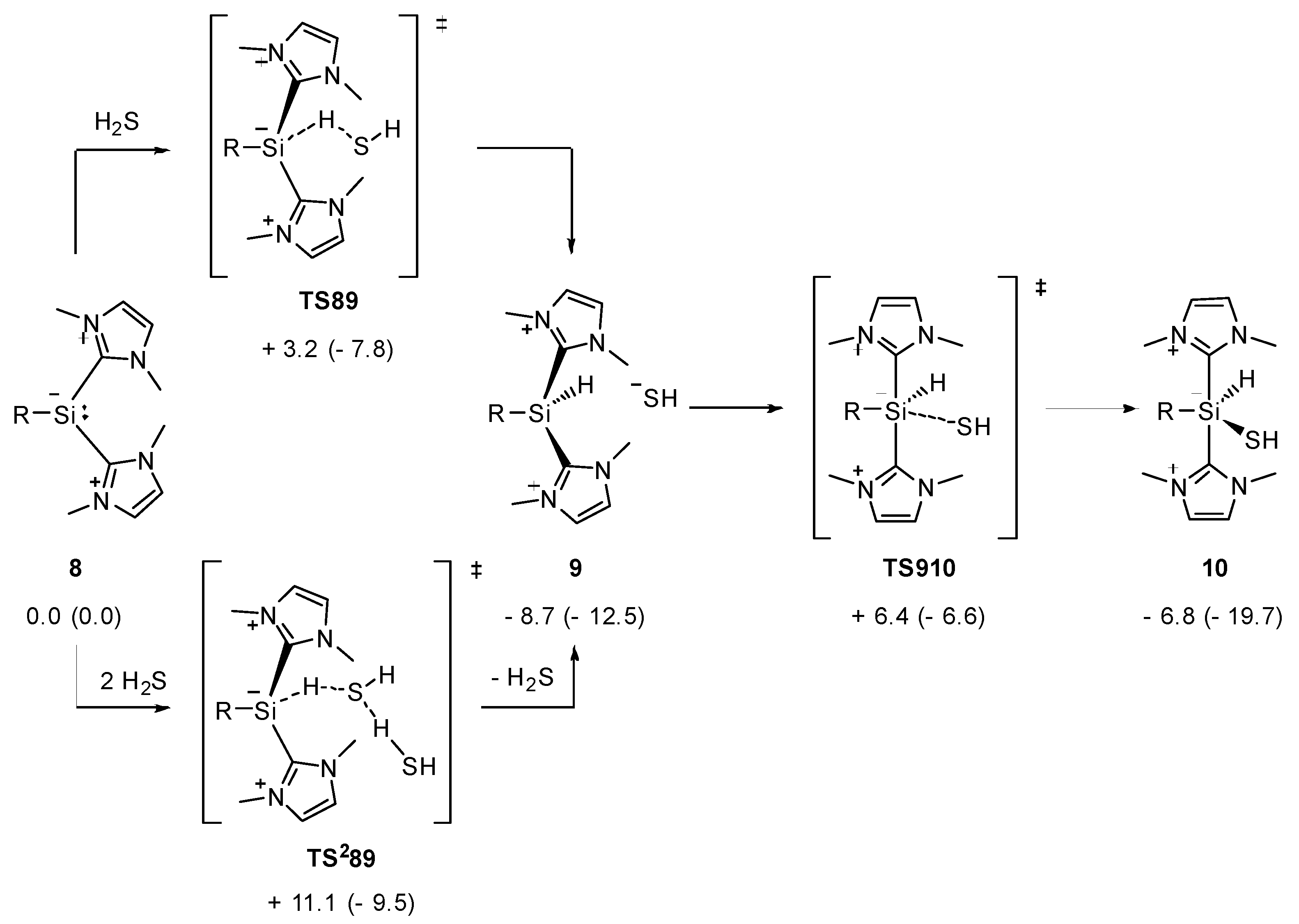
2.1. Reaction of Silyliumylidene A with H2S
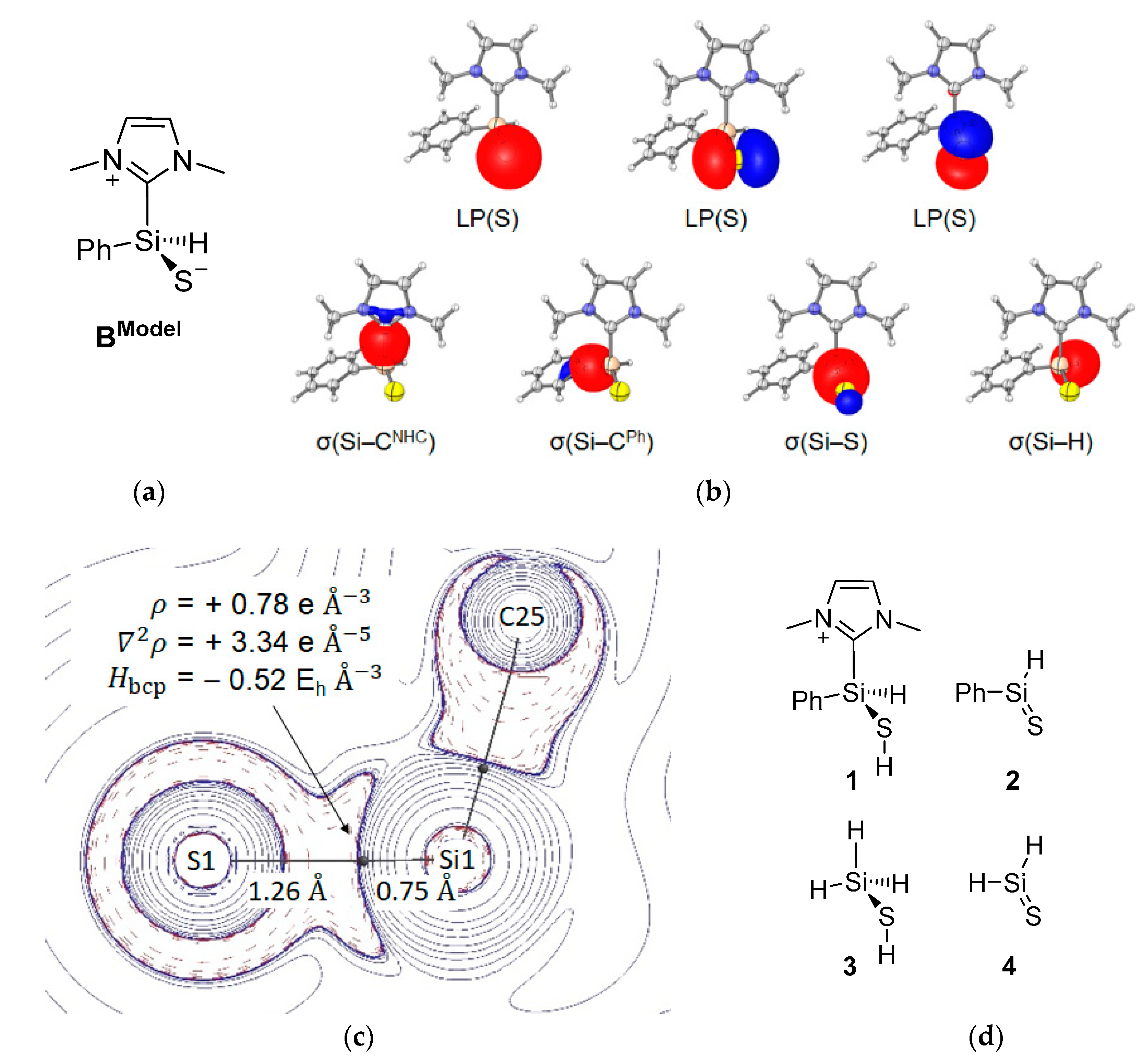
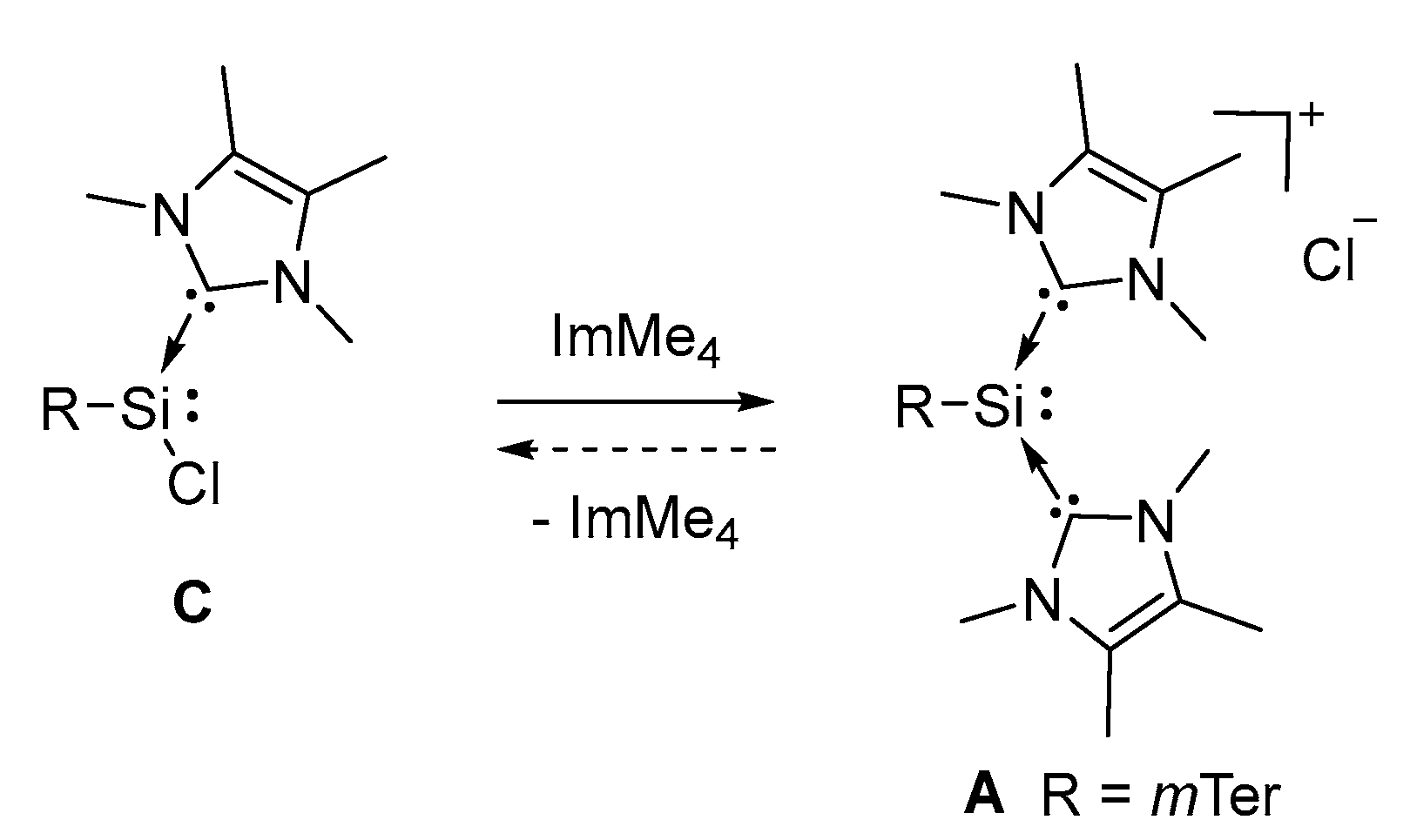
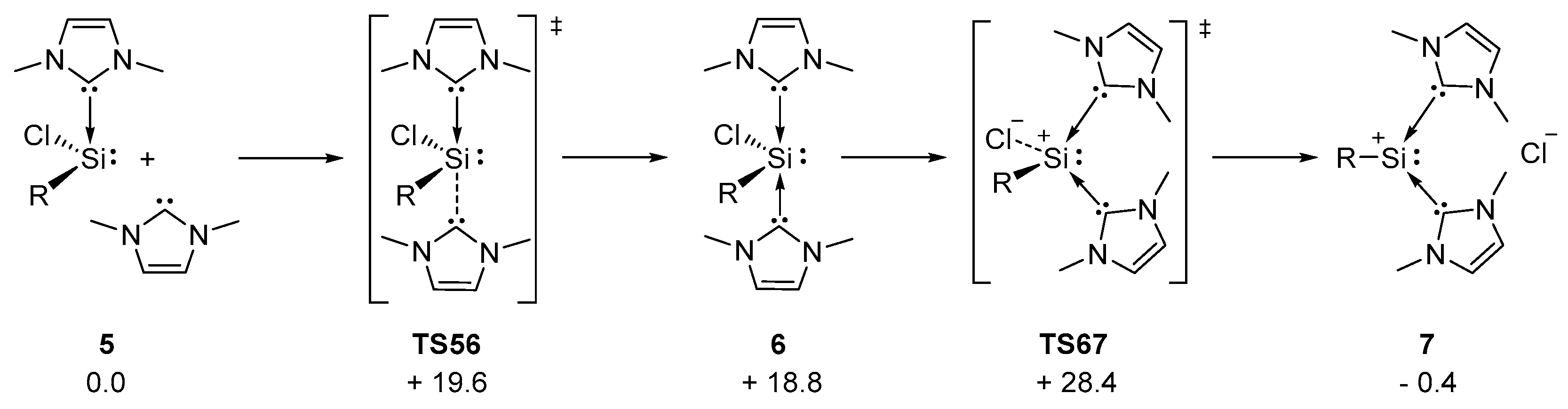
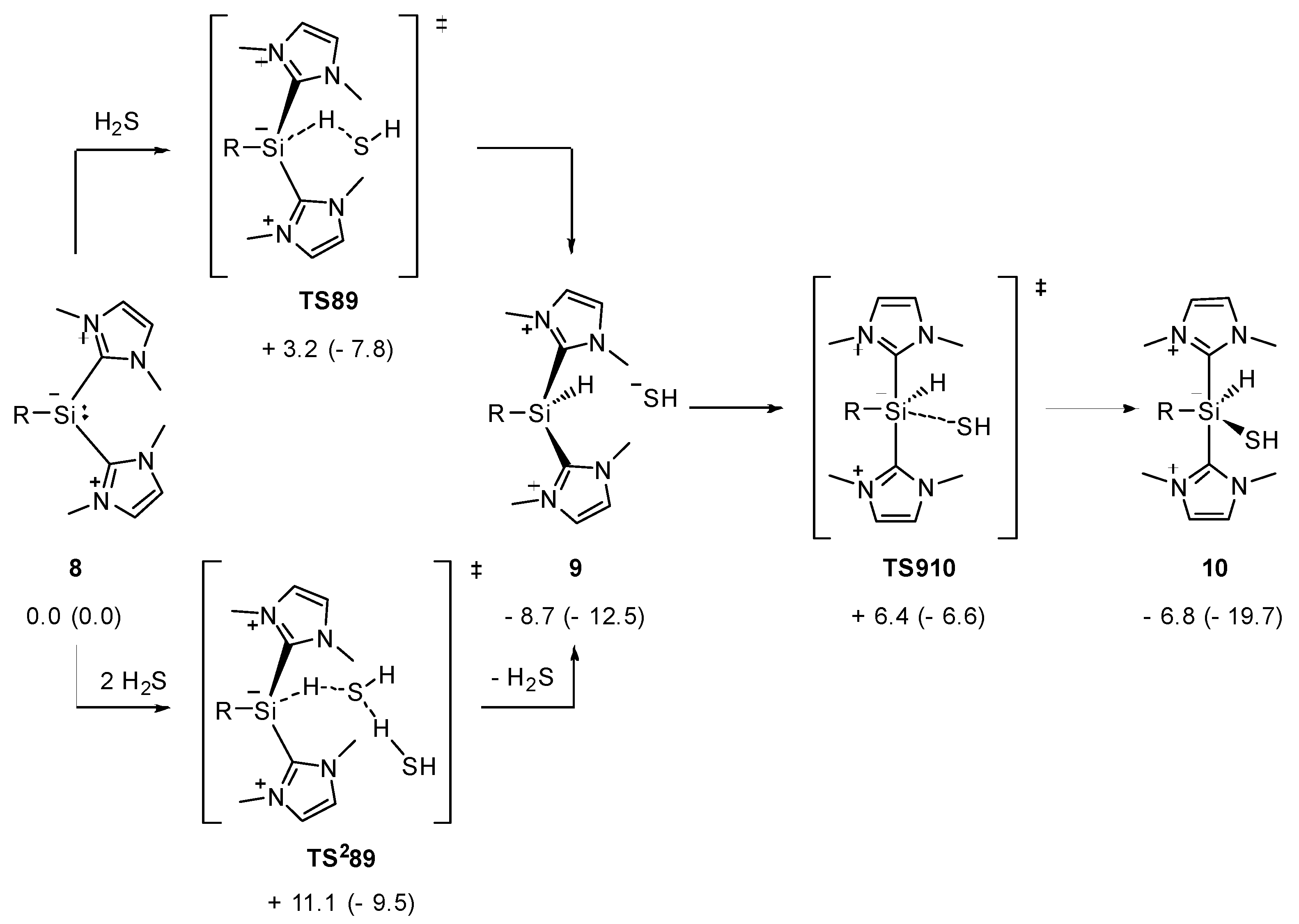
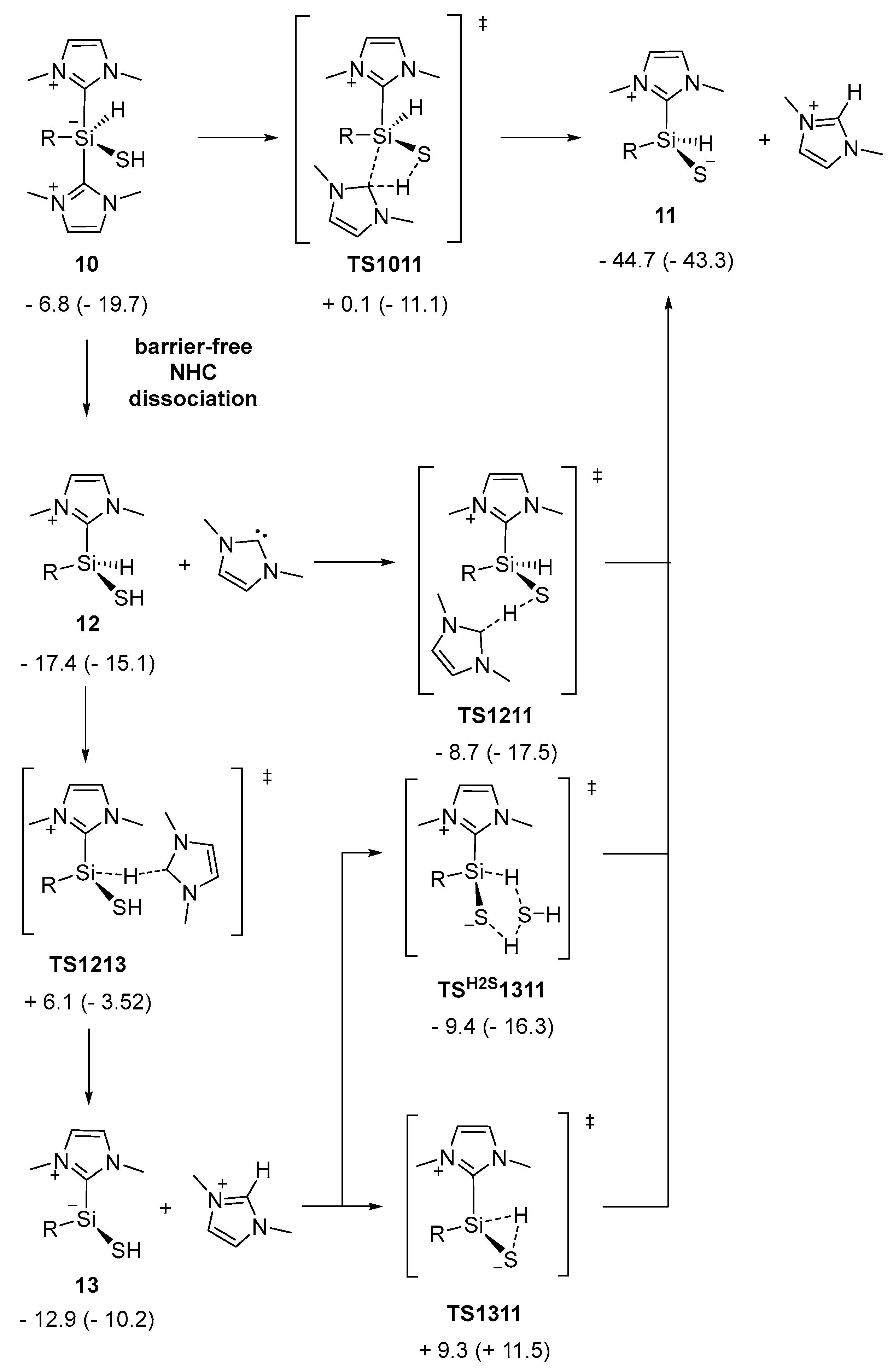
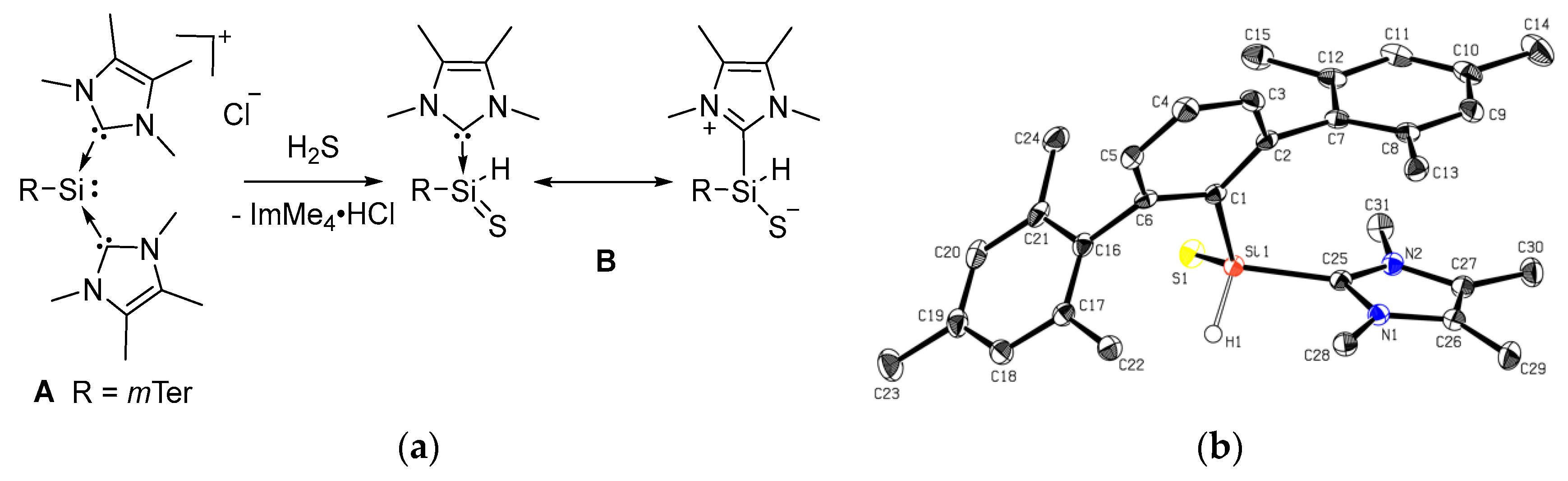
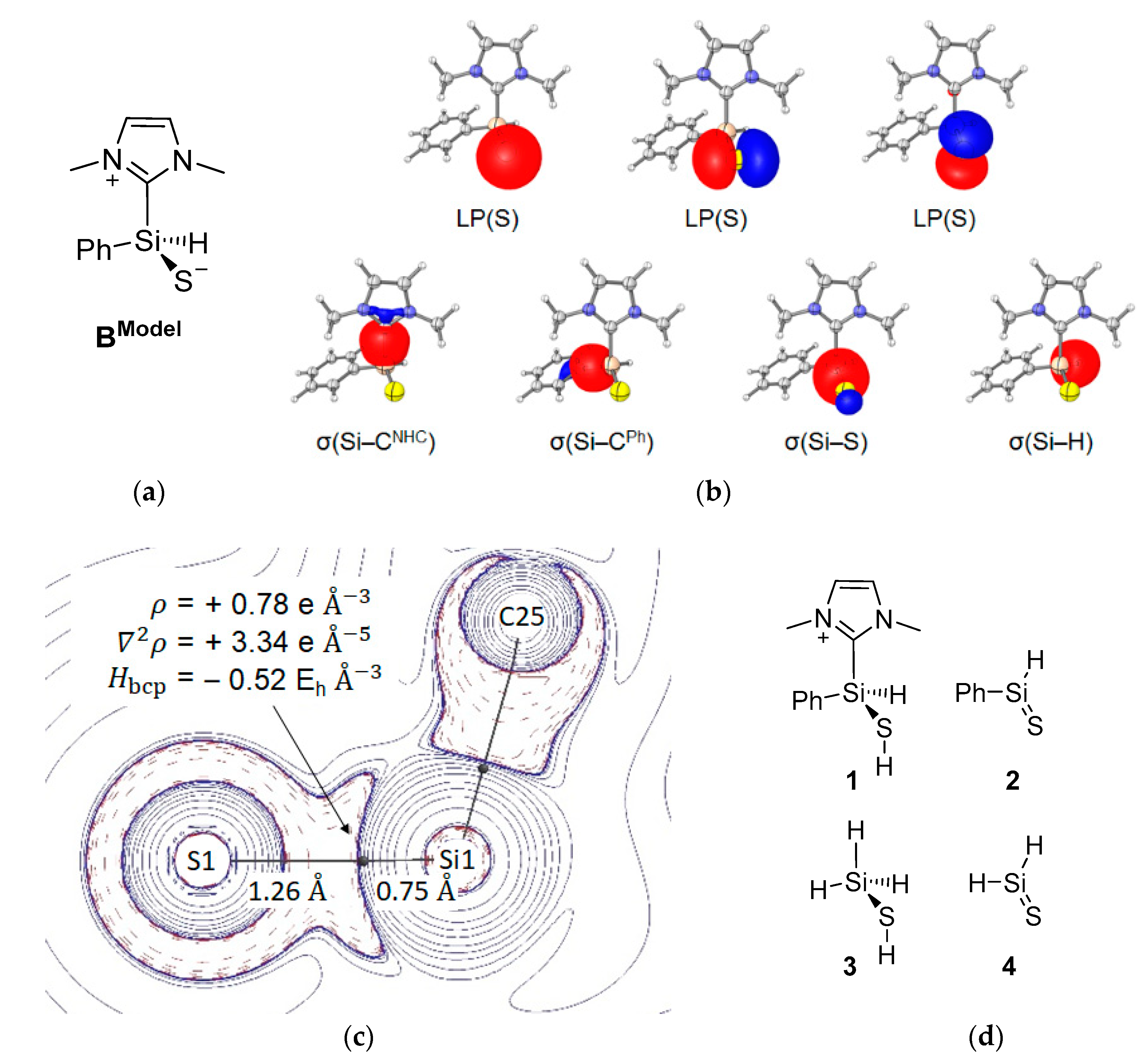
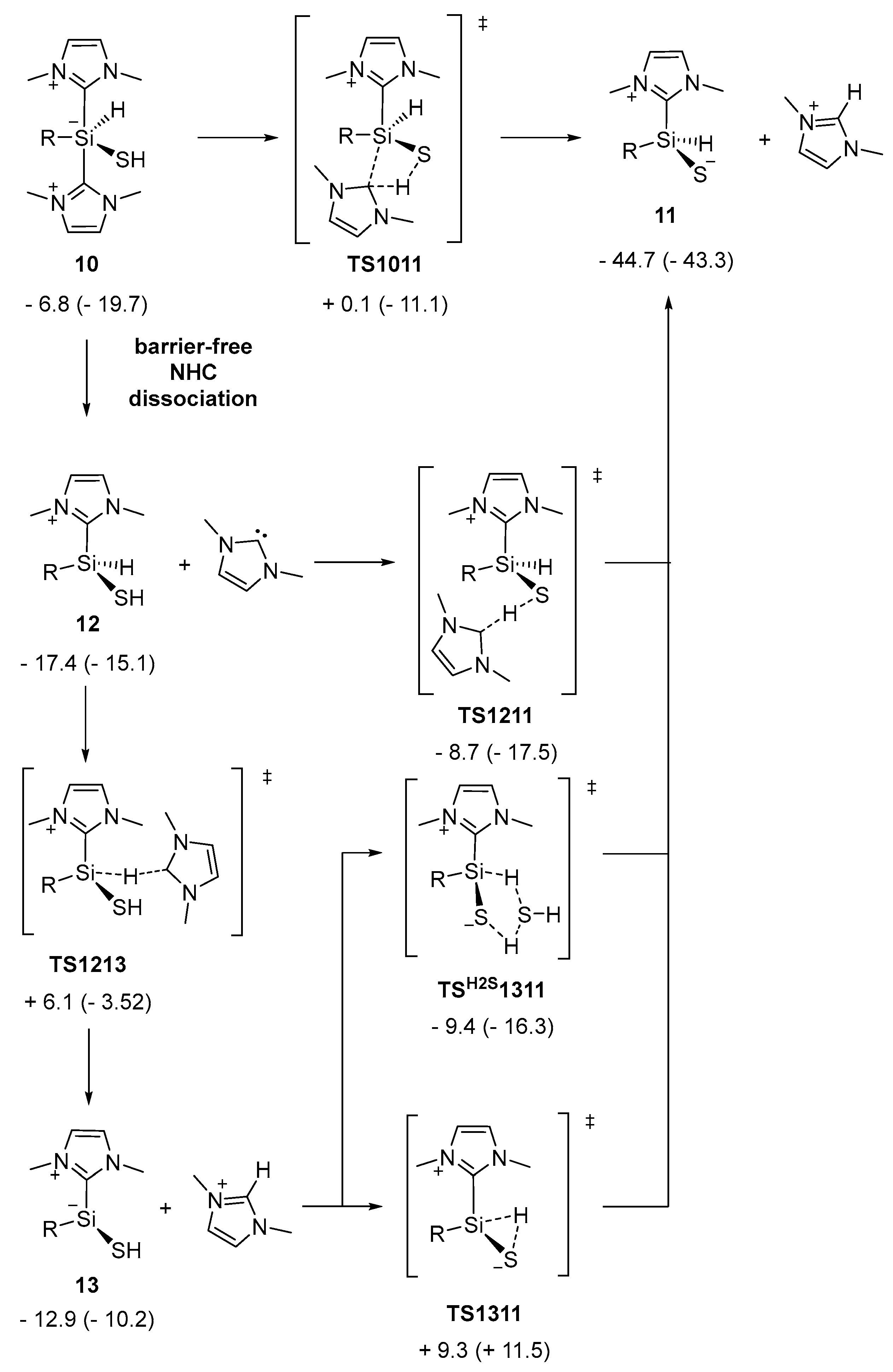
2.2. Mechanistic Investigations on the Reaction of Silyliumylidene A with H2S
3. Materials and Methods
3.1. General Methods and Instruments
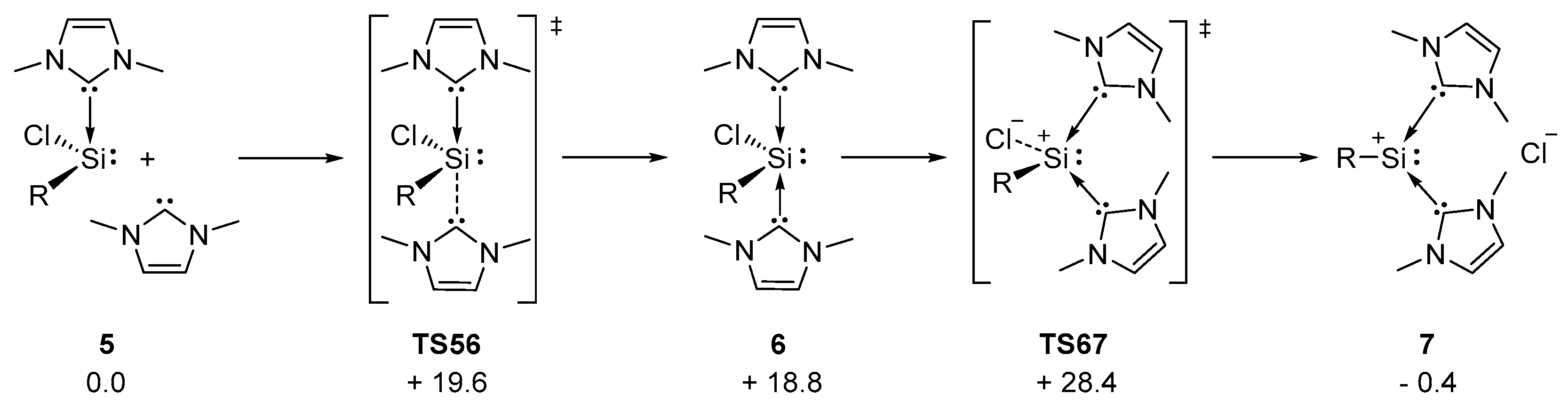
3.2. Improved and Upscaled Synthesis of Silyliumylidene A
3.3. Synthesis of Thiosilaaldehyde B
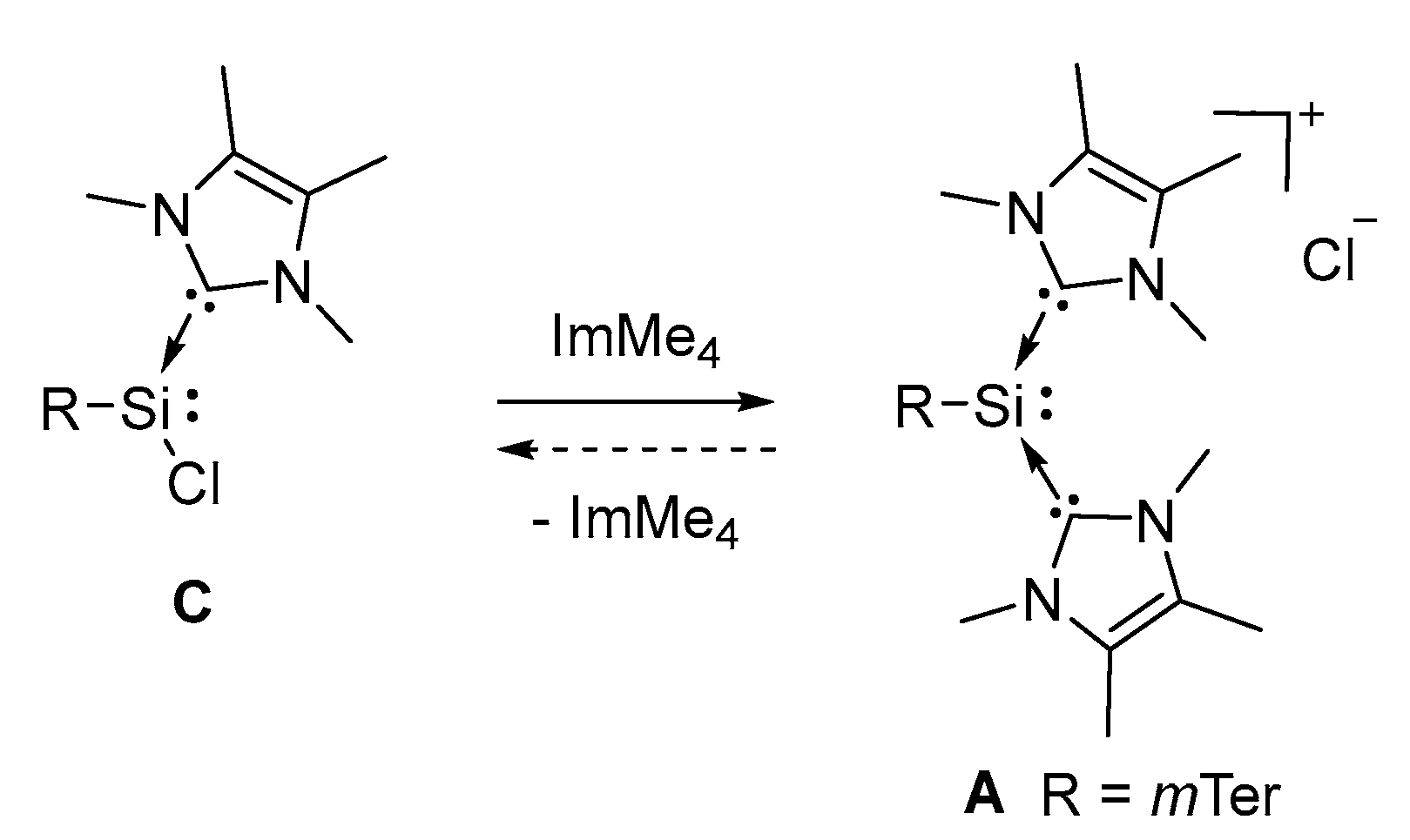
3.4. Conversion of Chlorosilylene C to Silyliumylidene A
3.5. DFT Calculations
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bayne, J.M.; Stephan, D.W. Phosphorus Lewis acids: Emerging reactivity and applications in catalysis. Chem. Soc. Rev. 2016, 45, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Hadlington, T.J.; Driess, M.; Jones, C. Low-valent group 14 element hydride chemistry: Towards catalysis. Chem. Soc. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.K.; Roesky, H.W. Group 14 Hydrides with Low Valent Elements for Activation of Small Molecules. Acc. Chem. Res. 2012, 45, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.M.D.; Rivard, E. Pushing Chemical Boundaries with N-Heterocyclic Olefins (NHOs): From Catalysis to Main Group Element Chemistry. Acc. Chem. Res. 2017, 50, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Saha, S.; Sen, S.S. Compounds with Low-Valent p-Block Elements for Small Molecule Activation and Catalysis. ChemCatChem 2016, 8, 486–501. [Google Scholar] [CrossRef]
- Yao, S.; Xiong, Y.; Driess, M. Zwitterionic and Donor-Stabilized N-Heterocyclic Silylenes (NHSis) for Metal-Free Activation of Small Molecules. Organometallics 2011, 30, 1748–1767. [Google Scholar] [CrossRef]
- Alvarado-Beltran, I.; Rosas-Sanchez, A.; Baceiredo, A.; Saffon-Merceron, N.; Branchadell, V.; Kato, T. A Fairly Stable Crystalline Silanone. Angew. Chem. Int. Ed. 2017, 56, 10481–10485. [Google Scholar] [CrossRef] [PubMed]
- Arz, M.I.; Geiß, D.; Straßmann, M.; Schnakenburg, G.; Filippou, A.C. Silicon(i) chemistry: The NHC-stabilised silicon(i) halides Si2X2(Idipp)2 (X = Br, I) and the disilicon(i)-iodido cation [Si2(I)(Idipp)2]+. Chem. Sci. 2015, 6, 6515–6524. [Google Scholar] [CrossRef]
