Functionalized o-Quinones: Concepts, Achievements and Prospects



Abstract
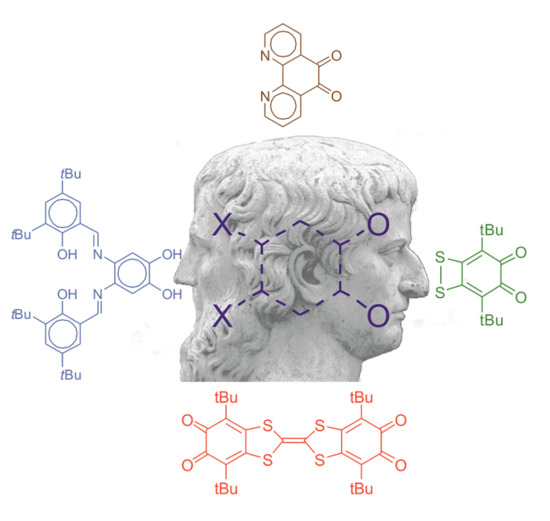
:
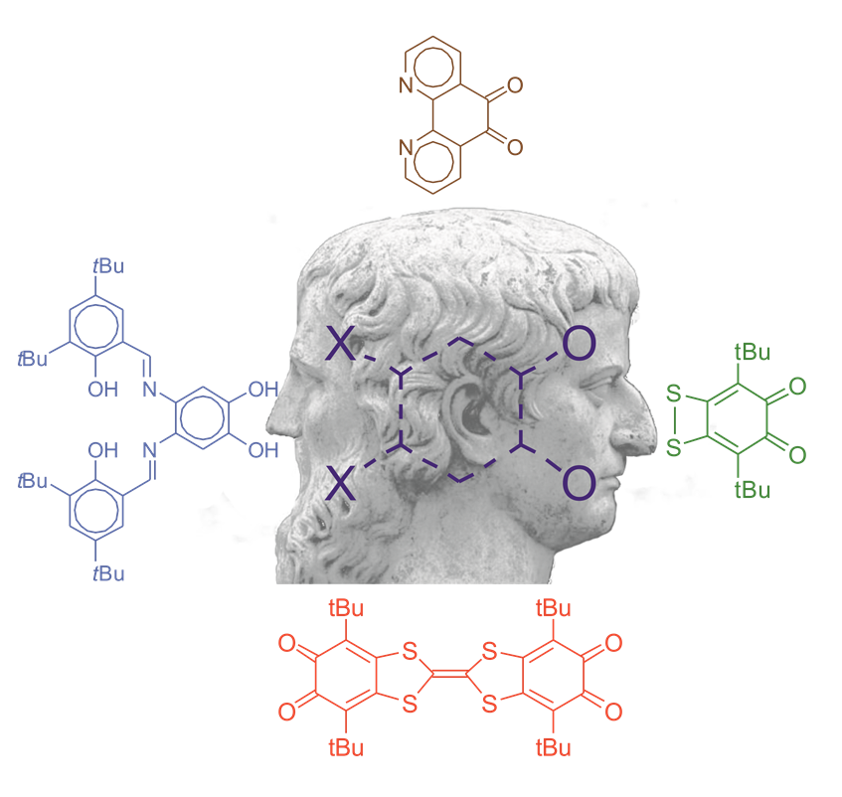
1. Introduction
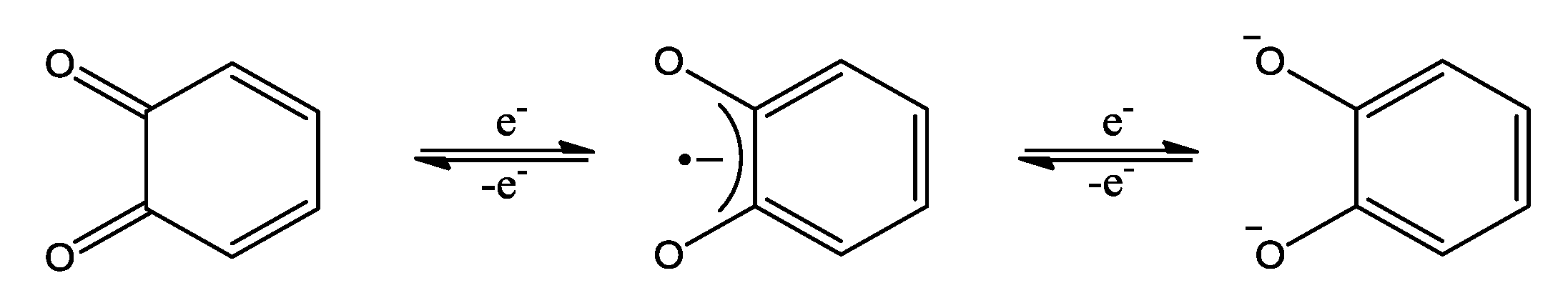
