Unexpected Formation and Structural Characterization of a Dinuclear Sodium Half-Sandwich Complex


{kind=link}
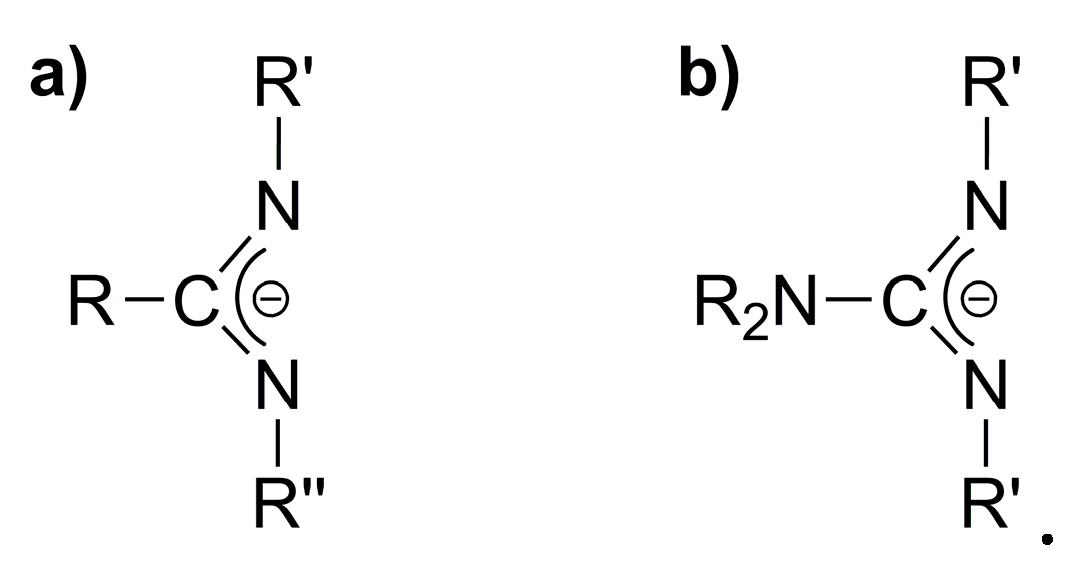{kind=link}
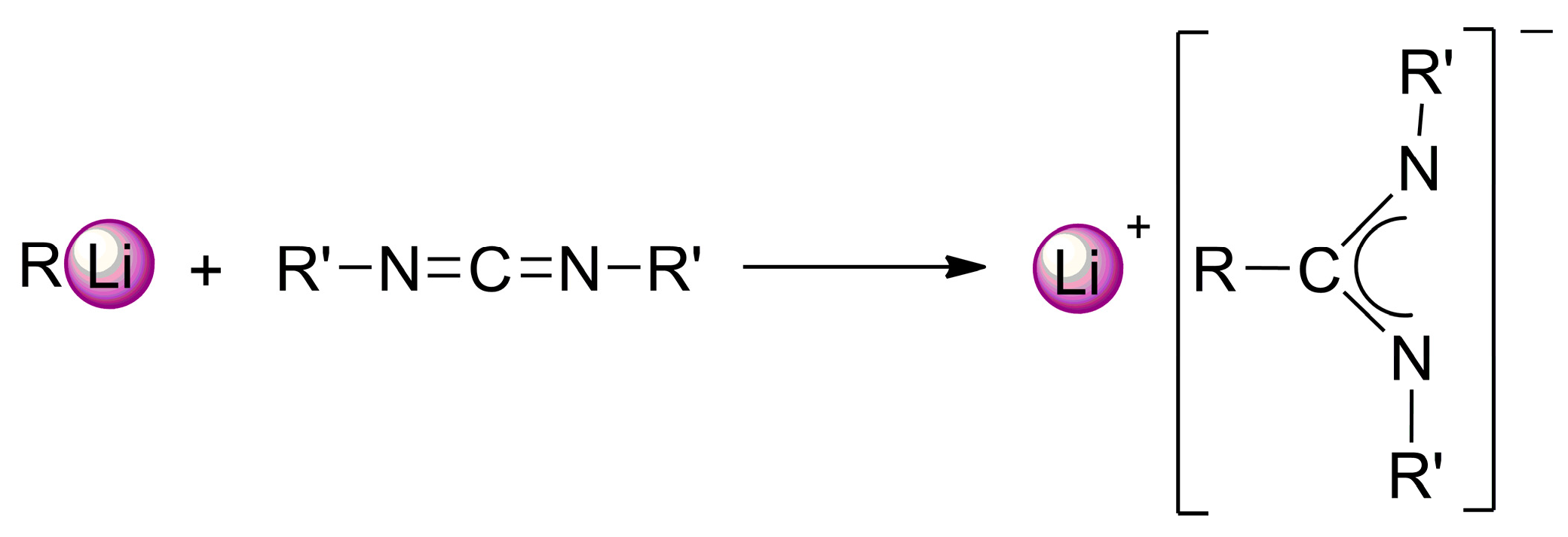{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
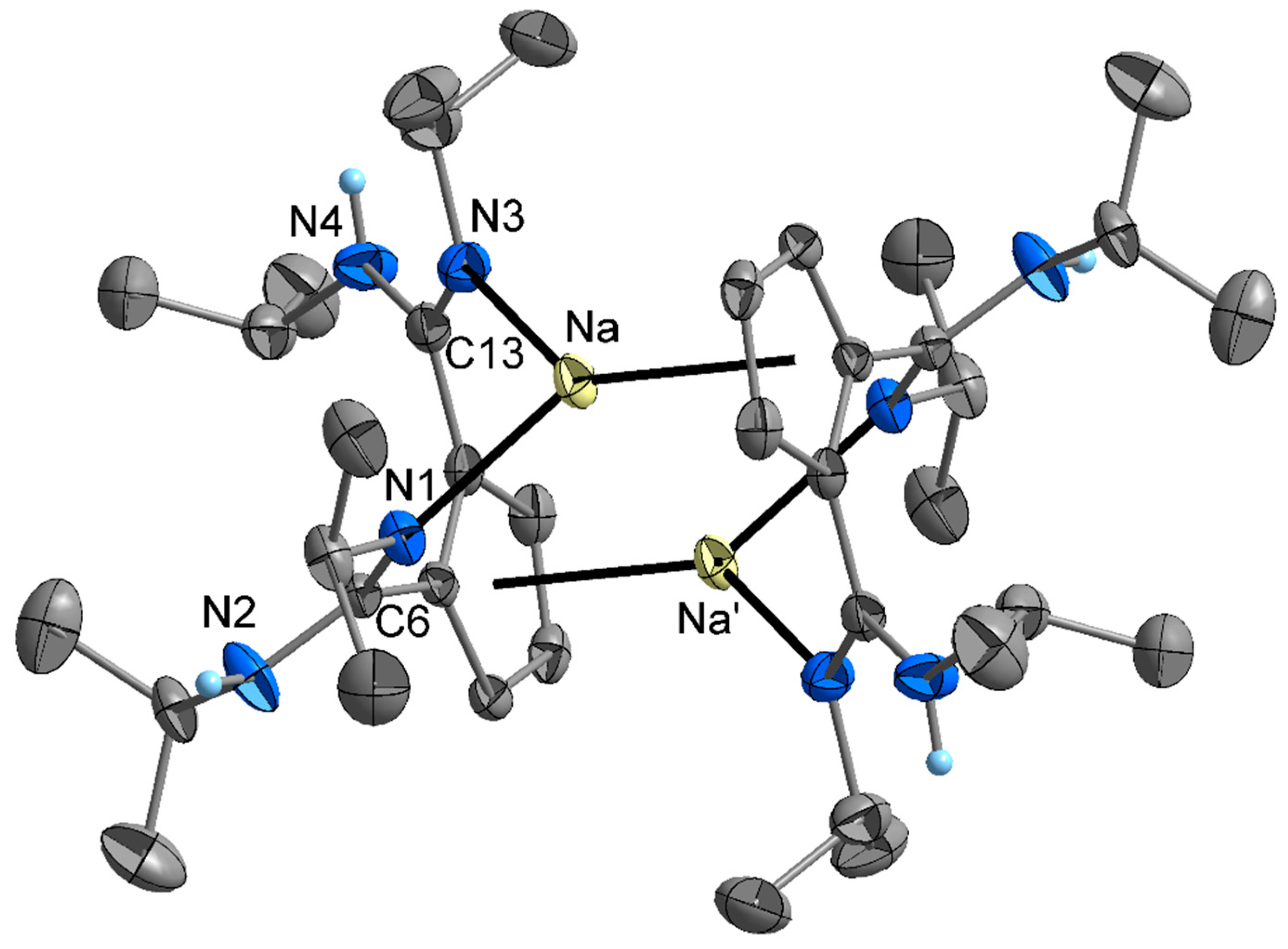
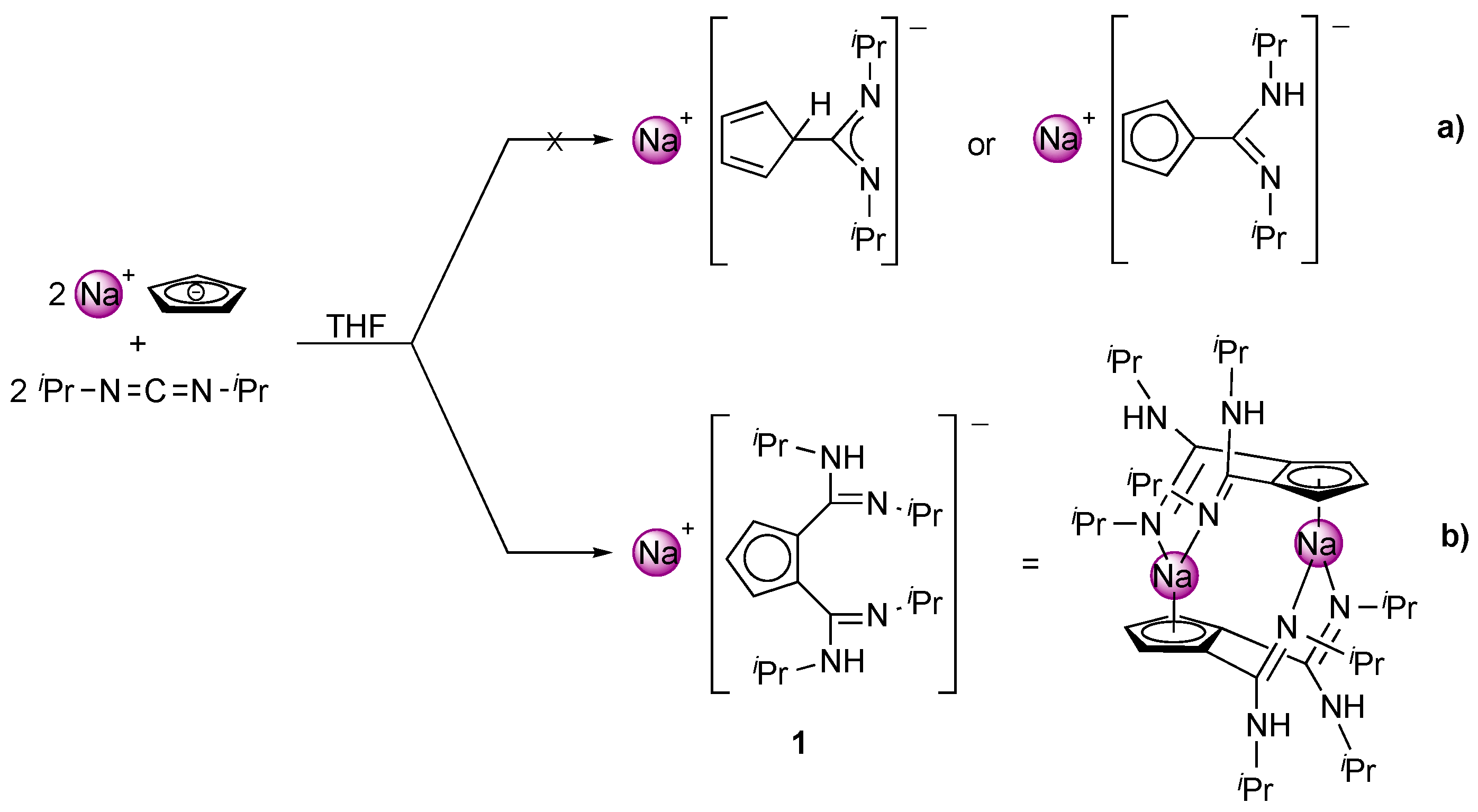
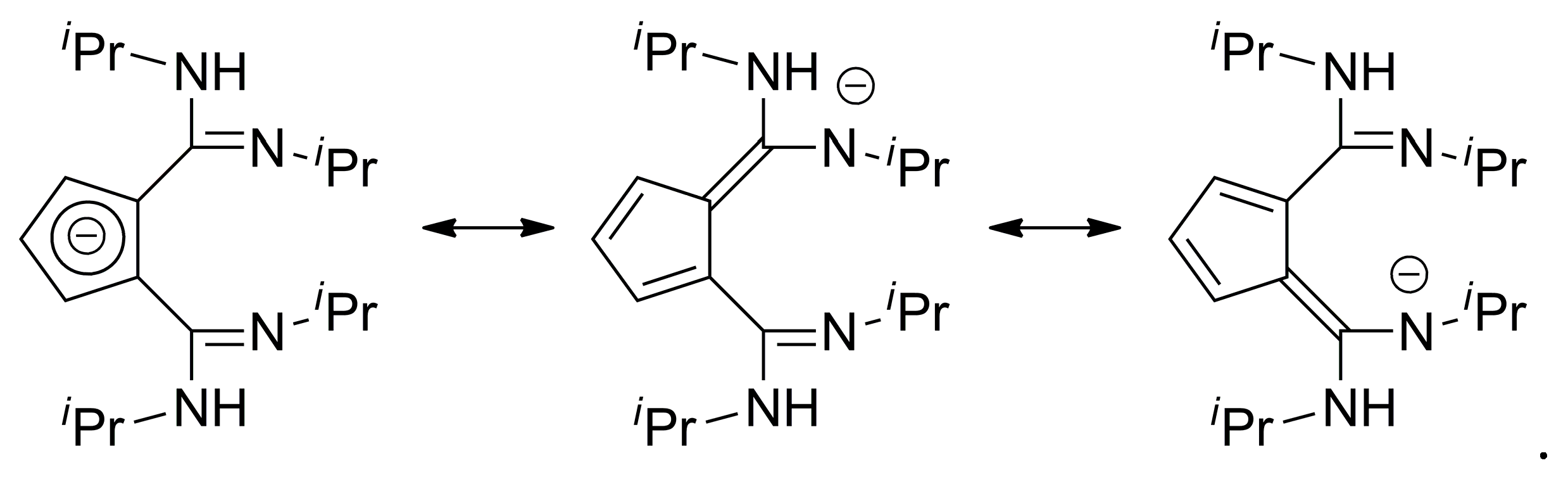
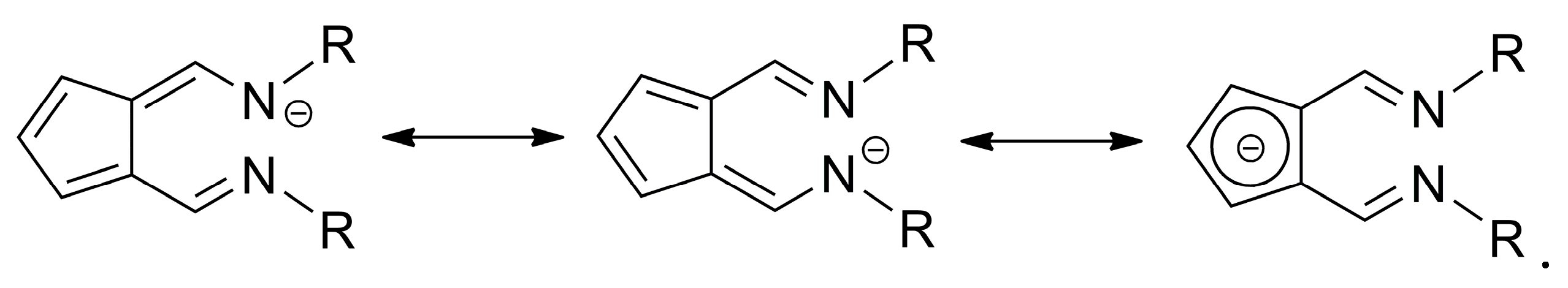
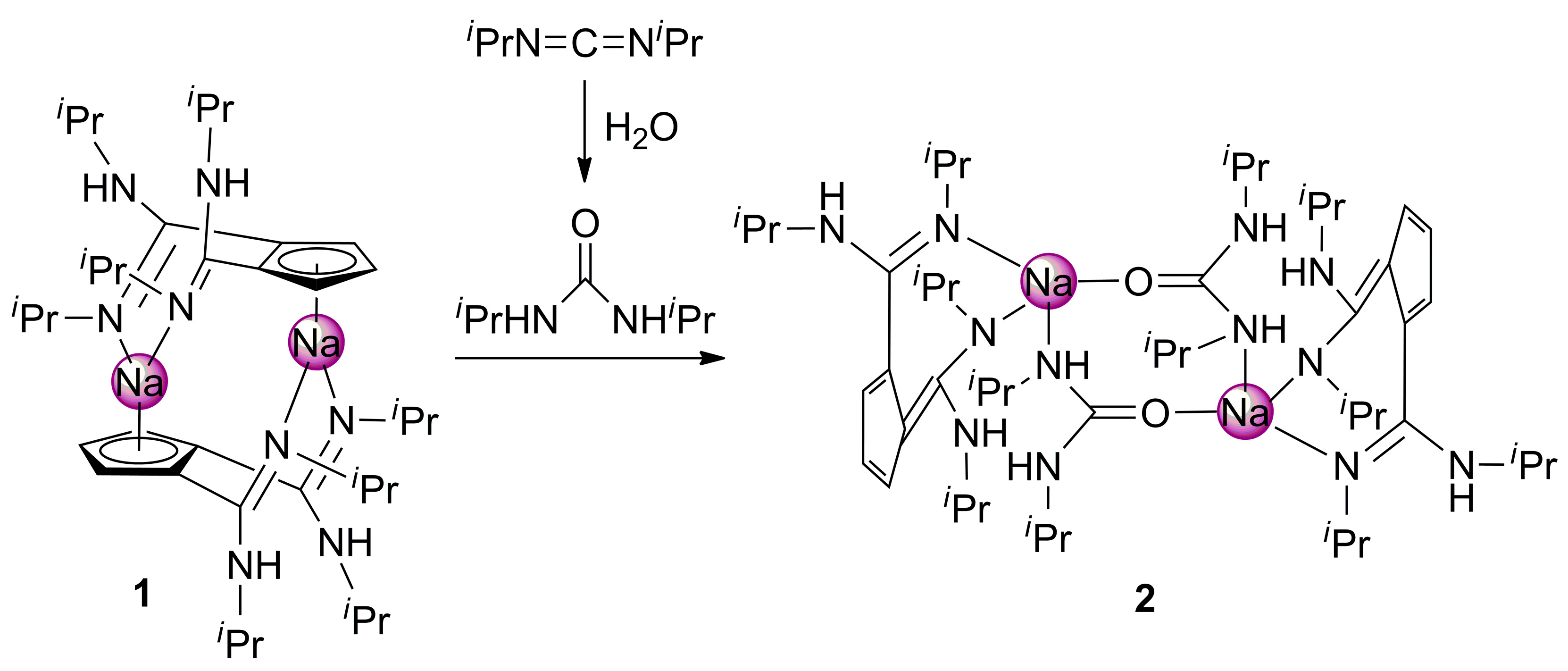
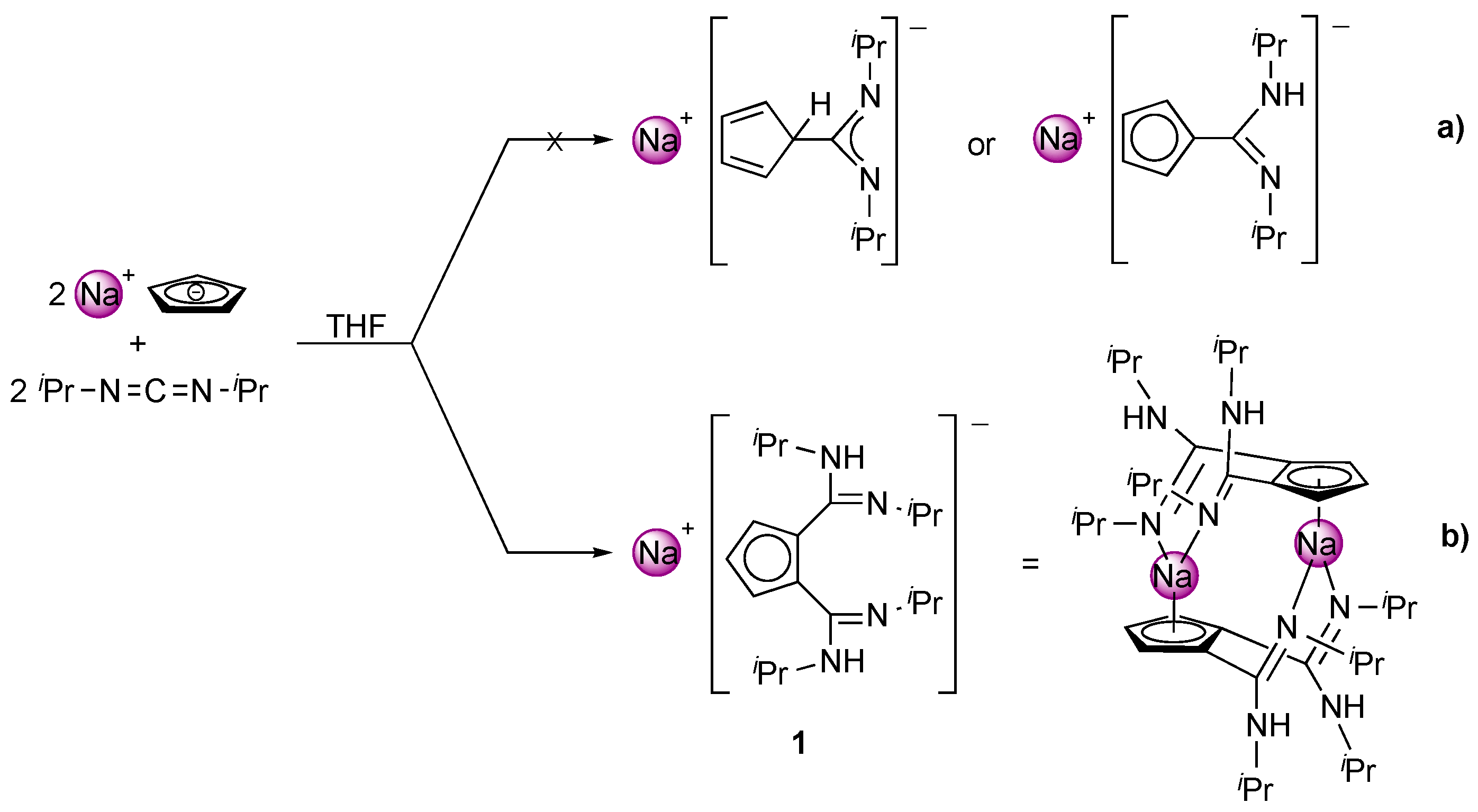
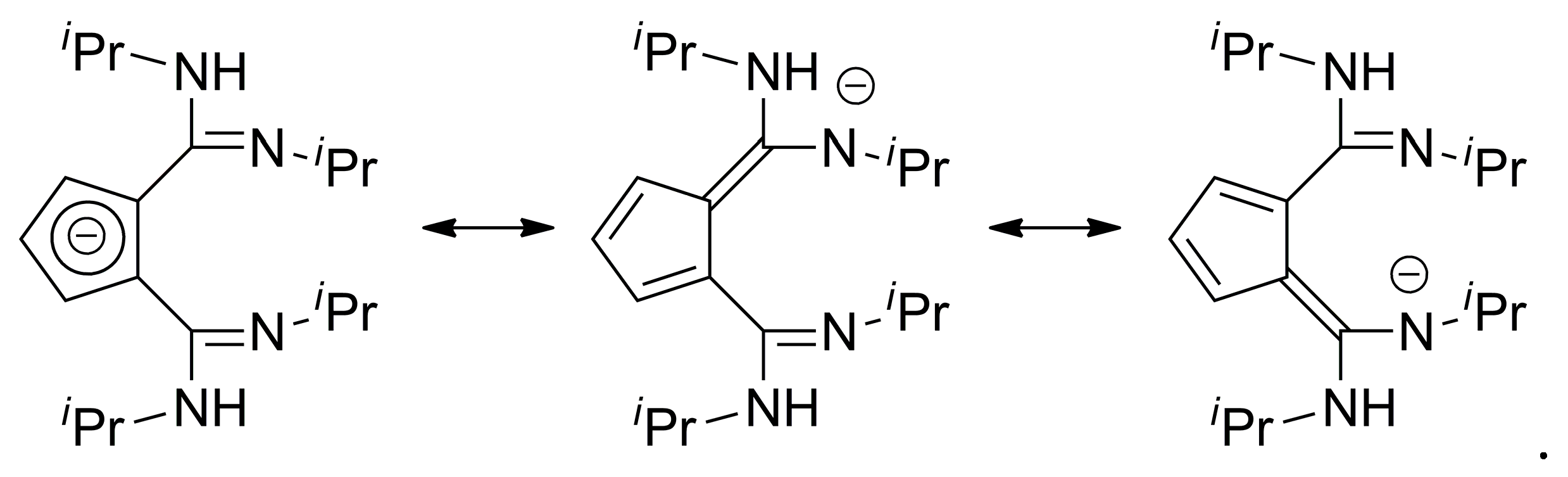
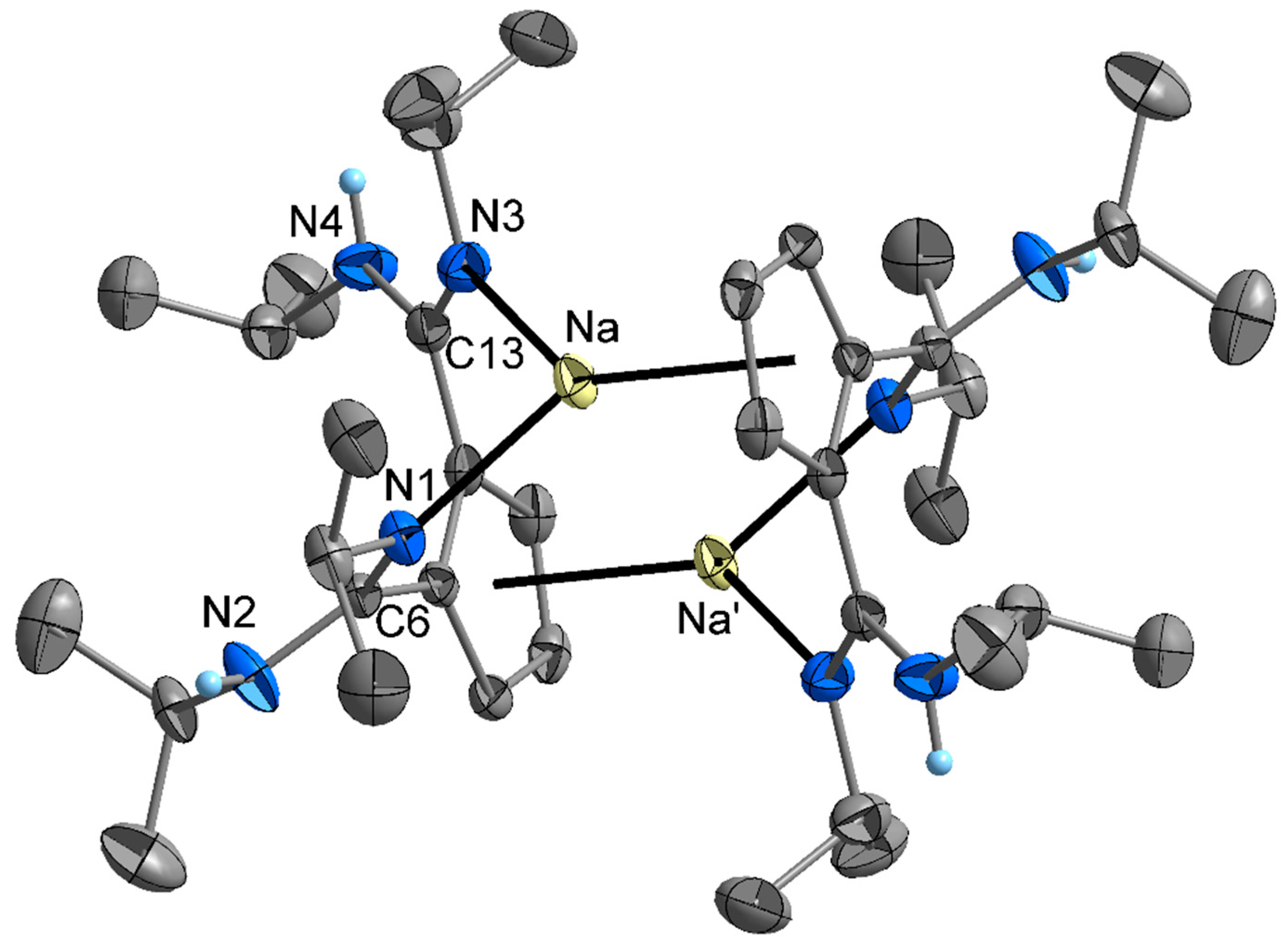
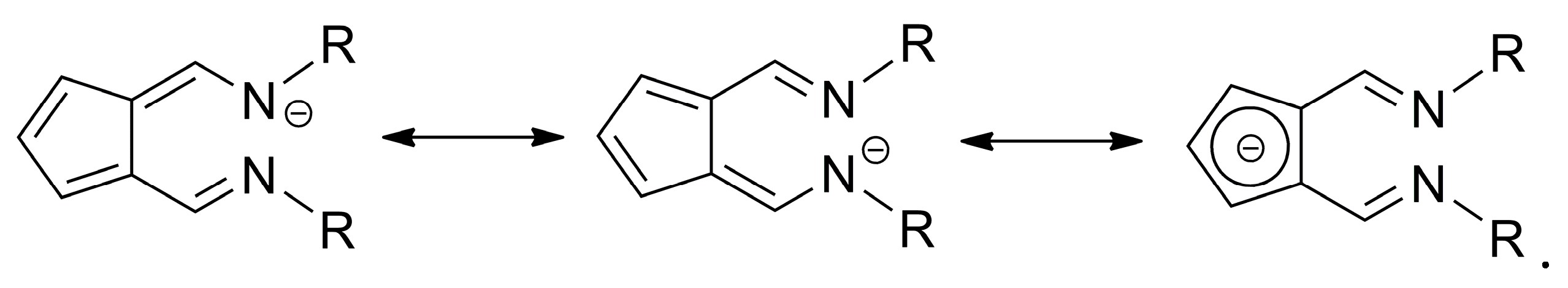
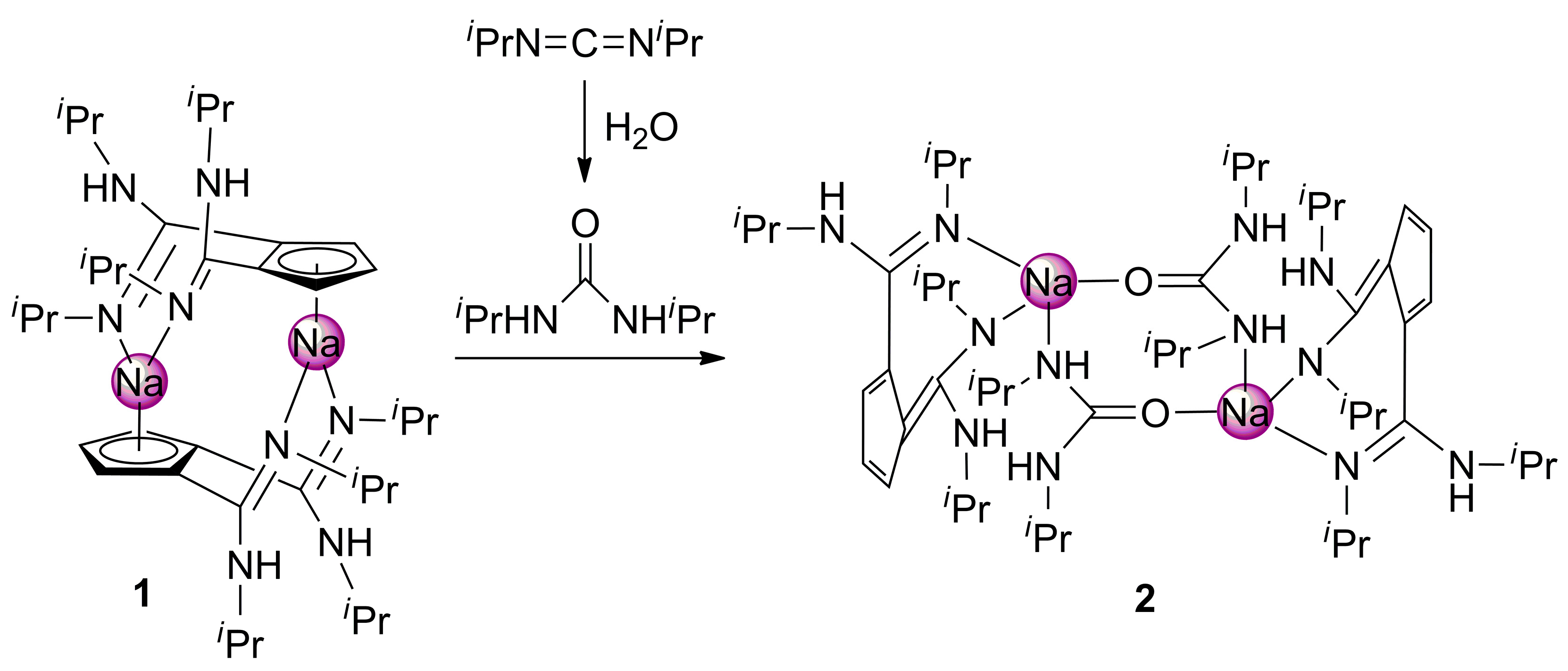
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. General Procedures
4.2. Synthesis of [NaC5H3{C(NHiPr)(=NiPr)}2-1,2]2 (1)
4.3. X-ray Crystallography
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kissounko, D.A.; Zabalov, M.V.; Brusova, G.P.; Lemenovskii, D.A. Principal trends in the chemistry of amidinate complexes of main-group and transition elements. Russ. Chem. Rev. 2006, 75, 351–374. [Google Scholar] [CrossRef]
- Edelmann, F.T. Advances in the coordination chemistry of amidinate and guanidinate ligands. Adv. Organomet. Chem. 2008, 57, 183–352. [Google Scholar]