- Arz, M.I.; Schnakenburg, G.; Meyer, A.; Schiemann, O.; Filippou, A.C. The Si2H radical supported by two N-heterocyclic carbenes. Chem. Sci. 2016, 7, 4973–4979. [Google Scholar] [CrossRef]
- Boehme, C.; Frenking, G. Electronic Structure of Stable Carbenes, Silylenes, and Germylenes. J. Am. Chem. Soc. 1996, 118, 2039–2046. [Google Scholar] [CrossRef]
- Burchert, A.; Müller, R.; Yao, S.; Schattenberg, C.; Xiong, Y.; Kaupp, M.; Driess, M. Taming Silicon Congeners of CO and CO2: Synthesis of Monomeric SiII and SiIV Chalcogenide Complexes. Angew. Chem. Int. Ed. 2017, 56, 6298–6301. [Google Scholar] [CrossRef] [PubMed]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and Structure of a Stable Silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar] [CrossRef]
- Ghana, P.; Arz, M.I.; Das, U.; Schnakenburg, G.; Filippou, A.C. Si=Si Double Bonds: Synthesis of an NHC-Stabilized Disilavinylidene. Angew. Chem. Int. Ed. 2015, 54, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Kira, M.; Ishida, S.; Iwamoto, T.; Kabuto, C. The First Isolable Dialkylsilylene. J. Am. Chem. Soc. 1999, 121, 9722–9723. [Google Scholar] [CrossRef]
- Mondal, K.C.; Roesky, H.W.; Schwarzer, M.C.; Frenking, G.; Tkach, I.; Wolf, H.; Kratzert, D.; Herbst-Irmer, R.; Niepotter, B.; Stalke, D. Conversion of a Singlet Silylene to a stable Biradical. Angew. Chem. Int. Ed. 2013, 52, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Mondal, K.C.; Roy, S.; Dittrich, B.; Andrada, D.M.; Frenking, G.; Roesky, H.W. A Triatomic Silicon(0) Cluster Stabilized by a Cyclic Alkyl(amino) Carbene. Angew. Chem. Int. Ed. 2016, 55, 3158–3161. [Google Scholar] [CrossRef] [PubMed]
- Nieder, D.; Yildiz, C.B.; Jana, A.; Zimmer, M.; Huch, V.; Scheschkewitz, D. Dimerization of a marginally stable disilenyl germylene to tricyclic systems: Evidence for reversible NHC-coordination. Chem. Commun. 2016, 52, 2799–2802. [Google Scholar] [CrossRef] [PubMed]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Stable Two-Coordinate Acyclic Silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [Google Scholar] [CrossRef] [PubMed]