2. OO~NN Systems
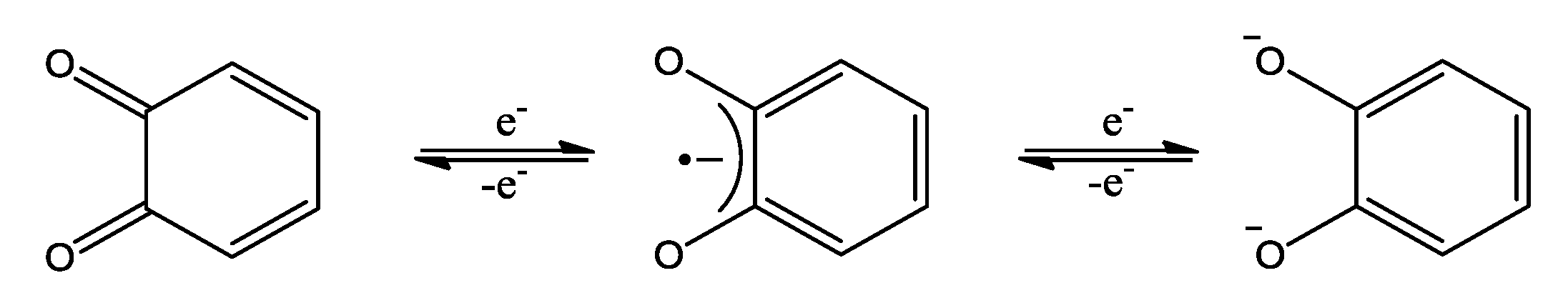
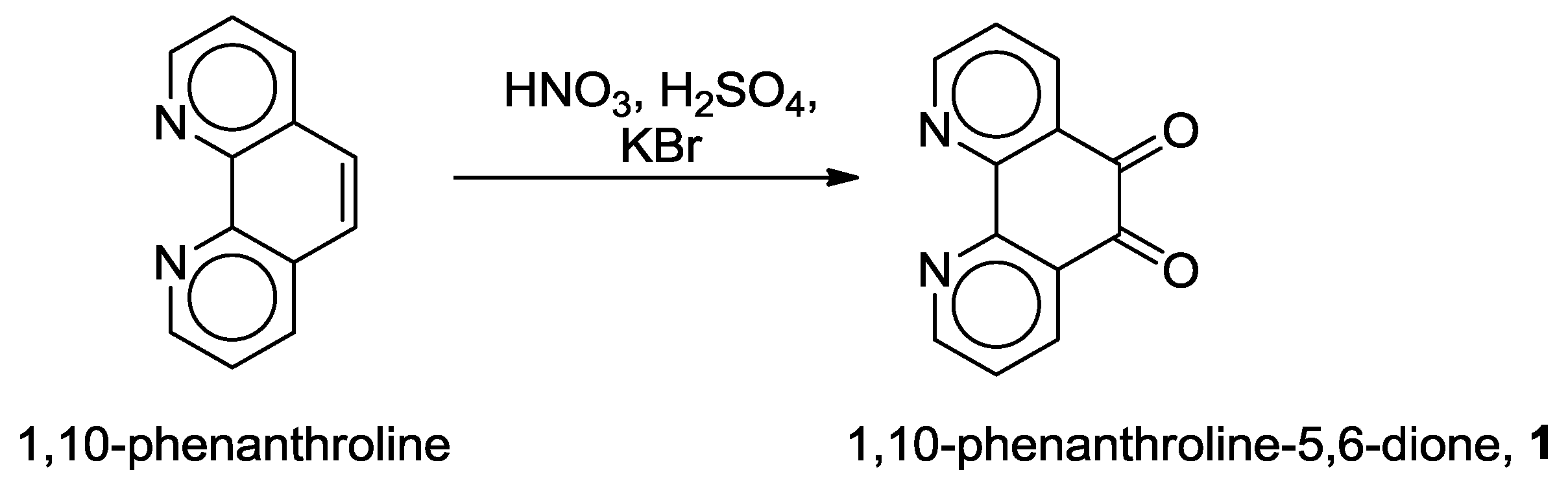
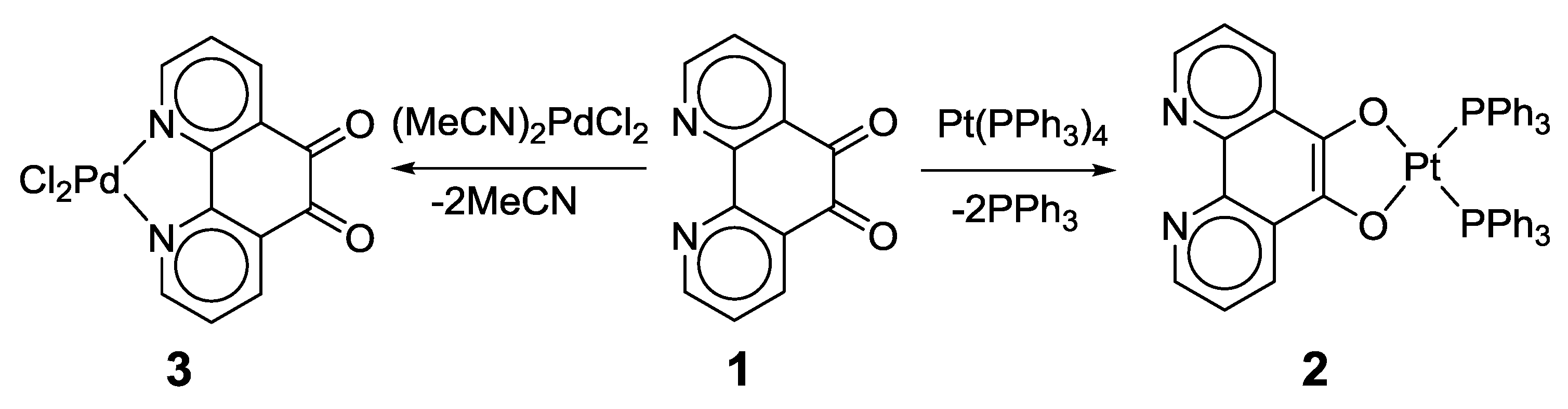
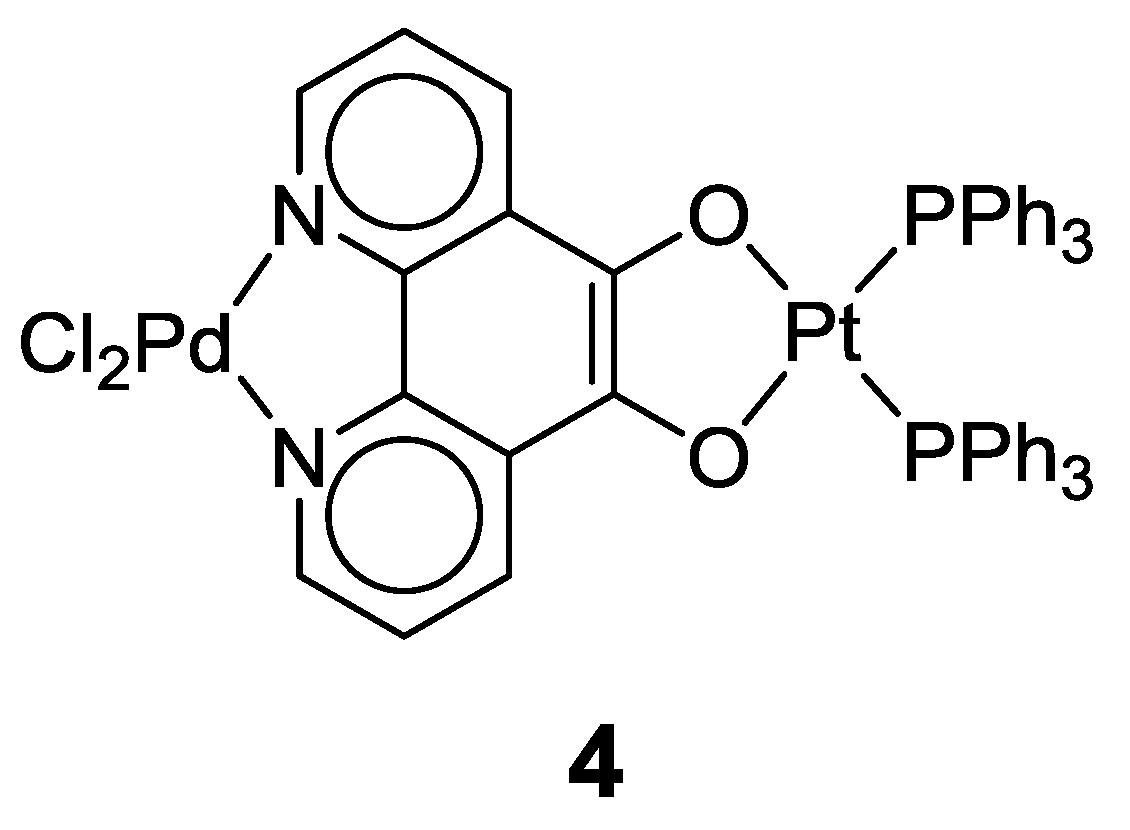