- Edelmann, F.T. Lanthanide Amidinates and guanidinates: From laboratory curiosities to efficient homogeneous catalysts and precursors for rare-earth oxide thin films. Chem. Soc. Rev. 2009, 38, 2253–2268. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.P. Bicyclic-guanidines, -guanidinates and -guanidinium salts: Wide ranging applications from a simple family of molecules. Chem. Commun. 2009, 25, 3659–3676. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, A.A. Guanidinate and amidopyridinate rare-earth complexes: Towards highly reactive alkyl and hydrido species. Coord. Chem. Rev. 2010, 254, 1327–1347. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Abdou, H.E., Jr.; Fackler, J.P. Coordination chemistry of gold(II) with amidinate, thiolate and ylide ligands. Coord. Chem. Rev. 2010, 254, 1253–1259. [Google Scholar] [CrossRef]
- Edelmann, F.T. Lanthanide amidinates and guanidinates in catalysis and materials science: A continuing success story. Chem. Soc. Rev. 2012, 41, 7657–7672. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, F.T. Recent progress in the chemistry of metal amidinates and guanidinates: Syntheses, catalysis and materials. Adv. Organomet. Chem. 2013, 61, 55–374. [Google Scholar]
- Zhang, Y.-Y.; Lin, Y.-J.; Shi, X.-C.; Jin, G.-X. Organometallic macrocycles and cages based on bis(amidinate) ligands. Pure Appl. Chem. 2014, 86, 953–965. [Google Scholar] [CrossRef]
- Jones, C. Bulky guanidinates for the stabilization of low oxidation state metallacycles. Coord. Chem. Rev. 2010, 254, 1273–1289. [Google Scholar] [CrossRef]
- Chlupatý, T.; Růžička, A. Hybrid amidinates and guanidinates of main group metals. Coord. Chem. Rev. 2016, 314, 103–113. [Google Scholar] [CrossRef]
- Tacke, R.; Ribbeck, T. Bis(amidinato)- and bis(guanidinato)silylenes and silylenes with one sterically demanding amidinato or guanidinato ligand: Synthesis and reactivity. Dalton Trans. 2017, 46, 13628–13659. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, H.; Kondo, H.; Hayashida, T.; Yamaguchi, Y.; Gondo, M.; Masuda, S.; Miyazaki, K.; Matsubara, K.; Kirchner, K. Chemistry of coordinatively unsaturated organoruthenium amidinates as entry to homogeneous catalysis. Coord. Chem. Rev. 2003, 245, 177–190. [Google Scholar] [CrossRef]
- Edelmann, F.T. Homogeneous catalysis using lanthanide amidinates and guanidinates. Struct. Bond. 2010, 137, 109–163. [Google Scholar]