- Rekken, B.D.; Brown, T.M.; Fettinger, J.C.; Tuononen, H.M.; Power, P.P. Isolation of a Stable, Acyclic, Two-Coordinate Silylene. J. Am. Chem. Soc. 2012, 134, 6504–6507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Xie, Y.; Wei, P.; Schaefer, H.F., III; Schleyer, P.V.R.; Robinson, G.H. Stabilization of elusive silicon oxides. Nat. Chem. 2015, 7, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Wendel, D.; Reiter, D.; Porzelt, A.; Altmann, P.J.; Inoue, S.; Rieger, B. Silicon and Oxygen’s Bond of Affection: An Acyclic Three-Coordinate Silanone and Its Transformation to an Iminosiloxysilylene. J. Am. Chem. Soc. 2017, 139, 17193–17198. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yao, S.; Inoue, S.; Epping, J.D.; Driess, M. A Cyclic Silylone (“Siladicarbene”) with an Electron-Rich Silicon(0) Atom. Angew. Chem. Int. Ed. 2013, 52, 7147–7150. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yao, S.; Inoue, S.; Irran, E.; Driess, M. The Elusive Silyliumylidene [ClSi:]+ and Silathionium [ClSi=S]+ Cations Stabilized by Bis(Iminophosphorane) Chelate Ligand. Angew. Chem. Int. Ed. 2012, 51, 10074–10077. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sekiguchi, A.; Driess, M. An N-Heterocyclic Carbene−Disilyne Complex and Its Reactivity toward ZnCl2. J. Am. Chem. Soc. 2010, 132, 14061–14063. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Huch, V.; Rzepa, H.S.; Scheschkewitz, D. Equilibrium between a cyclotrisilene and an isolable base adduct of a disilenyl silylene. Nat. Chem. 2013, 5, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Chernov, O.; Schnakenburg, G. SiBr2(Idipp): A Stable N-Heterocyclic Carbene Adduct of Dibromosilylene. Angew. Chem. Int. Ed. 2009, 48, 5687–5690. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Lebedev, Y.N.; Chernov, O.; Straßmann, M.; Schnakenburg, G. Silicon(II) Coordination Chemistry: N-Heterocyclic Carbene Complexes of Si2+ and SiI+. Angew. Chem. Int. Ed. 2013, 52, 6974–6978. [Google Scholar] [CrossRef] [PubMed]
- Ghadwal, R.S.; Pröpper, K.; Dittrich, B.; Jones, P.G.; Roesky, H.W. Neutral Pentacoordinate Silicon Fluorides Derived from Amidinate, Guanidinate, and Triazapentadienate Ligands and Base-Induced Disproportionation of Si2Cl6 to Stable Silylenes. Inorg. Chem. 2011, 50, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Ghadwal, R.S.; Roesky, H.W.; Merkel, S.; Henn, J.; Stalke, D. Lewis Base Stabilized Dichlorosilylene. Angew. Chem. Int. Ed. 2009, 48, 5683–5686. [Google Scholar] [CrossRef] [PubMed]