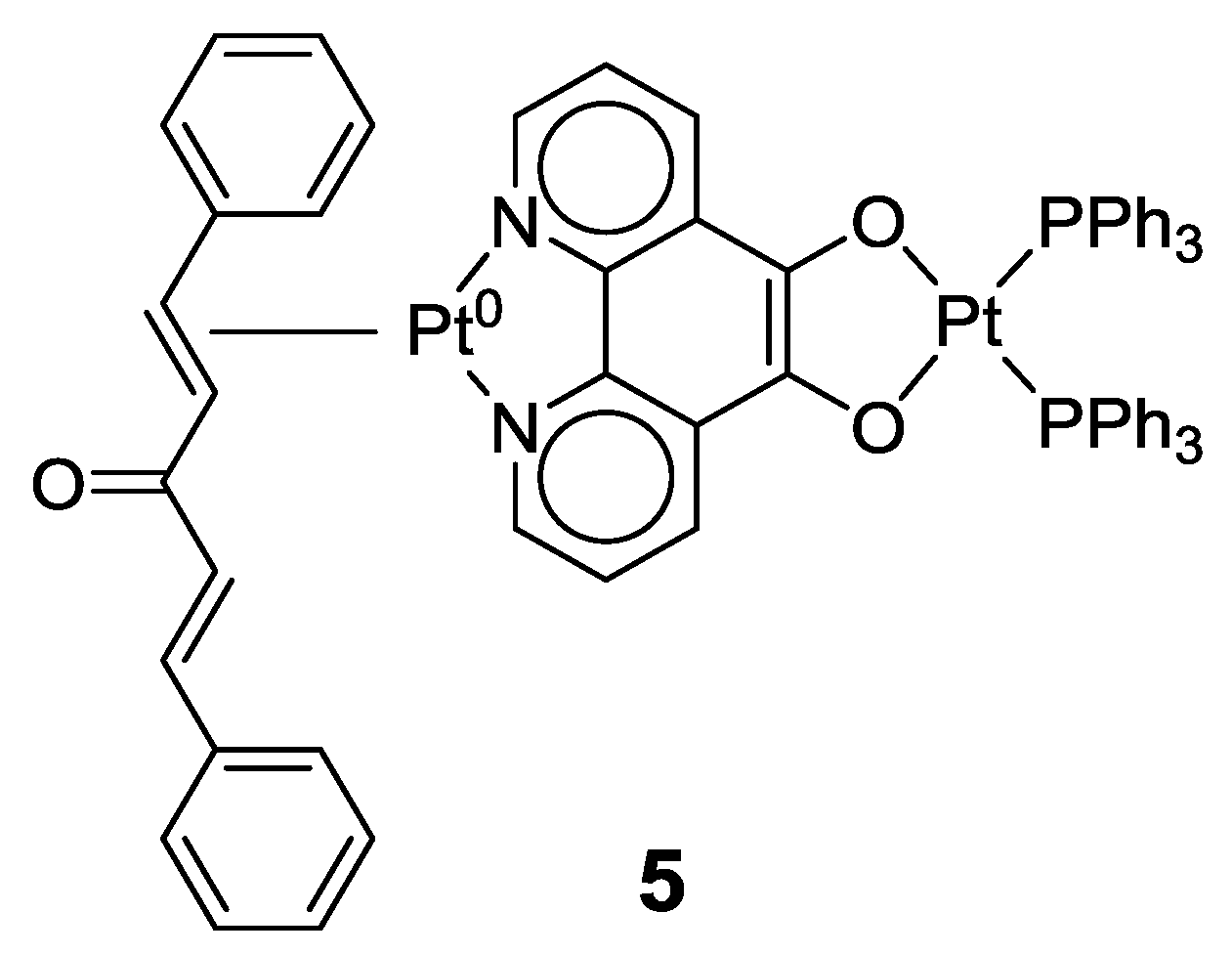
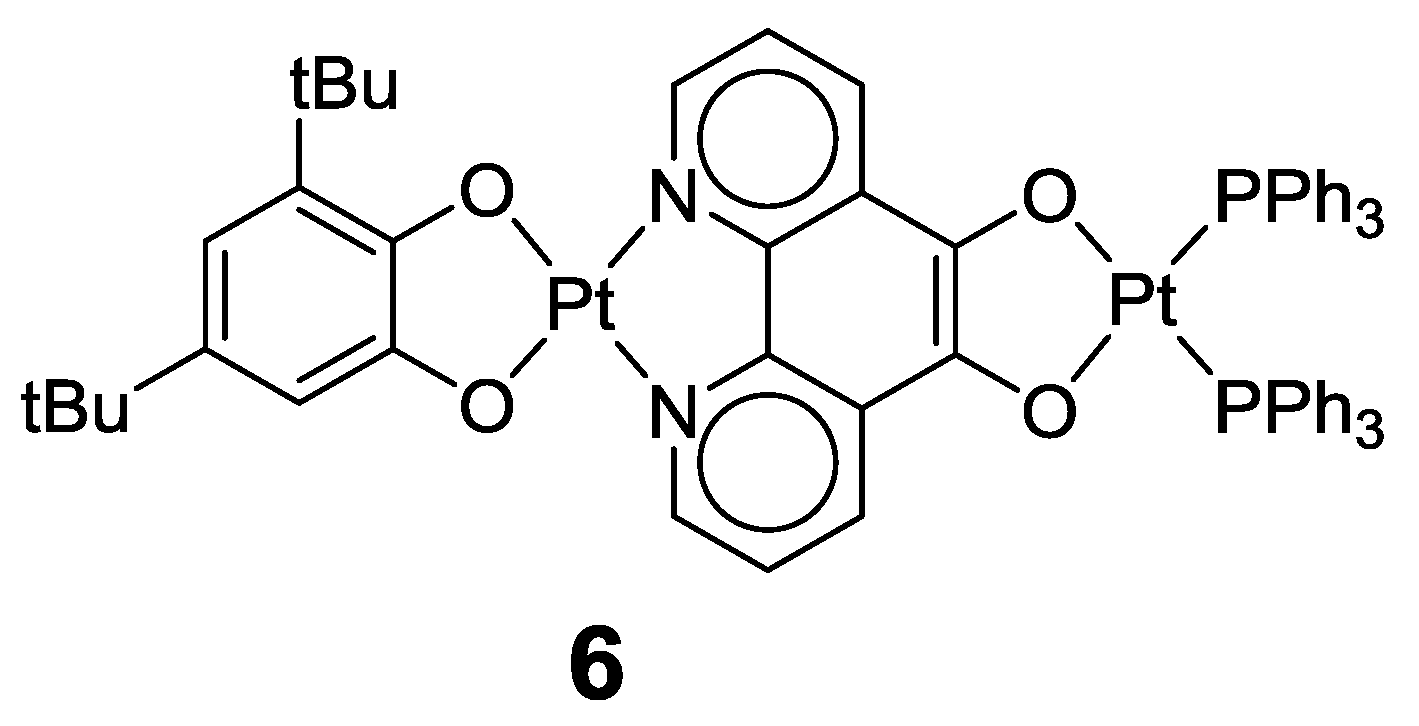
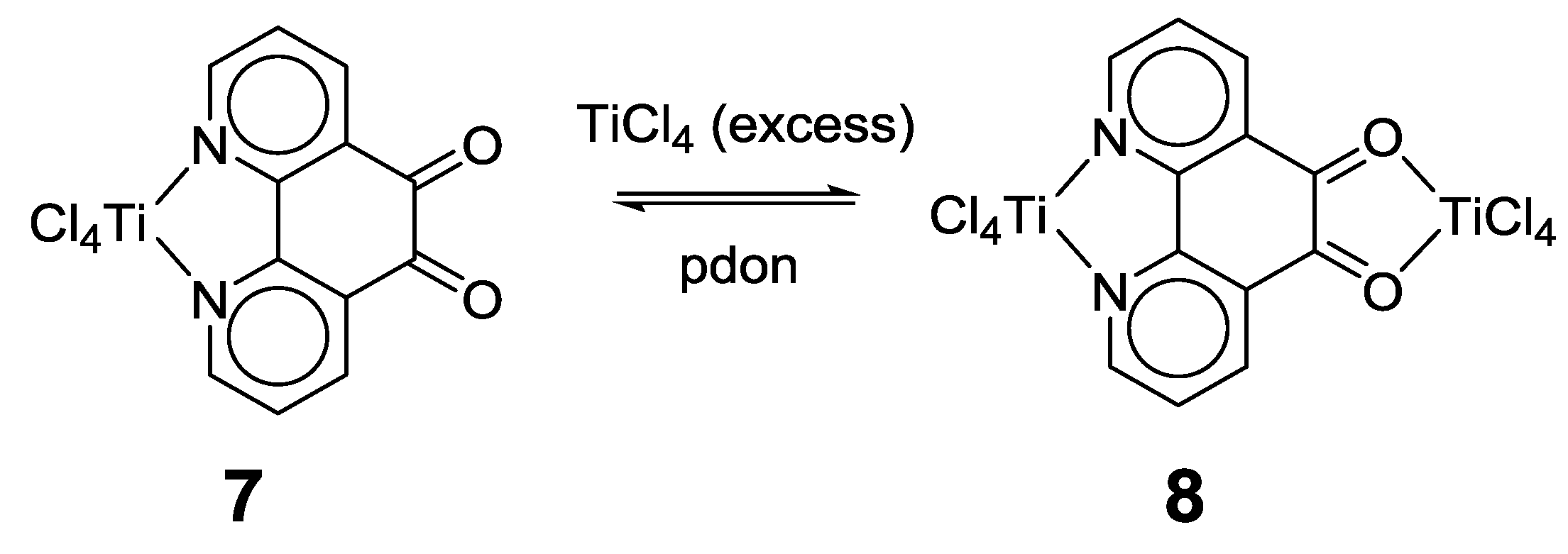
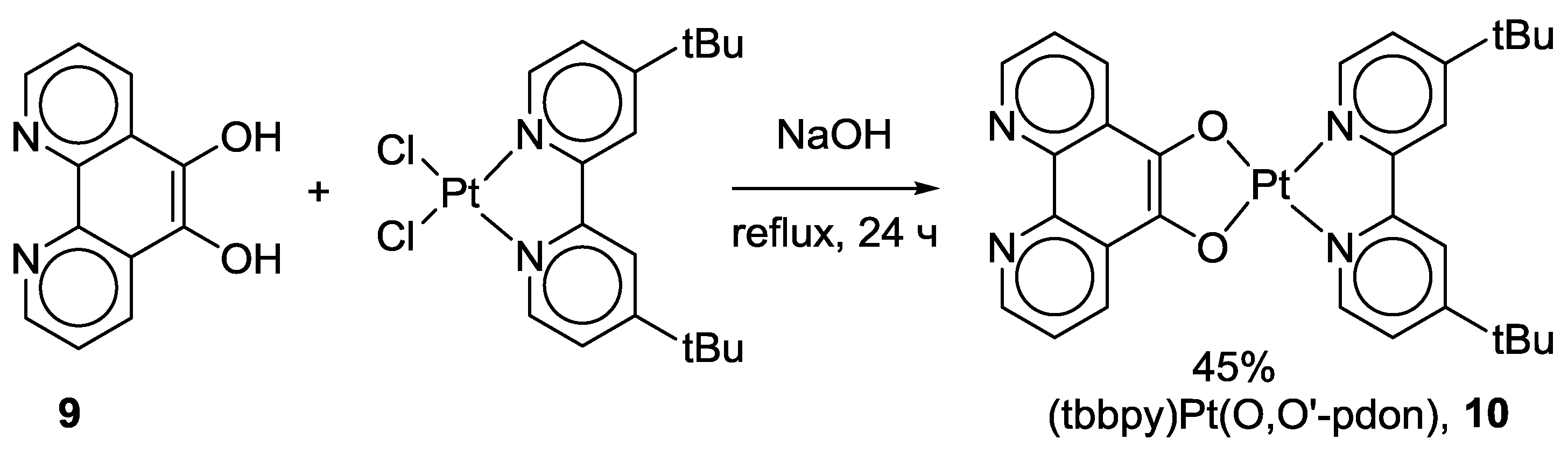
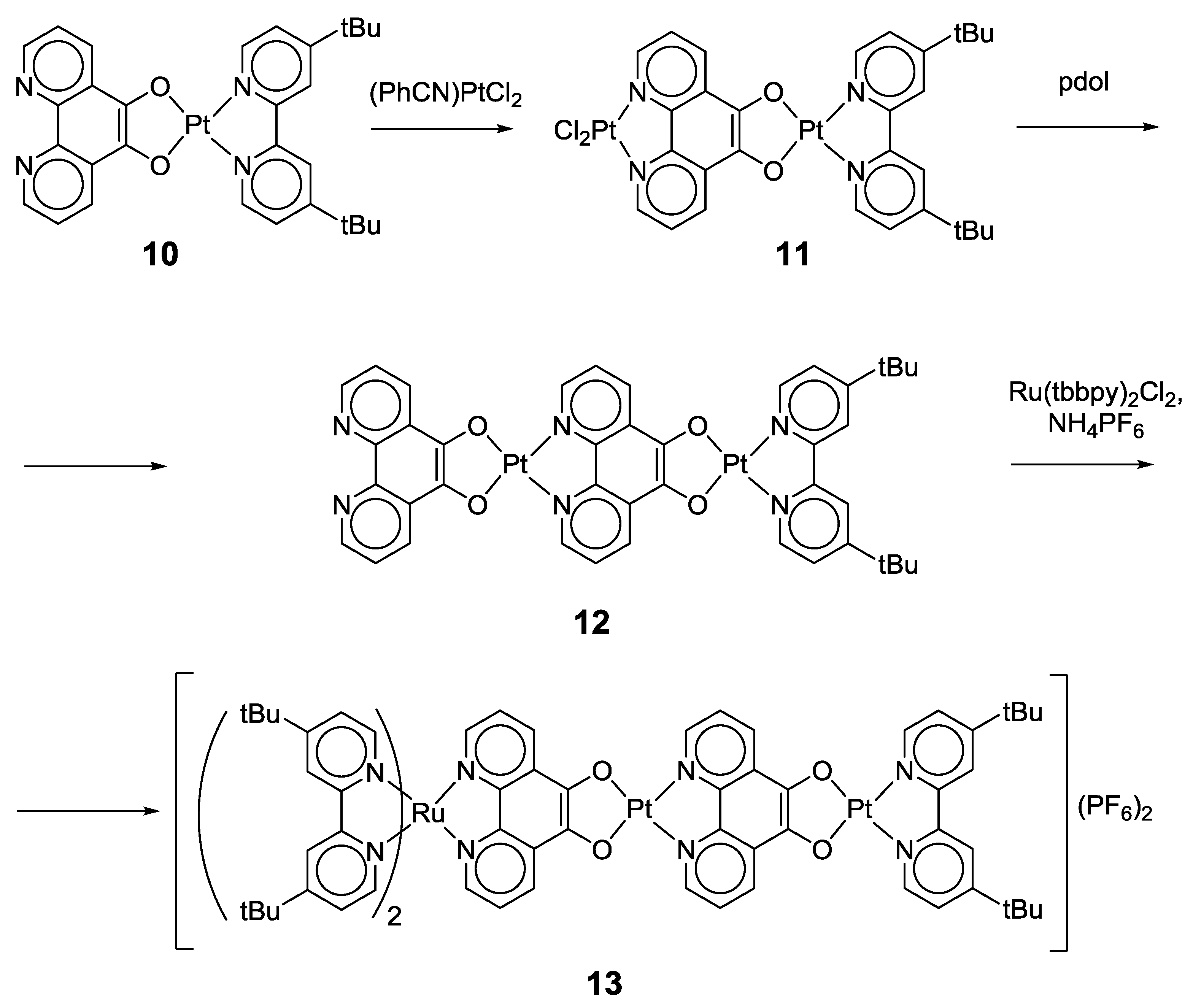