- Collins, S. Polymerization catalysis with transition metal amidinate and related complexes. Coord. Chem. Rev. 2011, 255, 118–138. [Google Scholar] [CrossRef]
- Elkin, T.; Eisen, M.S. Amidinate group 4 complexes in the polymerization of olefins. Catal. Sci. Technol. 2015, 5, 82–95. [Google Scholar] [CrossRef]
- Barry, S.T. Amidinates, guanidinates and iminopyrrolidinates: Understanding precursor thermolysis to design a better ligand. Coord. Chem. Rev. 2013, 257, 3192–3201. [Google Scholar] [CrossRef]
- Devi, A. “Old Chemistries” for new applications: Perspectives for development of precursors for MOCVD and ALD applications. Coord. Chem. Rev. 2013, 257, 3332–3384. [Google Scholar] [CrossRef]
- Koponen, S.E.; Gordon, P.G.; Barry, S.T. Principles of precursor design for vapour deposition methods. Polyhedron 2016, 108, 59–66. [Google Scholar] [CrossRef]
- Lichtenberg, C.; Adelhardt, M.; Wörle, M.; Büttner, T.; Meyer, K.; Grützmacher, H. Mono- and Dinuclear Neutral and Cationic Iron(II) Compounds Supported by an Amidinato-diolefin Ligand: Characterization and Catalytic Application. Organometallics 2015, 34, 3079–3089. [Google Scholar] [CrossRef]