- Rivard, E. Donor-acceptor chemistry in the main group. Dalton Trans. 2014, 43, 8577–8586. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, J.I.; Meyer, L.; Nadj, A.; Diefenbach, M.; Holthausen, M.C. Unraveling the Amine-Induced Disproportionation Reaction of Perchlorinated Silanes—A DFT Study. Chem. Eur. J. 2016, 22, 14328–14335. [Google Scholar] [CrossRef] [PubMed]
- Sinhababu, S.; Kundu, S.; Paesch, A.N.; Herbst-Irmer, R.; Stalke, D.; Fernández, I.; Frenking, G.; Stückl, A.C.; Schwederski, B.; Kaim, W.; et al. A Route to Base Coordinate Silicon Difluoride and the Silicon Trifluoride Radical. Chem. Eur. J. 2018, 24, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, J.I.; Scheibel, M.G.; Diefenbach, M.; Neumeyer, F.; Würtele, C.; Kulminskaya, N.; Linser, R.; Auner, N.; Schneider, S.; Holthausen, M.C. A Disilene Base Adduct with a Dative Si–Si Single Bond. Angew. Chem. Int. Ed. 2016, 55, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Tillmann, J.; Meyer, L.; Schweizer, J.I.; Bolte, M.; Lerner, H.W.; Wagner, M.; Holthausen, M.C. Chloride-Induced Aufbau of Perchlorinated Cyclohexasilanes from Si2Cl6: A Mechanistic Scenario. Chem. Eur. J. 2014, 20, 9234–9239. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Wegner, F.; Nadj, A.; Bolte, M.; Auner, N.; Wagner, M.; Holthausen, M.C.; Lerner, H.W. The Perchlorinated Silanes Si2Cl6 and Si3Cl8 as Sources of SiCl2. Chem. Eur. J. 2011, 17, 4715–4719. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P.P. Learning from silylenes and supersilylenes. In Organosilicon Chemistry VI: From Molecules to Materials, 1; Auner, N., Weis, J., Eds.; Wiley-VCH: Weinheim, Germany, 2005; Volume 2, pp. 10–24. ISBN 9783527618224. [Google Scholar]
- Müller, T. Stability, Reactivity, and Strategies for the Synthesis of Silyliumylidenes, RSi:+. A Computational Study. Organometallics 2010, 29, 1277–1283. [Google Scholar] [CrossRef]
- Jutzi, P.; Mix, A.; Rummel, B.; Schoeller, W.W.; Neumann, B.; Stammler, H.-G. The (Me5C5)Si+ Cation: A Stable Derivative of HSi+. Science 2004, 305, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Driess, M.; Yao, S.; Brym, M.; van Wüllen, C. Low-Valent Silicon Cations with Two-Coordinate Silicon and Aromatic Character. Angew. Chem. Int. Ed. 2006, 45, 6730–6733. [Google Scholar] [CrossRef] [PubMed]
- Hudnall, T.W.; Ugarte, R.A.; Perera, T.A. Main group complexes with N-Heterocyclic carbenes: Bonding, stabilization and applications in catalysis. In N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools (2); The Royal Society of Chemistry: London, UK, 2017; pp. 178–237. ISBN 978-1-78262-423-3. [Google Scholar]