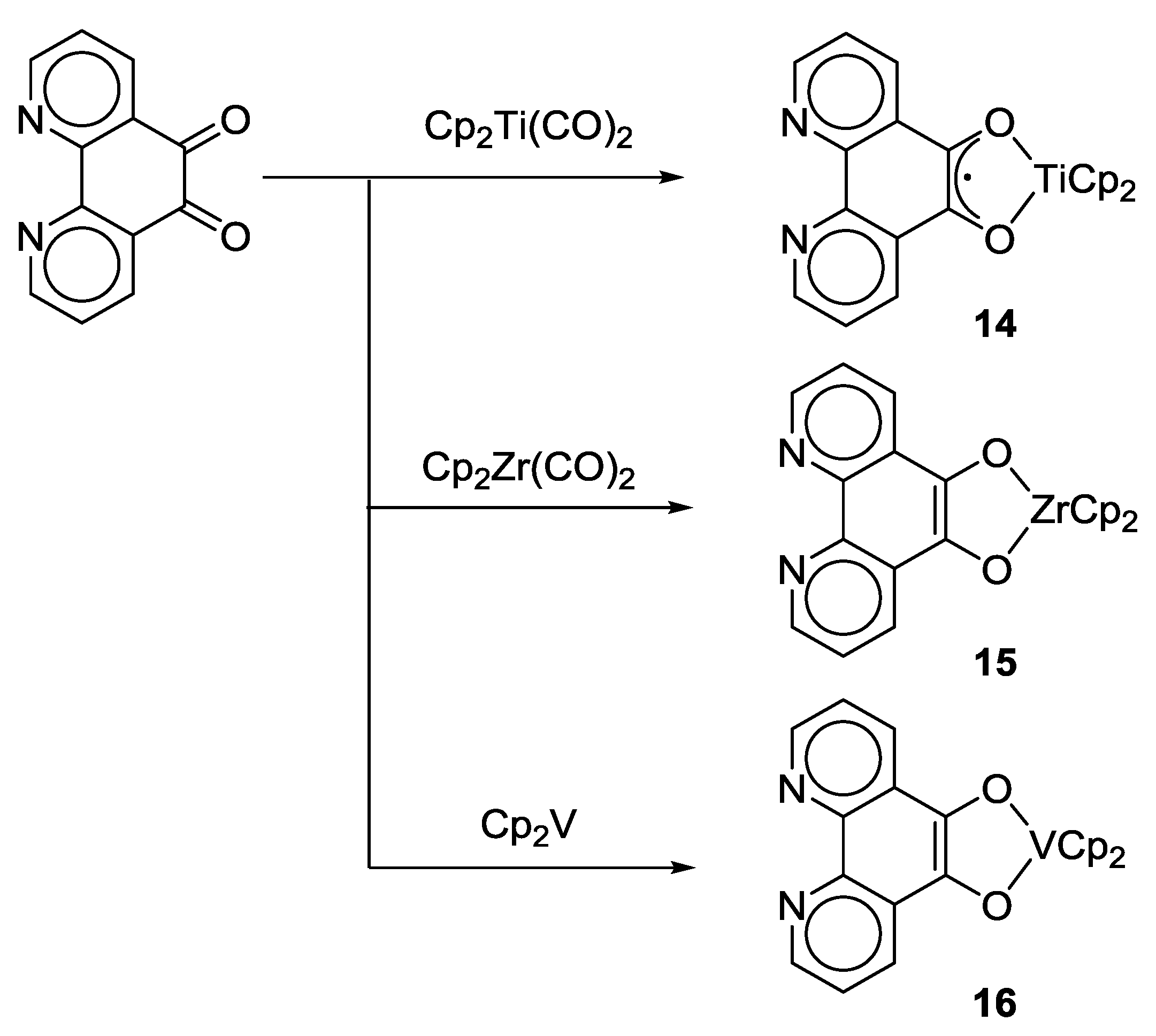
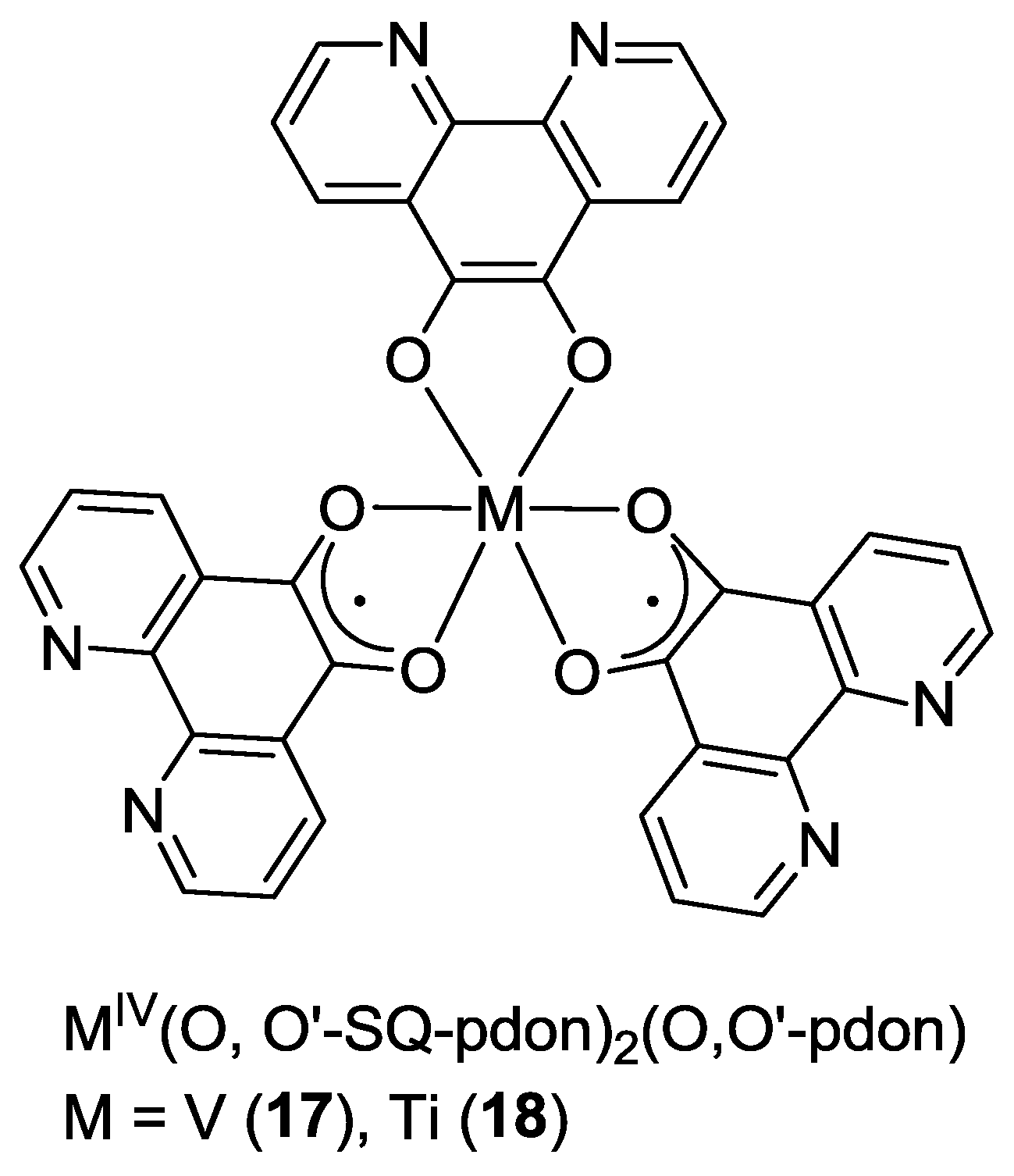
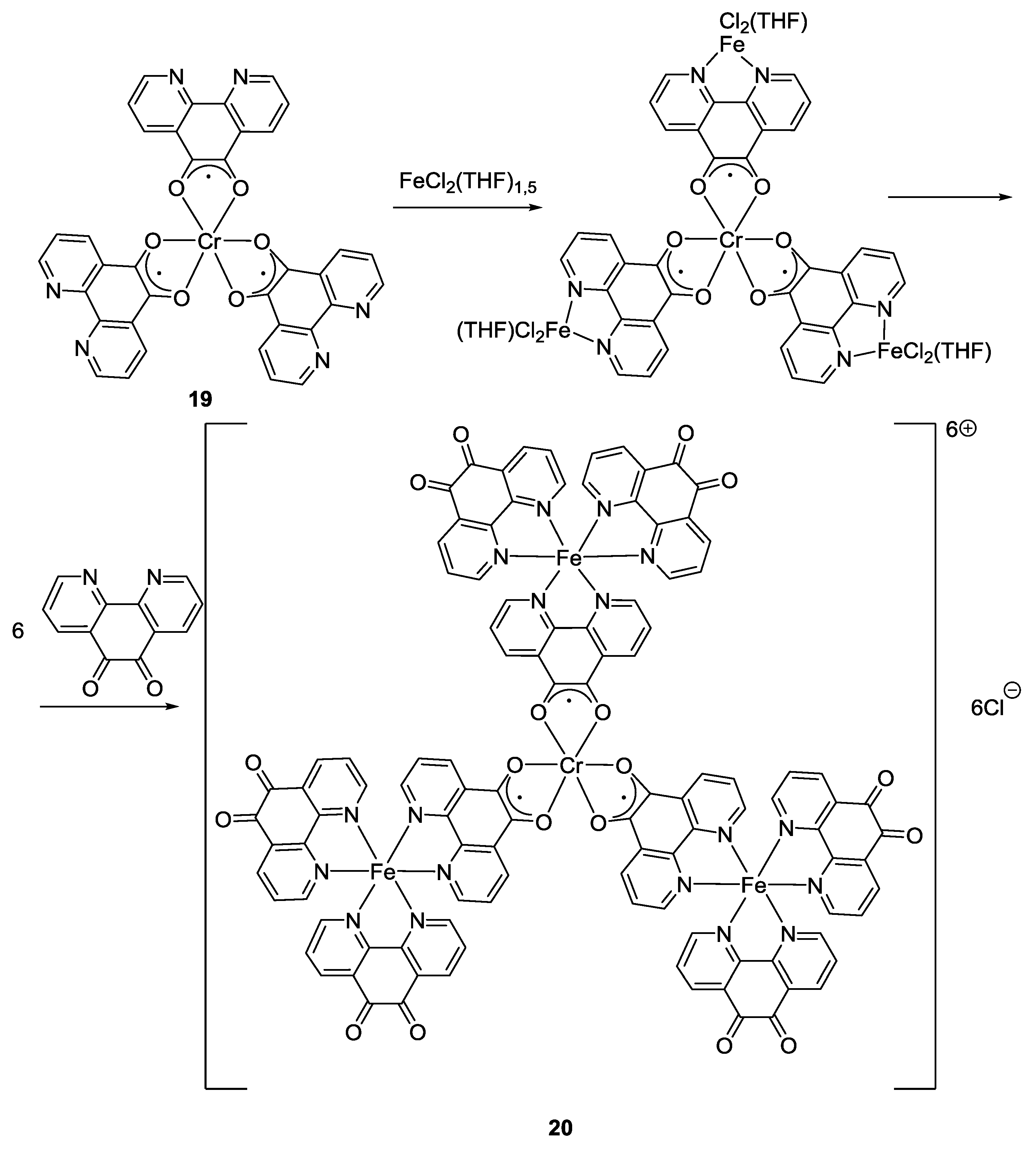
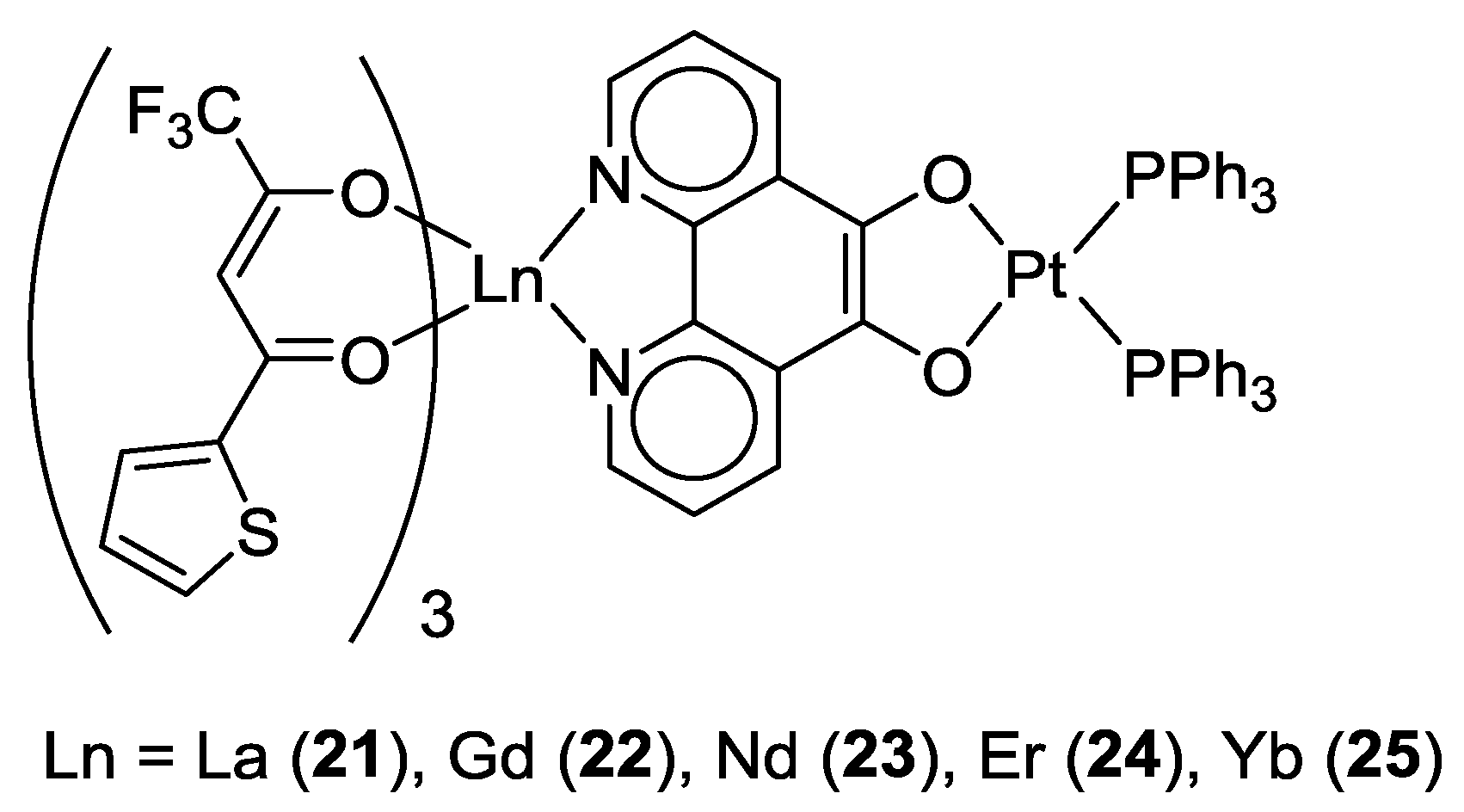
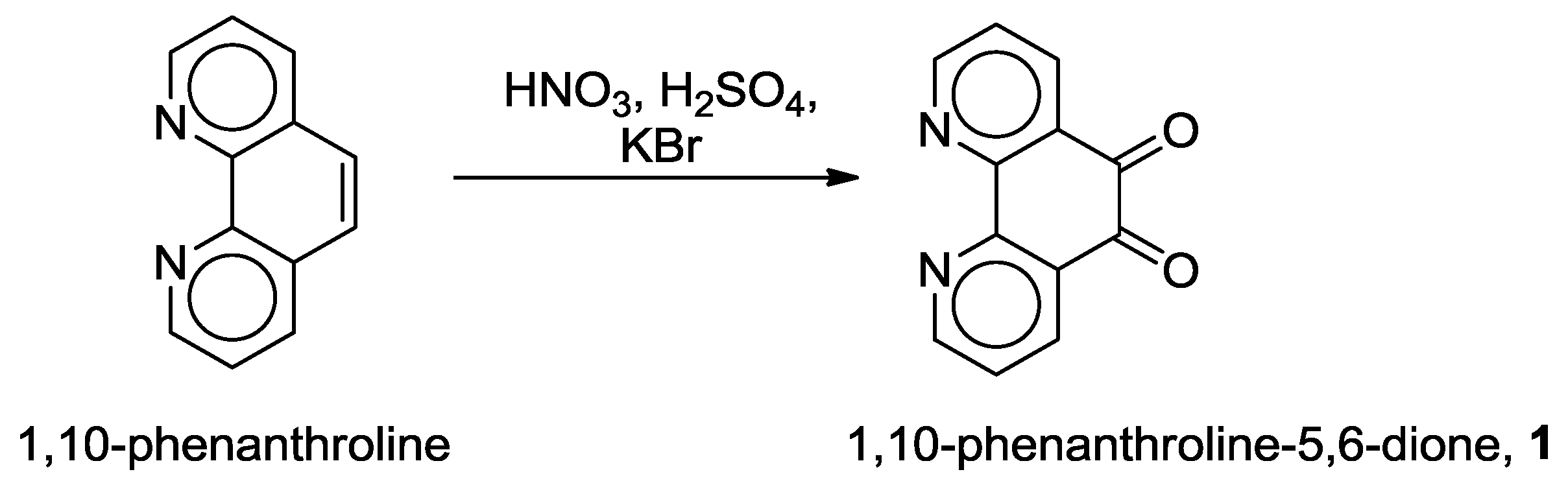