- Cole, M.L.; Jones, C.; Junk, P.C. Ether and crown ether adduct complexes of sodium and potassium cyclopentadienide and methylcyclopentadienide—Molecular structures of [Na(dme)Cp]∞, [K(dme)0.5Cp]∞, [Na(15-crown-5)Cp], [Na(18-crown-6)CpMe] and the “naked Cp–” complex [K(15-crown-5)2][Cp]. J. Chem. Soc. Dalton Trans. 2002, 6, 896–905. [Google Scholar] [CrossRef]
- Ursillo, S.; Can, D.; N’Dongo, H.W.P.; Schmutz, P.; Spingler, B.; Alberto, R. Cyclopentadienyl Chemistry in Water: Synthesis and Properties of Bifunctionalized [(η5-C5H3{COOR}2)M(CO)3] (M = Re and 99mTc) Complexes. Organometallics 2014, 33, 6945–6952. [Google Scholar] [CrossRef]
- Hafner, K.; Vöpel, K.H.; Ploss, G.; König, C. Cyclisch konjugierte 5- und 7-Ringsysteme, I. Synthesen und Reaktionen von Fulvenaldehyden. Justus Liebigs Ann. Chem. 1963, 661, 52–75. [Google Scholar] [CrossRef]
- Hafner, K.; Vöpel, K.H.; Ploss, G.; König, C. 6-(Dimethylamino)fulvene. Org. Synth. 1967, 47, 52–53. [Google Scholar]
- Etkin, N.; Ong, C.M.; Stephan, D.W. Synthesis of 1,2-Cyclopentadienyl Diimine Anions and Their Zirconium Complexes. Organometallics 1998, 17, 3656–3660. [Google Scholar] [CrossRef]