- Melaimi, M.; Jazzar, R.; Soleilhavoup, M.; Bertrand, G. Cyclic (Alkyl)(amino)carbenes (CAACs): Recent Developments. Angew. Chem. Int. Ed. 2017, 56, 10046–10068. [Google Scholar] [CrossRef] [PubMed]
- Agou, T.; Hayakawa, N.; Sasamori, T.; Matsuo, T.; Hashizume, D.; Tokitoh, N. Reactions of Diaryldibromodisilenes with N-Heterocyclic Carbenes: Formation of Formal Bis-NHC Adducts of Silyliumylidene Cations. Chem. Eur. J. 2014, 20, 9246–9249. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.U.; Szilvási, T.; Inoue, S. A facile access to a novel NHC-stabilized silyliumylidene ion and C-H activation of phenylacetylene. Chem. Commun. 2014, 50, 12619–12622. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, N.; Sadamori, K.; Mizutani, S.; Agou, T.; Sugahara, T.; Sasamori, T.; Tokitoh, N.; Hashizume, D.; Matsuo, T. Synthesis and Characterization of N-Heterocyclic Carbene-Coordinated Silicon Compounds Bearing a Fused-Ring Bulky Eind Group. Inorganics 2018, 6, 30. [Google Scholar] [CrossRef]
- Li, Y.; Chan, Y.-C.; Li, Y.; Purushothaman, I.; De, S.; Parameswaran, P.; So, C.-W. Synthesis of a Bent 2-Silaallene with a Perturbed Electronic Structure from a Cyclic Alkyl(amino) Carbene-Diiodosilylene. Inorg. Chem. 2016, 55, 9091–9098. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, Y.-C.; Leong, B.-X.; Li, Y.; Richards, E.; Purushothaman, I.; De, S.; Parameswaran, P.; So, C.-W. Trapping a Silicon(I) Radical with Carbenes: A Cationic cAAC–Silicon(I) Radical and an NHC–Parent-Silyliumylidene Cation. Angew. Chem. Int. Ed. 2017, 56, 7573–7578. [Google Scholar] [CrossRef] [PubMed]
- Yeong, H.-X.; Xi, H.-W.; Li, Y.; Lim, K.H.; So, C.-W. A Silyliumylidene Cation Stabilized by an Amidinate Ligand and 4-Dimethylaminopyridine. Chem. Eur. J. 2013, 19, 11786–11790. [Google Scholar] [CrossRef] [PubMed]
- Leszczyńska, K.; Mix, A.; Berger, R.J.F.; Rummel, B.; Neumann, B.; Stammler, H.-G.; Jutzi, P. The Pentamethylcyclopentadienylsilicon(II) Cation as a Catalyst for the Specific Degradation of Oligo(ethyleneglycol) Diethers. Angew. Chem. Int. Ed. 2011, 50, 6843–6846. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A.; Inoue, S.; Präsang, C.; Driess, M. Steering S–H and N–H Bond Activation by a Stable N-Heterocyclic Silylene: Different Addition of H2S, NH3, and Organoamines on a Silicon(II) Ligand versus Its Si(II)→Ni(CO)3 Complex. J. Am. Chem. Soc. 2010, 132, 3038–3046. [Google Scholar] [CrossRef] [PubMed]