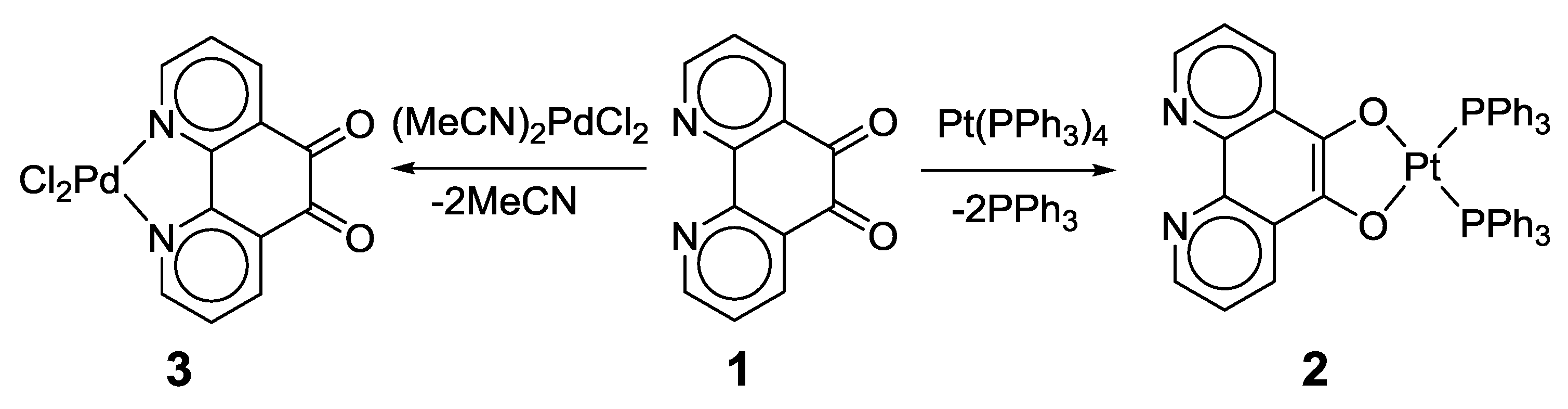
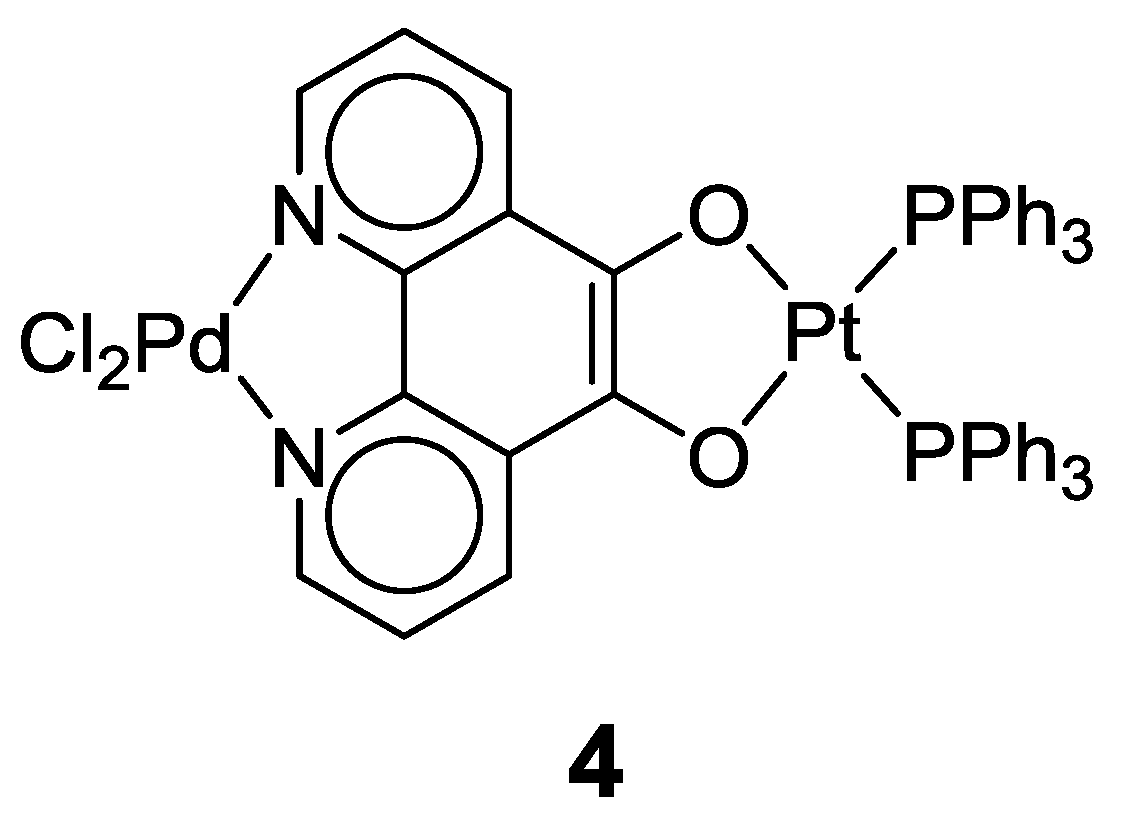
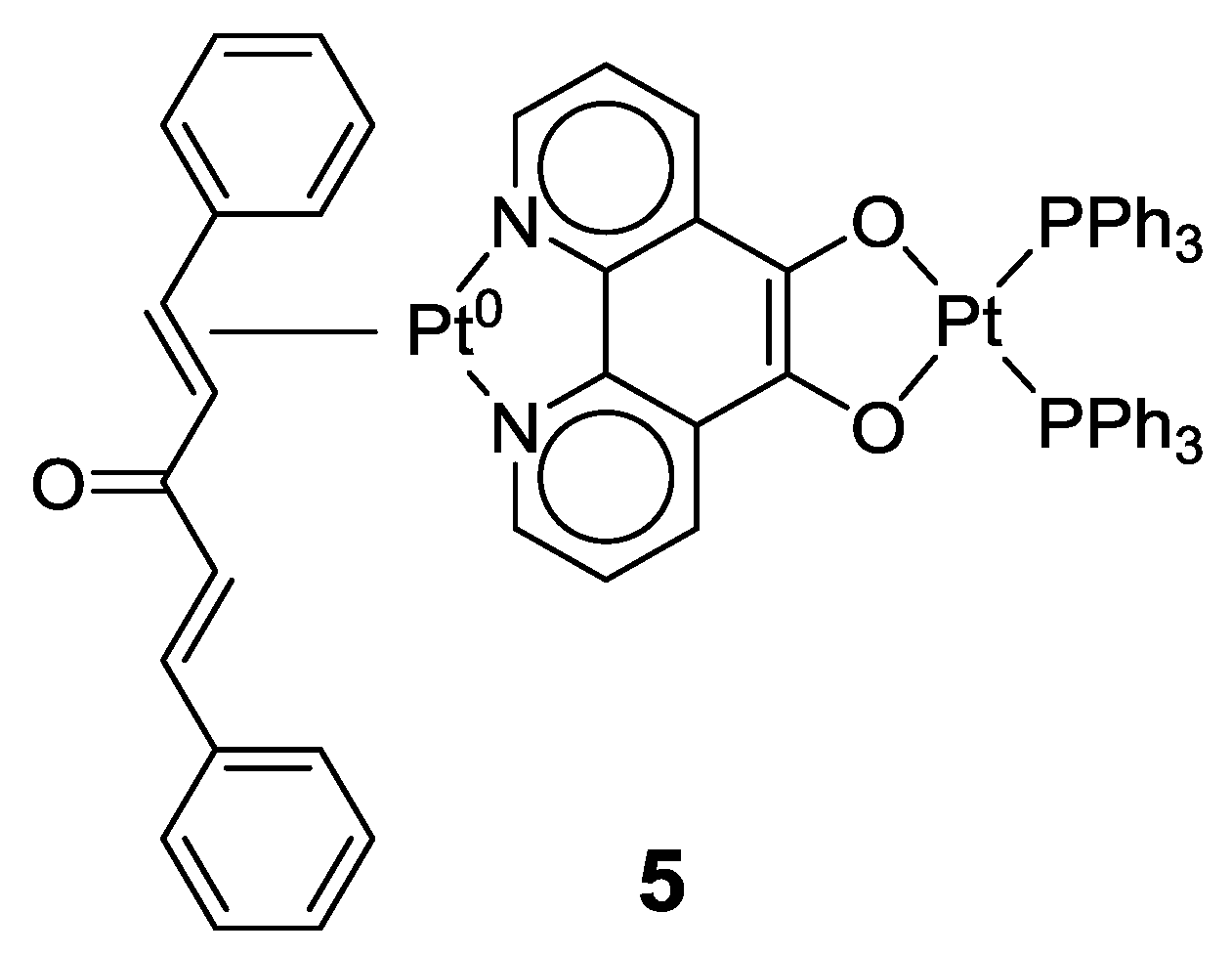
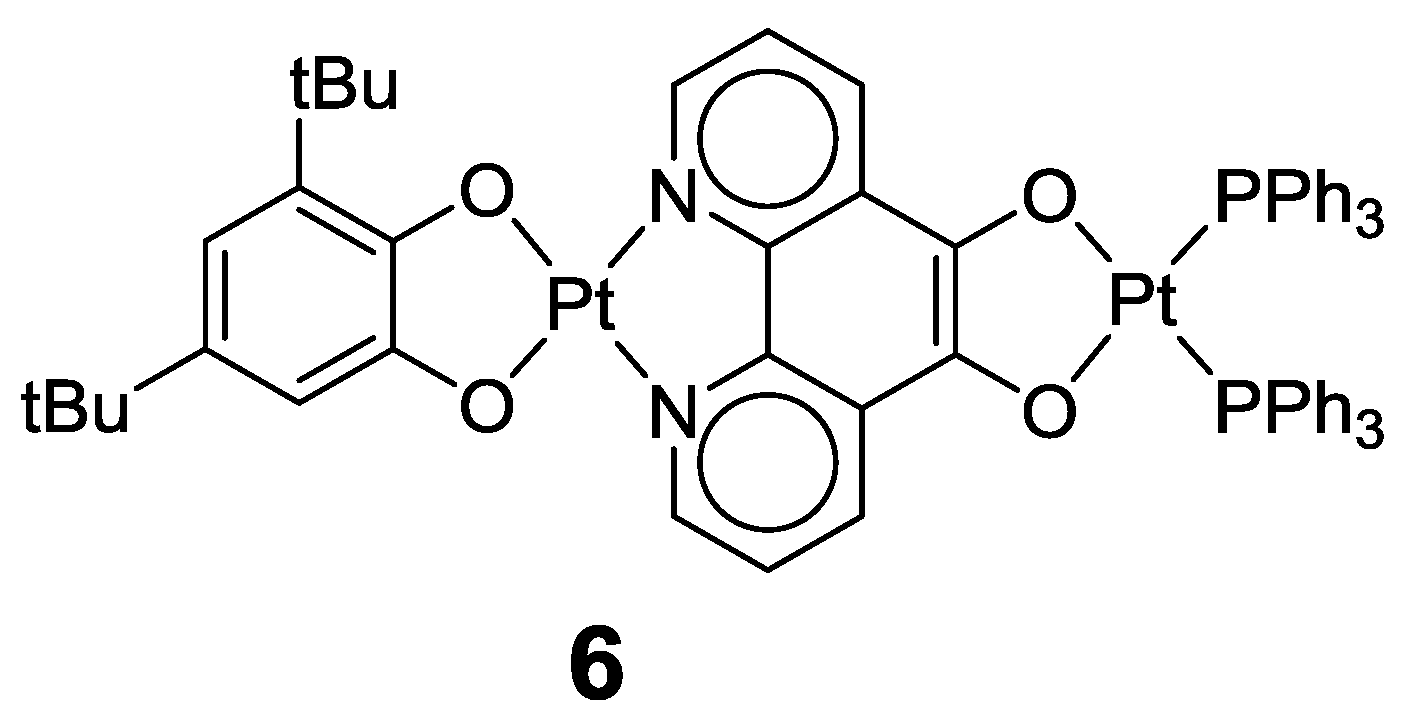
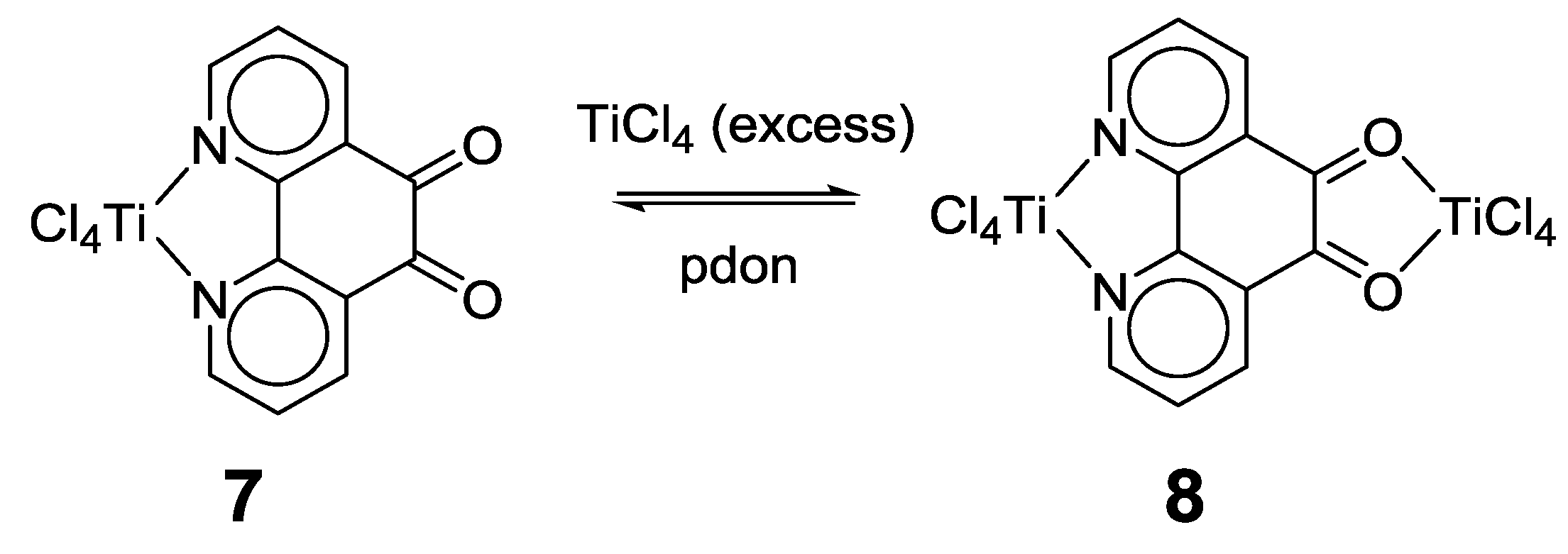