- Ong, C.M.; Stephan, D.W. 1,2-Cyclopentadienyl Diimine−Group 13 Complexes. Inorg. Chem. 1999, 38, 5189–5191. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.J.; Melchionna, M.; Parsons, S. Ambidentate Character of the 6-Aminofulvene-2-aldiminate Ligand Containing Both Diimine and Cyclopentadienyl Donors. Organometallics 2007, 26, 128–135. [Google Scholar] [CrossRef]
- Bailey, P.J.; Collins, A.; Haack, P.; Parsons, S.; Rahman, M.; Smith, D.; White, F.J. Palladium complexes of 6-aminofulvene-2-aldiminate (AFA) ligands. Dalton Trans. 2010, 39, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Willcocks, A.M.; Johnson, A.L.; Raithby, P.R.; Schiffers, S.; Warren, J.E. Bis(tert-butyl isocyanide-κC)[4-fluoro-N-({2-[N-(4-fluorophenyl)carboximidoyl]cyclopenta-2,4-dien-1-ylidene}methyl)anilinido-κ2N,N′]copper(I). Acta Crystallogr. Sect. C 2011, 67, m215–m217. [Google Scholar] [CrossRef] [PubMed]
- Willcocks, A.M.; Gilbank, A.; Richards, S.P.; Brayshaw, S.K.; Kingsley, A.J.; Odedra, R.; Johnson, A.L. Synthesis and Structure of 6-Aminofulvene-2-aldiminato Complexes. Inorg. Chem. 2011, 50, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.J.; Rahman, M.; Parsons, S.; Ahzar, M.R.; White, F.J. Metalloligands containing aminofulvene-aldiminate (AFA) ligands and their bimetallic complexes. Dalton Trans. 