- Präsang, C.; Stoelzel, M.; Inoue, S.; Meltzer, A.; Driess, M. Metal-Free Activation of EH3 (E=P, As) by an Ylide-like Silylene and Formation of a Donor-Stabilized Arsasilene with a HSi=AsH Subunit. Angew. Chem. Int. Ed. 2010, 49, 10002–10005. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Brym, M.; van Wüllen, C.; Driess, M. From a Stable Silylene to a Mixed-Valent Disiloxane and an Isolable Silaformamide–Borane Complex with Considerable Silicon–Oxygen Double-Bond Character. Angew. Chem. Int. Ed. 2007, 46, 4159–4162. [Google Scholar] [CrossRef] [PubMed]
- Szilvási, T.; Nyíri, K.; Veszprémi, T. Unique Insertion Mechanisms of Bis-dehydro-β-diketiminato Silylene. Organometallics 2011, 30, 5344–5351. [Google Scholar] [CrossRef]
- Filippou, A.C.; Chernov, O.; Blom, B.; Stumpf, K.W.; Schnakenburg, G. Stable N-Heterocyclic Carbene Adducts of Arylchlorosilylenes and Their Germanium Homologues. Chem. Eur. J. 2010, 16, 2866–2872. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Chernov, O.; Stumpf, K.W.; Schnakenburg, G. Metal–Silicon Triple Bonds: The Molybdenum Silylidyne Complex [Cp(CO)2Mo≡Si-R]. Angew. Chem. Int. Ed. 2010, 49, 3296–3300. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Szilvási, T.; Blom, B.; Driess, M. A Persistent 1,2-Dihydrophosphasilene Adduct. Angew. Chem. Int. Ed. 2015, 54, 15060–15063. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Szilvási, T.; Blom, B.; Irran, E.; Driess, M. From an Isolable Acyclic Phosphinosilylene Adduct to Donor-Stabilized Si=E Compounds (E=O, S, Se). Chem. Eur. J. 2015, 21, 18930–18933. [Google Scholar] [CrossRef] [PubMed]
- Arz, M.I.; Hoffmann, D.; Schnakenburg, G.; Filippou, A.C. NHC-stabilized Silicon(II) Halides: Reactivity Studies with Diazoalkanes and Azides. Z. Anorg. Allg. Chem. 2016, 642, 1287–1294. [Google Scholar] [CrossRef]
- Ahmad, S.U.; Szilvási, T.; Irran, E.; Inoue, S. An NHC-Stabilized Silicon Analogue of Acylium Ion: Synthesis, Structure, Reactivity, and Theoretical Studies. J. Am. Chem. Soc. 2015, 137, 5828–5836. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Wendel, D.; Ahmad, S.U.; Szilvasi, T.; Pothig, A.; Inoue, S. Chalcogen-atom transfer and exchange reactions of NHC-stabilized heavier silaacylium ions. Dalton Trans. 2017, 46, 16014–16018. [Google Scholar] [CrossRef] [PubMed]