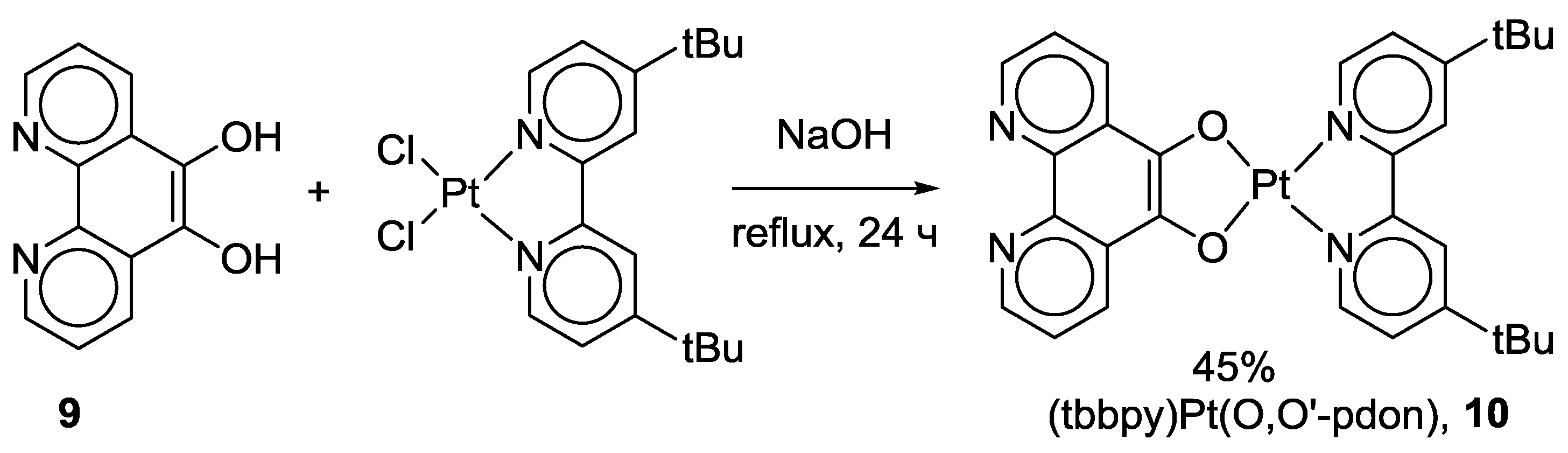
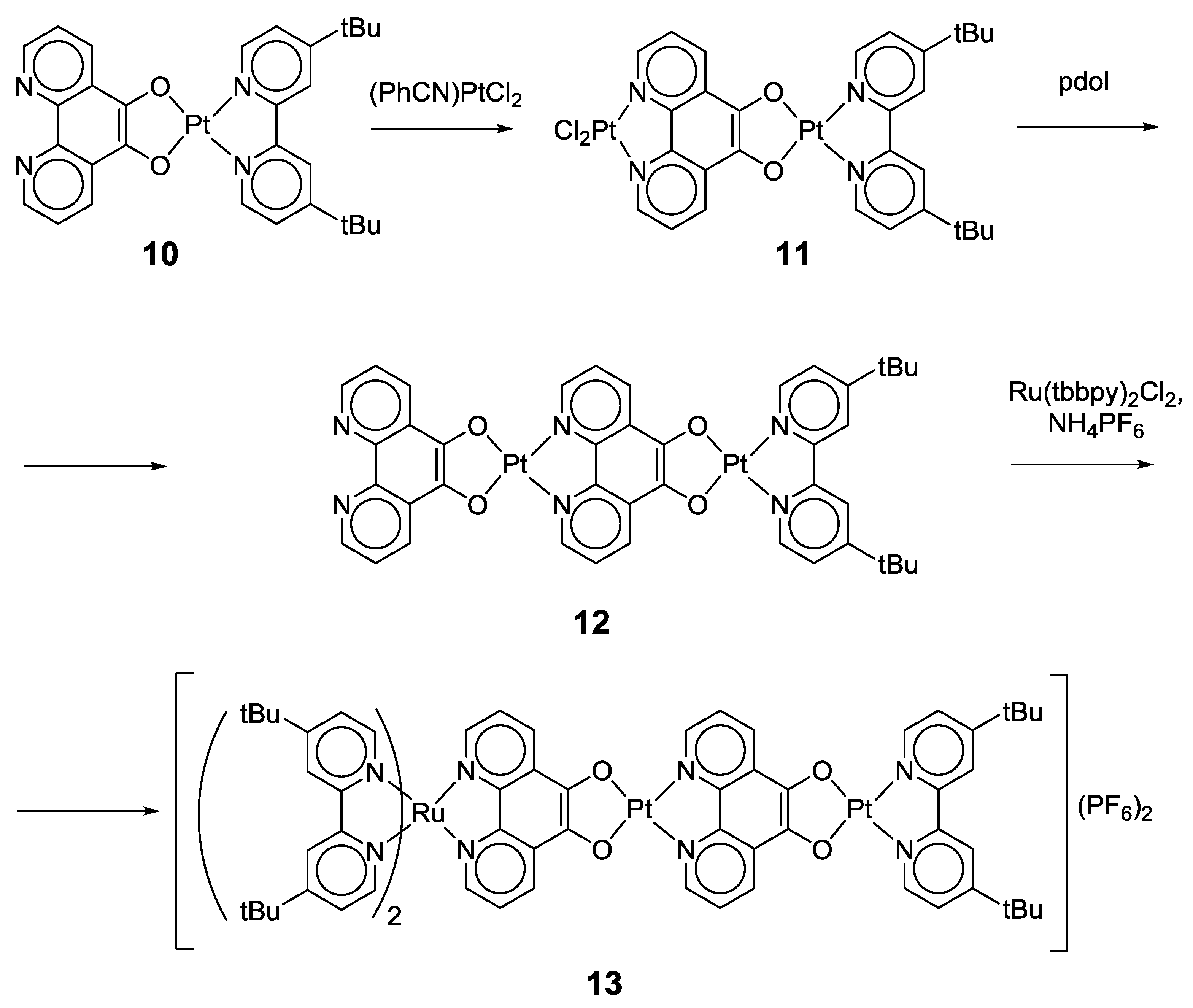
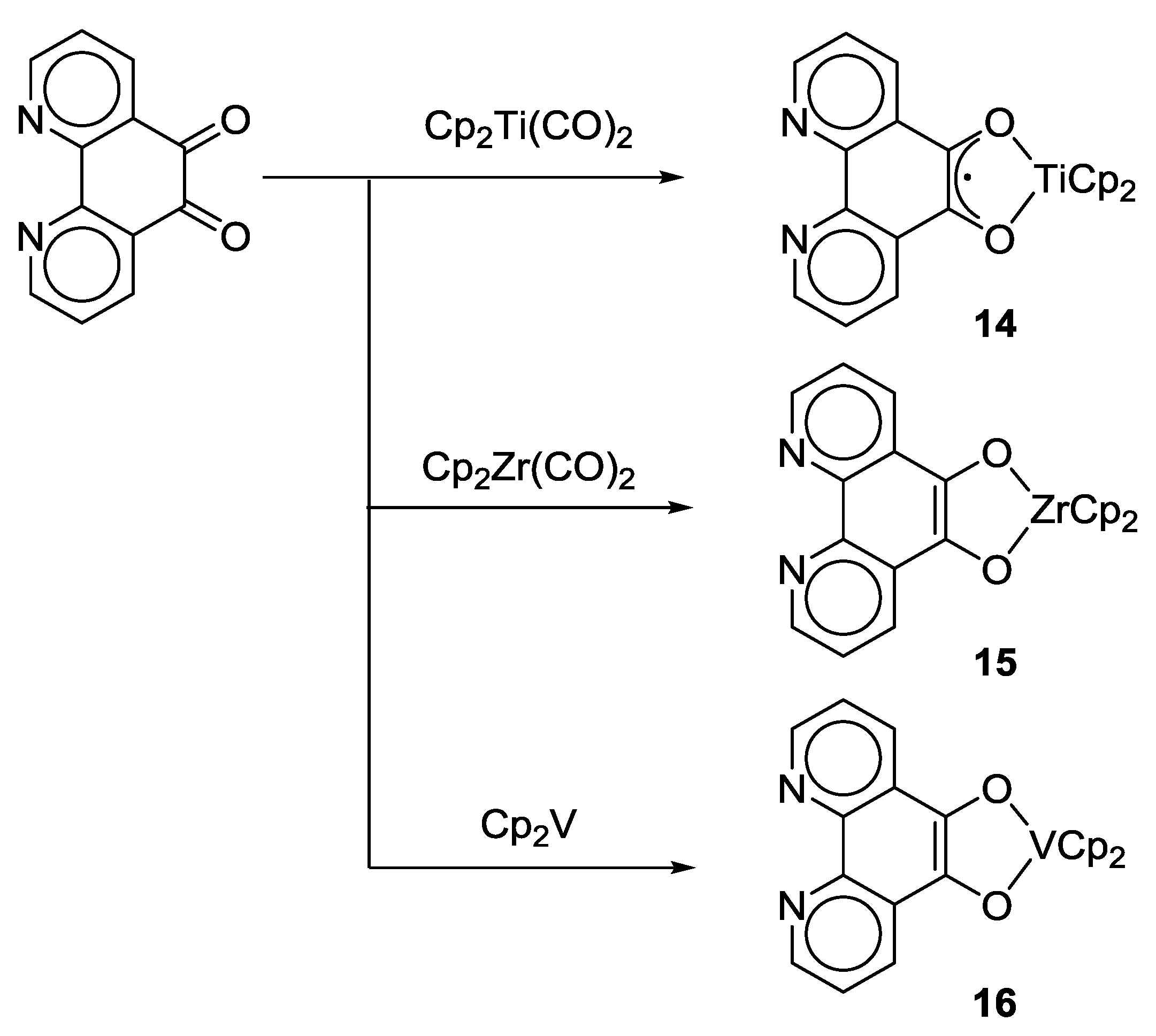
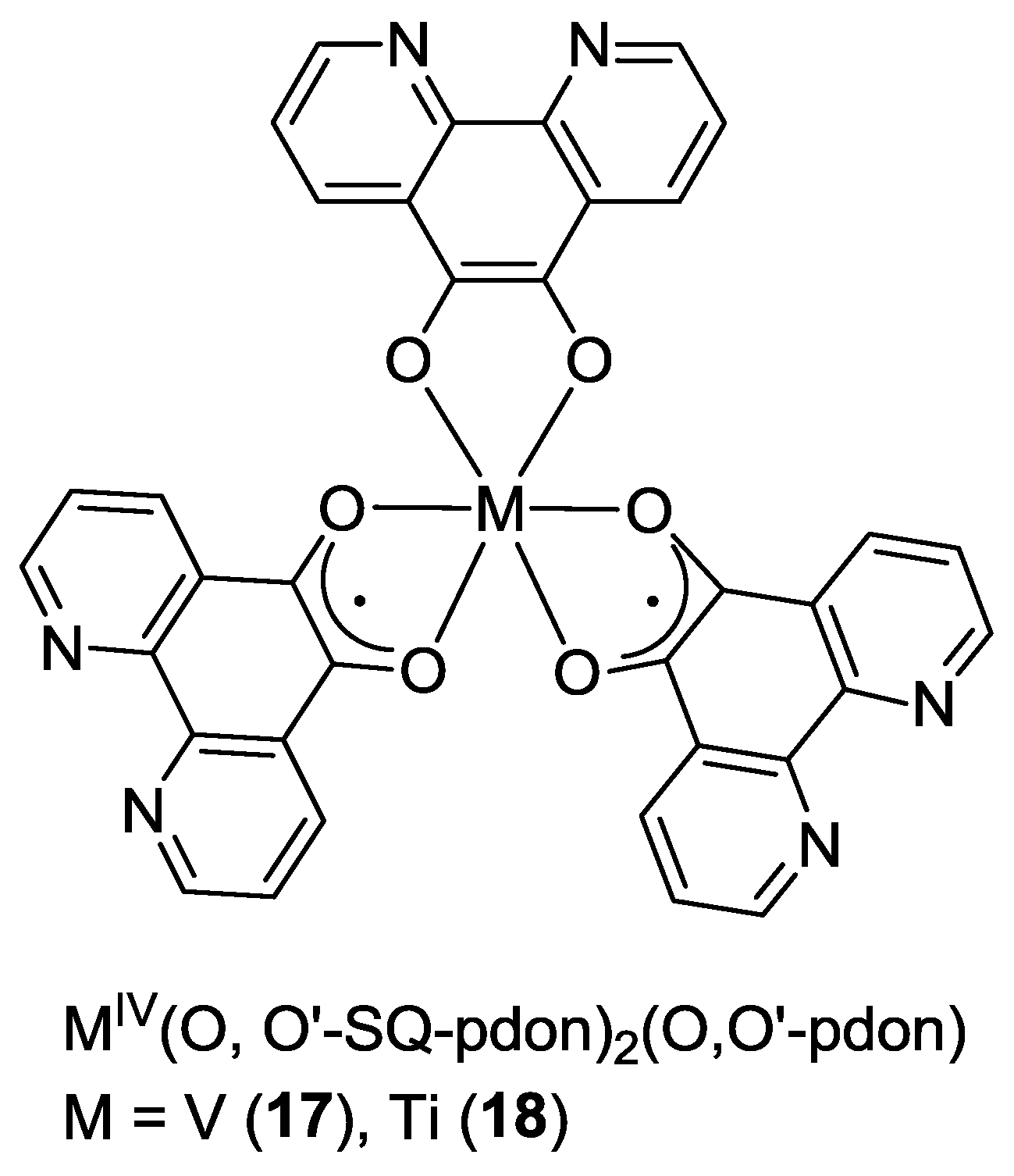
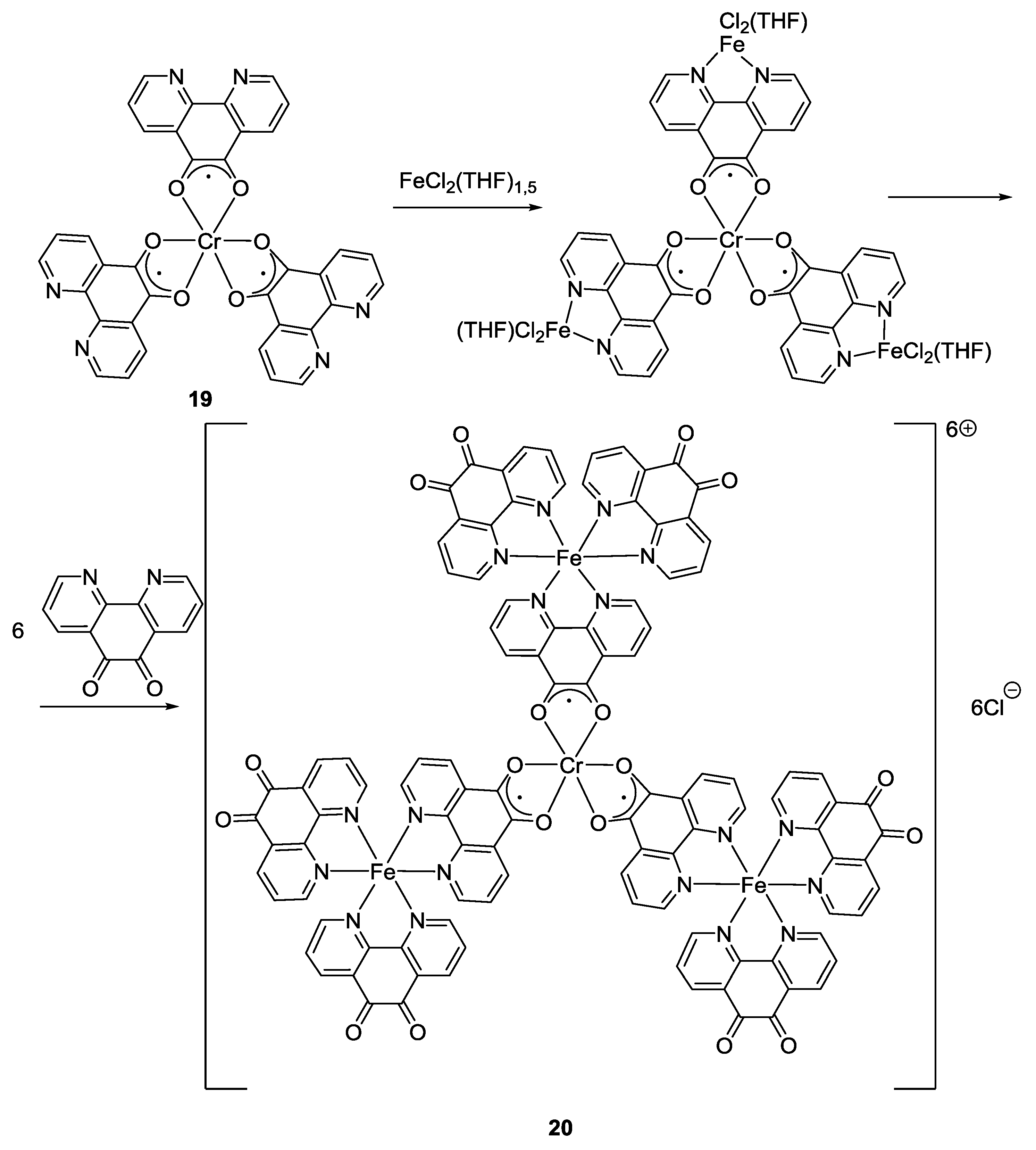