2013, 42, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.J.; Loroño-González, D.; Parsons, S. 6-Aminofulvene-2-aldimine, a novel class of ambidentate cyclopentadienyl/diimine ligand: Synthesis and characterisation of magnesium complexes. Chem. Commun. 2003, 12, 1426–1427. [Google Scholar] [CrossRef]
- Kuhn, N.; Steimann, M.; Weyers, G.; Henkel, G. l,3-Diisopropyl-4,5-dimethylimidazolium-2-N,N’-diisopropylamidinat, ein neuartiges Betain. Z. Naturforsch. 1999, 54, 434–440. [Google Scholar] [CrossRef]
- Márquez, A.; Àvila, E.; Urbaneja, C.; Àlvarez, E.; Palma, P.; Cámpora, J. Copper(I) Complexes of Zwitterionic Imidazolium-2-Amidinates, a Promising Class of Electroneutral, Amidinate-Type Ligands. Inorg. Chem. 2015, 54, 11007–11017. [Google Scholar] [CrossRef] [PubMed]
- Baishya, A.; Kumar, L.; Barman, M.K.; Biswal, H.S.; Nembenna, S. N-Heterocyclic Carbene–Carbodiimide (“NHC–CDI”) Adduct or Zwitterionic-Type Neutral Amidinate-Supported Magnesium(II) and Zinc(II) Complexes. Inorg. Chem. 2017, 56, 9535–9546. [Google Scholar] [CrossRef] [PubMed]
- Hart, W.P.; Shihua, D.; Rausch, M.D. The formation and reactions of (η5-carboxycyclopentadienyl)dicarbonylcobalt. J. Organomet. Chem. 1985, 282, 111–121. [Google Scholar] [CrossRef]
- Stoe, C. X-Area and X-Red; Stoe & Cie GmbH: Darmstadt, Germany, 2002. [Google Scholar]