- Baceiredo, A.; Kato, T. Multiple Bonds to Silicon (Recent Advances in the Chemistry of Silicon Containing Multiple Bonds). In Organosilicon Compounds: Theory and Experiment (Synthesis); Lee, V.Y., Ed.; Academic Press: London, UK, 2017; pp. 533–618. ISBN 978-0-12-801981-8. [Google Scholar]
- Lutters, D.; Merk, A.; Schmidtmann, M.; Müller, T. The Silicon Version of Phosphine Chalcogenides: Synthesis and Bonding Analysis of Stabilized Heavy Silaaldehydes. Inorg. Chem. 2016, 55, 9026–9032. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P.; Atsumi, M. Molecular Double-Bond Covalent Radii for Elements Li–E112. Chem. Eur. J. 2009, 15, 12770–12779. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Webb, R.L.; McElhill, E.A. The Electrical Effect of the Trifluoromethyl Group. J. Am. Chem. Soc. 1950, 72, 408–411. [Google Scholar] [CrossRef]
- Von Ragué Schleyer, P.; Kos, A.J. The importance of negative (anionic) hyperconjugation. Tetrahedron 1983, 39, 1141–1150. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990; ISBN 0198558651. [Google Scholar]
- Matta, C.F.; Boyd, R.J. An Introduction to the Quantum Theory of Atoms in Molecule; Wiley-VCH: Weinheim, Germany, 2007; ISBN 3527610707. [Google Scholar]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 238, 383–412. [Google Scholar] [CrossRef]
- Macchi, P.; Sironi, A. Interactions involving metals—From ‘Chemical Categories’ to QTAIM, and Backwards. In The Quantum Theory of Atoms in Molecules; Matta, C.F., Boyd, R.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E.; Slee, T.S.; Bader, R.F.W.; Lau, C.D.H.; Nguyen Dang, T.T.; MacDougall, P.J. Description of homoaromaticity in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5069–5075. [Google Scholar] [CrossRef]
- Mandado, M.; Mosquera, R.A.; Graña, A.M. On the effects of electron correlation and conformational changes on the distortion of the charge distribution in alkyl chains. Chem. Phys. Lett. 2002, 355, 529–537. [Google Scholar] [CrossRef]
- Simons, R.S.; Haubrich, S.T.; Mork, B.V.; Niemeyer, M.; Power, P.P. The Syntheses and Characterization of the Bulky Terphenyl Silanes and Chlorosilanes 2,6-Mes2C6H3SiCl3, 2,6-Trip2C6H3SiCl3, 2,6-Mes2C6H3SiHCl2, 2,6-Trip2C6H3SiHCl2, 2,6-Mes2C6H3SiH3, 2,6-Trip2C6H3SiH3and 2,6-Mes2C6H3SiCl2SiCl3. Main Group Chem. 1998, 2, 275–283. [Google Scholar] [CrossRef]