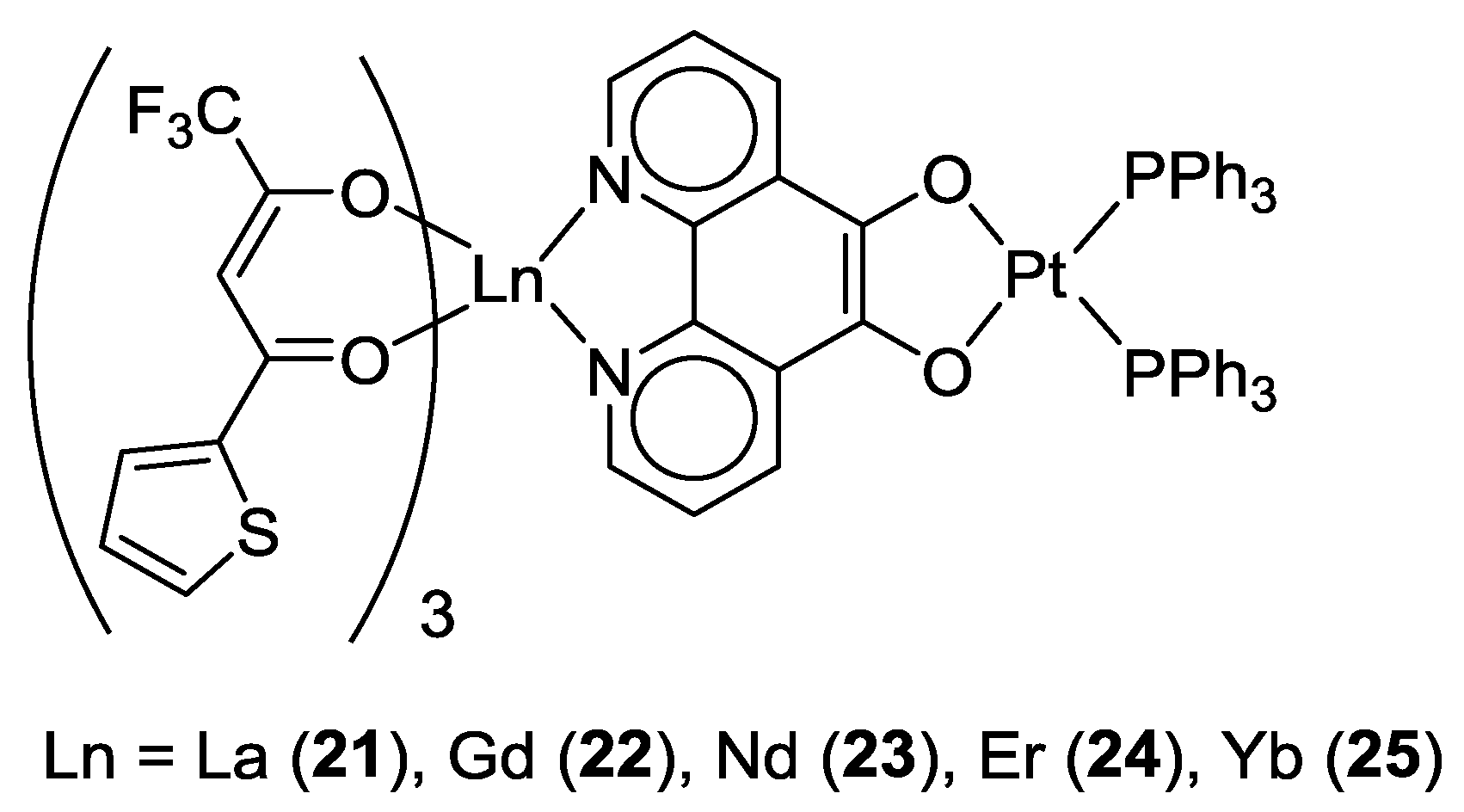
2.1. Coordination Properties of 1,10-Phenanthroline-5,6-dione
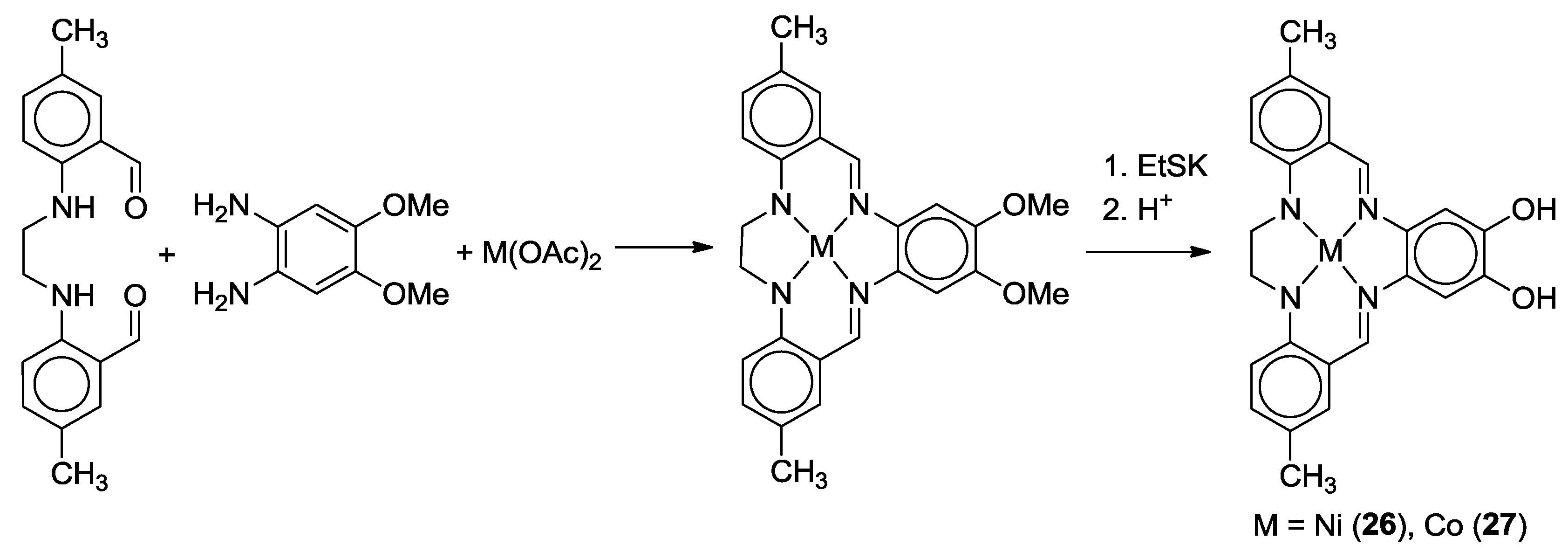
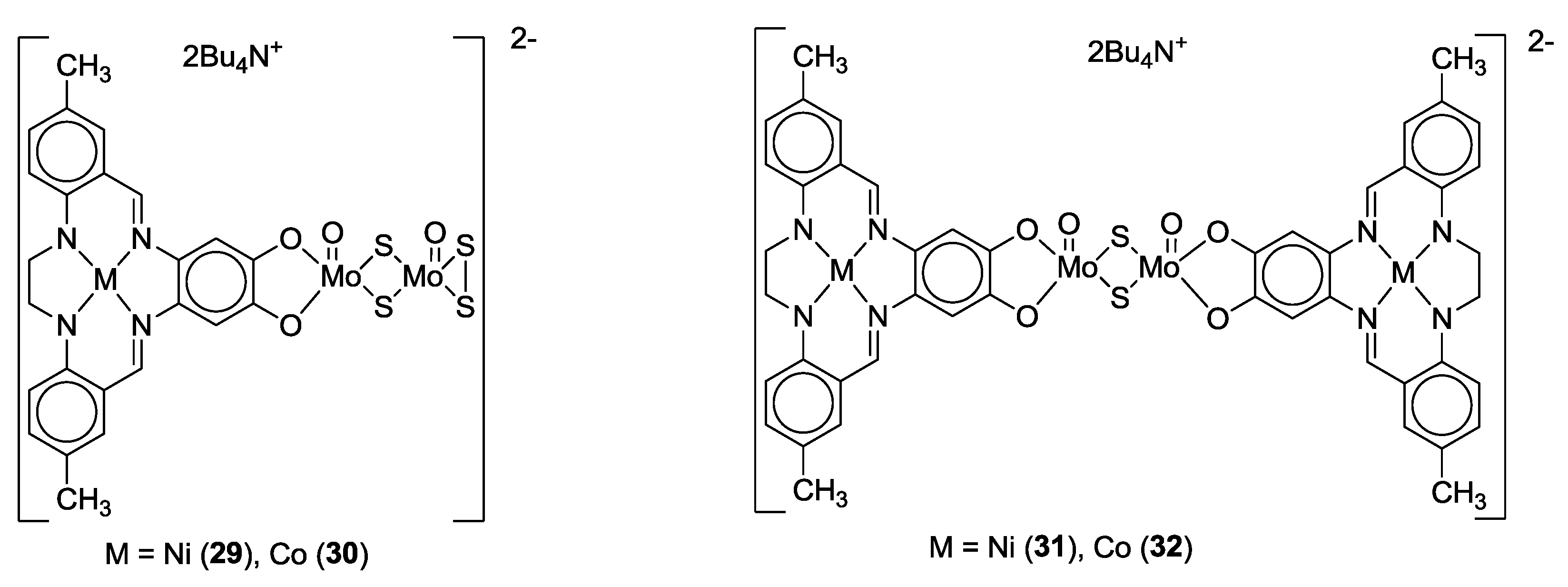