- Sheldrick, G.M. ShelXS, Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
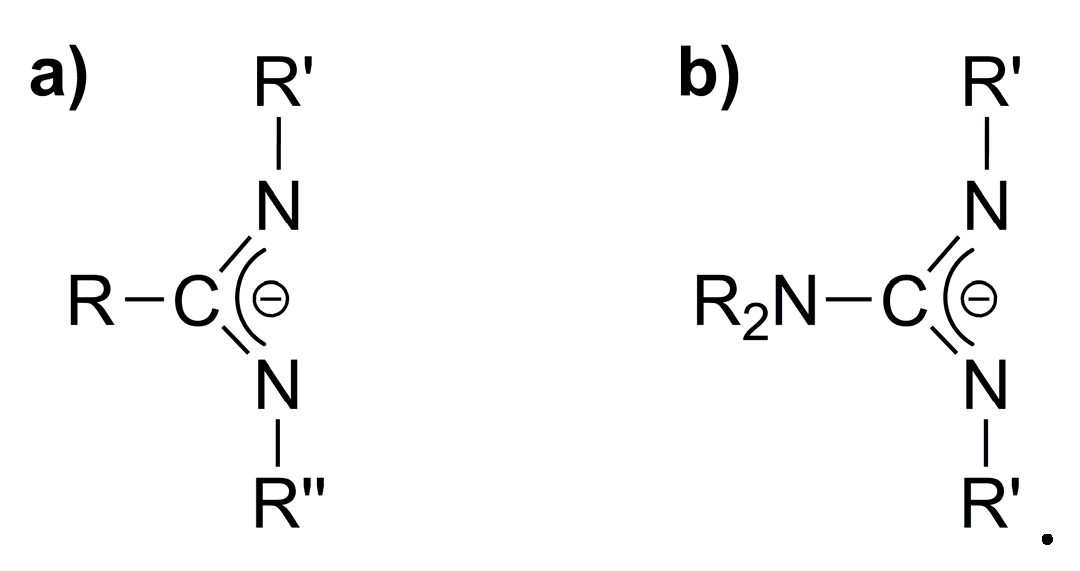
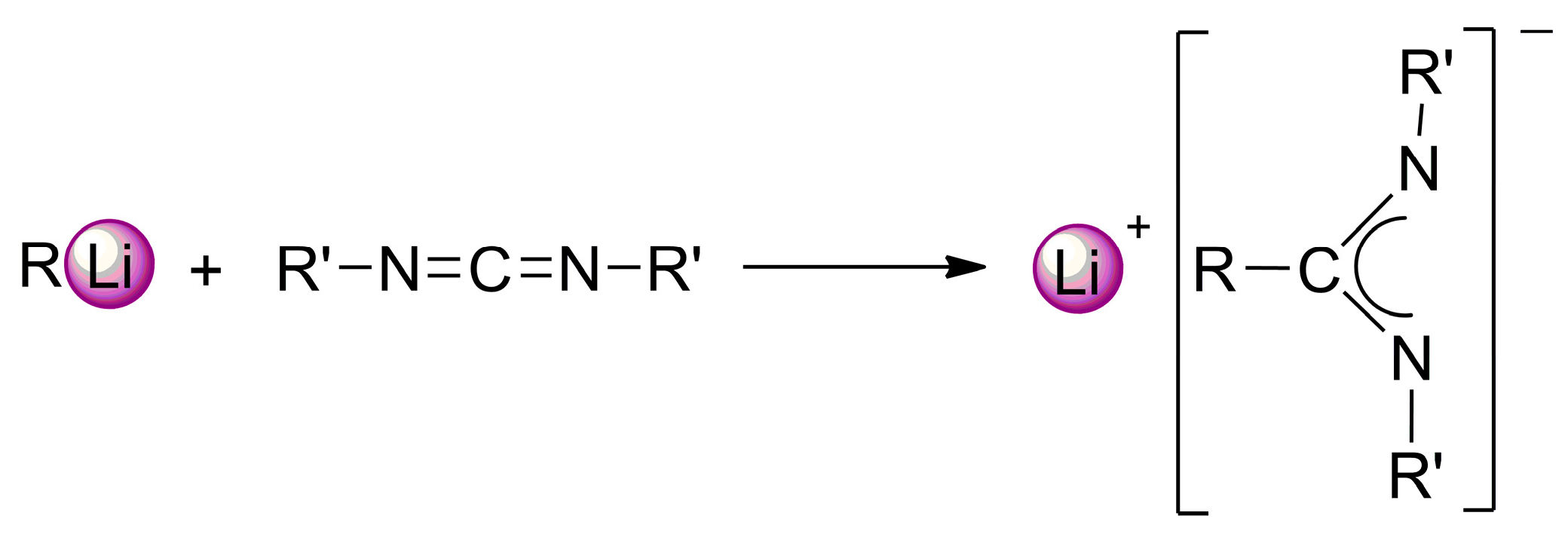
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harmgarth, N.; Liebing, P.; Hilfert, L.; Busse, S.; Edelmann, F.T. Unexpected Formation and Structural Characterization of a Dinuclear Sodium Half-Sandwich Complex. Inorganics 2018, 6, 47. https://doi.org/10.3390/inorganics6020047
Harmgarth N, Liebing P, Hilfert L, Busse S, Edelmann FT. Unexpected Formation and Structural Characterization of a Dinuclear Sodium Half-Sandwich Complex. Inorganics. 2018; 6(2):47. https://doi.org/10.3390/inorganics6020047
Chicago/Turabian StyleHarmgarth, Nicole, Phil Liebing, Liane Hilfert, Sabine Busse, and Frank T. Edelmann. 2018. "Unexpected Formation and Structural Characterization of a Dinuclear Sodium Half-Sandwich Complex" Inorganics 6, no. 2: 47. https://doi.org/10.3390/inorganics6020047
APA StyleHarmgarth, N., Liebing, P., Hilfert, L., Busse, S., & Edelmann, F. T. (2018). Unexpected Formation and Structural Characterization of a Dinuclear Sodium Half-Sandwich Complex. Inorganics, 6(2), 47. https://doi.org/10.3390/inorganics6020047