- Kuhn, N.; Kratz, T. Synthesis of Imidazol-2-ylidenes by Reduction of Imidazole-2(3H)-thiones. Synthesis 1993, 1993, 561–562. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Revision, D.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Houk, K.N. Integration Grid Errors for Meta-GGA-Predicted Reaction Energies: Origin of Grid Errors for the M06 Suite of Functionals. J. Chem. Theory Comput. 2010, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Glendening Eric, D.; Landis Clark, R.; Weinhold, F. Natural bond orbital methods. WIRES Ccomput. Mol. Sci. 2011, 2, 1–42. [Google Scholar] [CrossRef]
- Glendening Eric, D.; Landis Clark, R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Chem. Theory Comput. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll (Version 17. 01. 25); TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- Andrienko, G.A. ChemCraf—graphical software for visualization of quantum chemistry computations. Available online: http://www.chemcraftprog.com (accessed on 3 January 2015).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | dSi–S [Å] | dSi–bcp [Å] | dbcp–S [Å] | ρbcp [eÅ−3] | ∇2ρbcp [eÅ−5] | Hbcp [EhÅ−3] | εbcp | δSi,S |
|---|---|---|---|---|---|---|---|---|
| BModel | 2.00 | 0.75 | 1.26 | 0.78 | 3.33 | −0.52 | 0.01 | 0.78 |
| 1 | 2.13 | 0.77 | 1.36 | 0.66 | 1.36 | −0.43 | 0.12 | 0.56 |
| 2 | 1.95 | 0.73 | 1.22 | 0.83 | 5.24 | −0.55 | 0.21 | 1.15 |
| 3 | 2.14 | 0.78 | 1.37 | 0.64 | 1.36 | −0.40 | 0.10 | 0.57 |
| 4 | 1.94 | 0.73 | 1.21 | 0.83 | 5.39 | −0.56 | 0.23 | 1.25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porzelt, A.; Schweizer, J.I.; Baierl, R.; Altmann, P.J.; Holthausen, M.C.; Inoue, S. S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions. Inorganics 2018, 6, 54. https://doi.org/10.3390/inorganics6020054
Porzelt A, Schweizer JI, Baierl R, Altmann PJ, Holthausen MC, Inoue S. S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions. Inorganics. 2018; 6(2):54. https://doi.org/10.3390/inorganics6020054
Chicago/Turabian StylePorzelt, Amelie, Julia I. Schweizer, Ramona Baierl, Philipp J. Altmann, Max C. Holthausen, and Shigeyoshi Inoue. 2018. "S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions" Inorganics 6, no. 2: 54. https://doi.org/10.3390/inorganics6020054
APA StylePorzelt, A., Schweizer, J. I., Baierl, R., Altmann, P. J., Holthausen, M. C., & Inoue, S. (2018). S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions. Inorganics, 6(2), 54. https://doi.org/10.3390/inorganics6020054