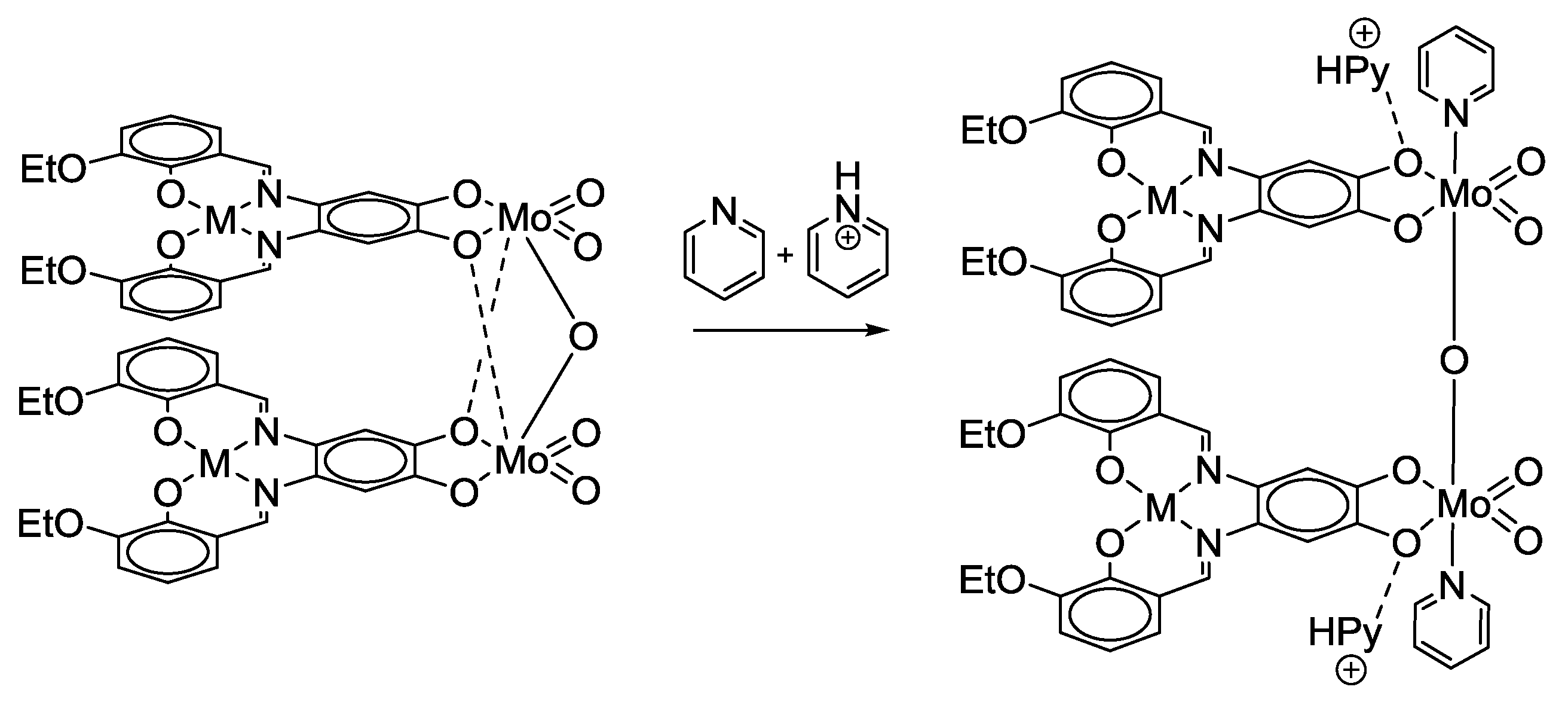
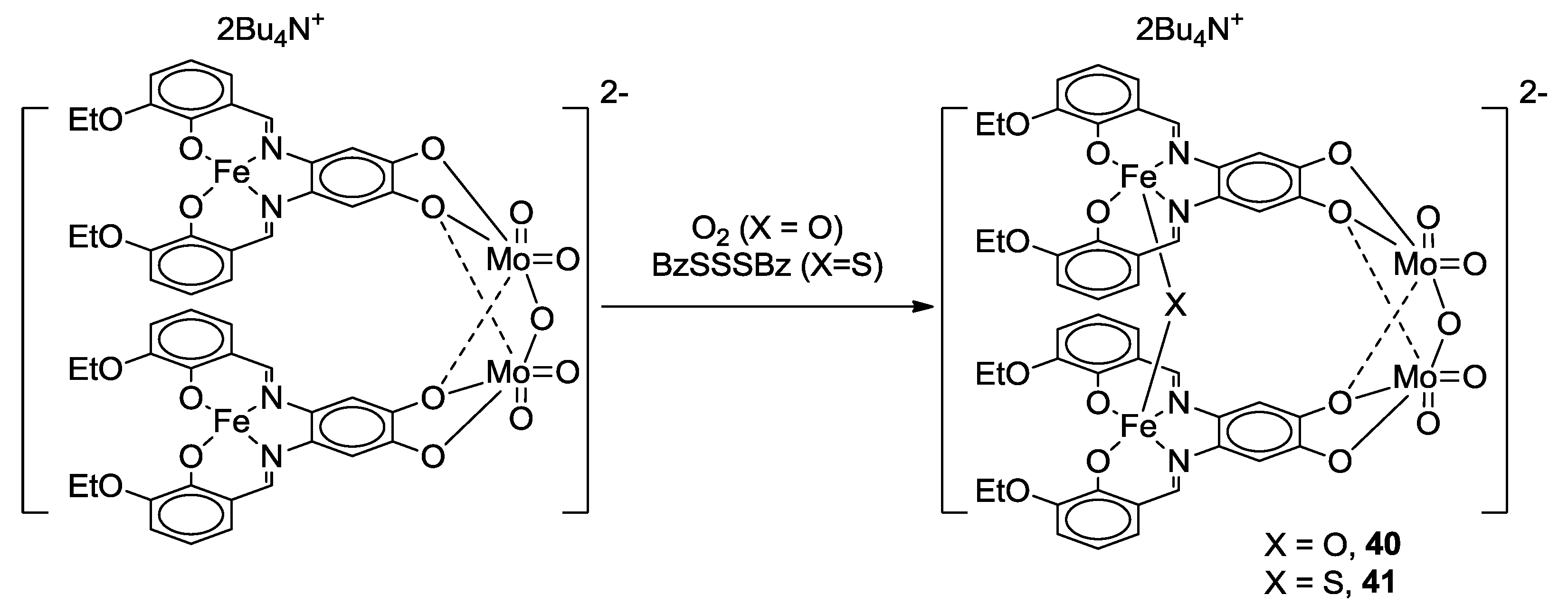
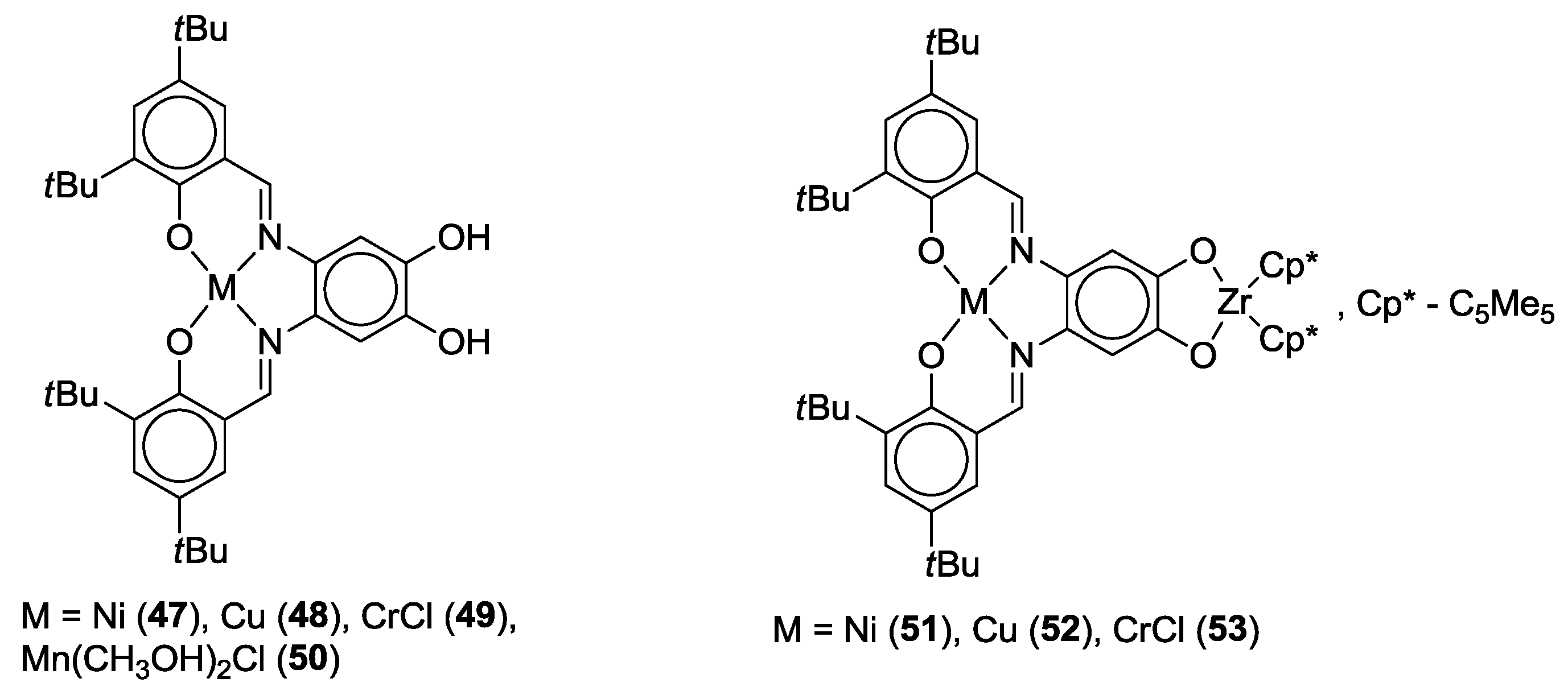
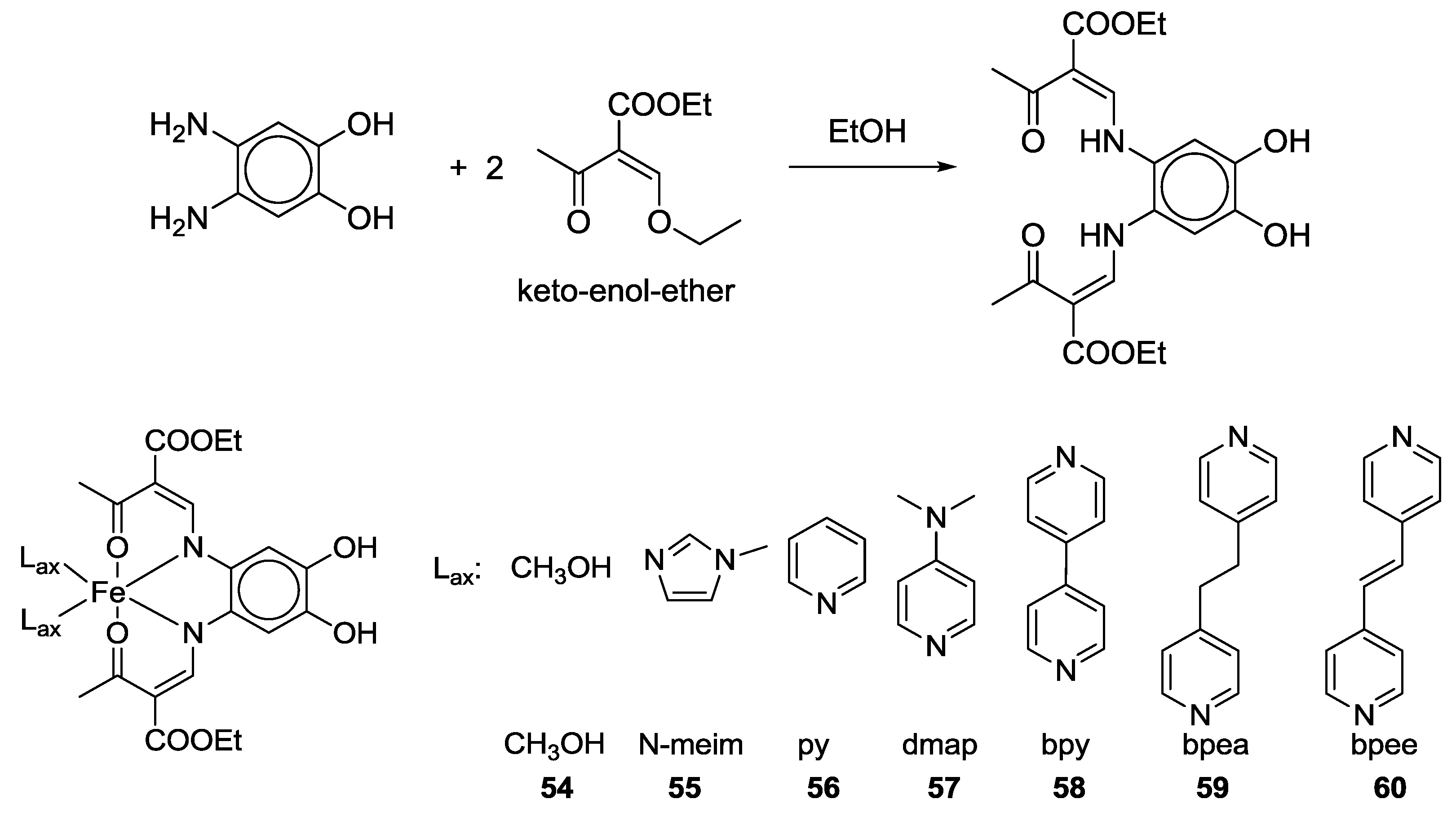
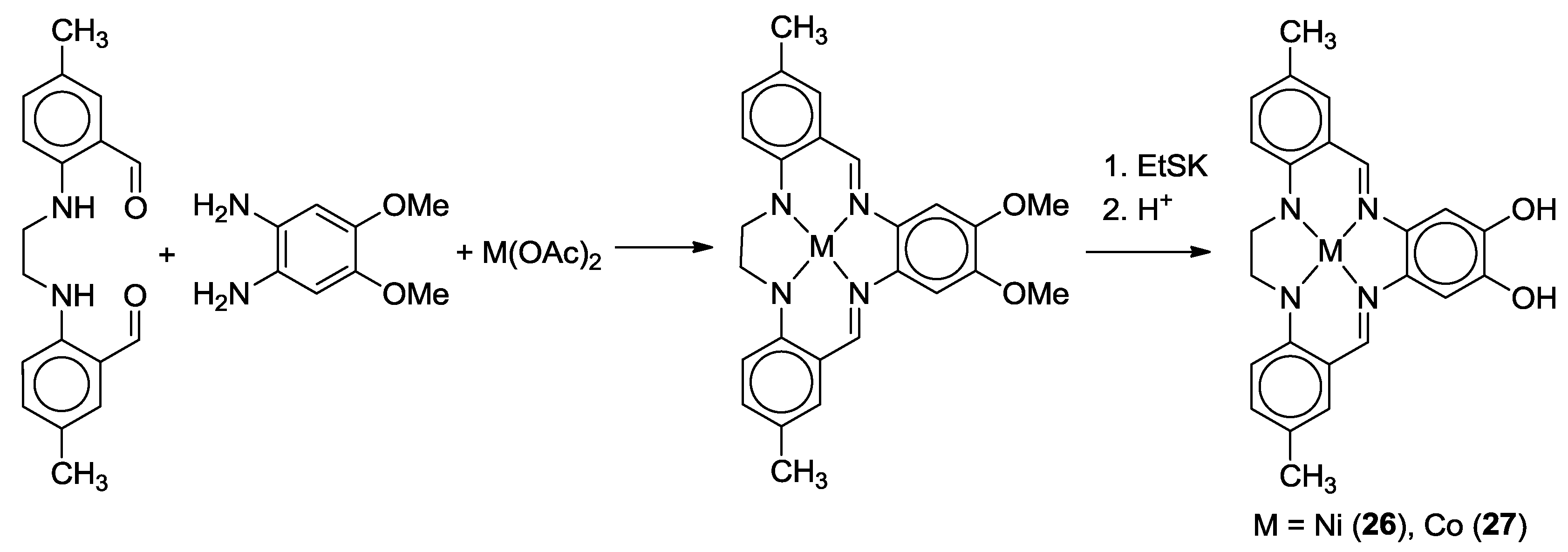
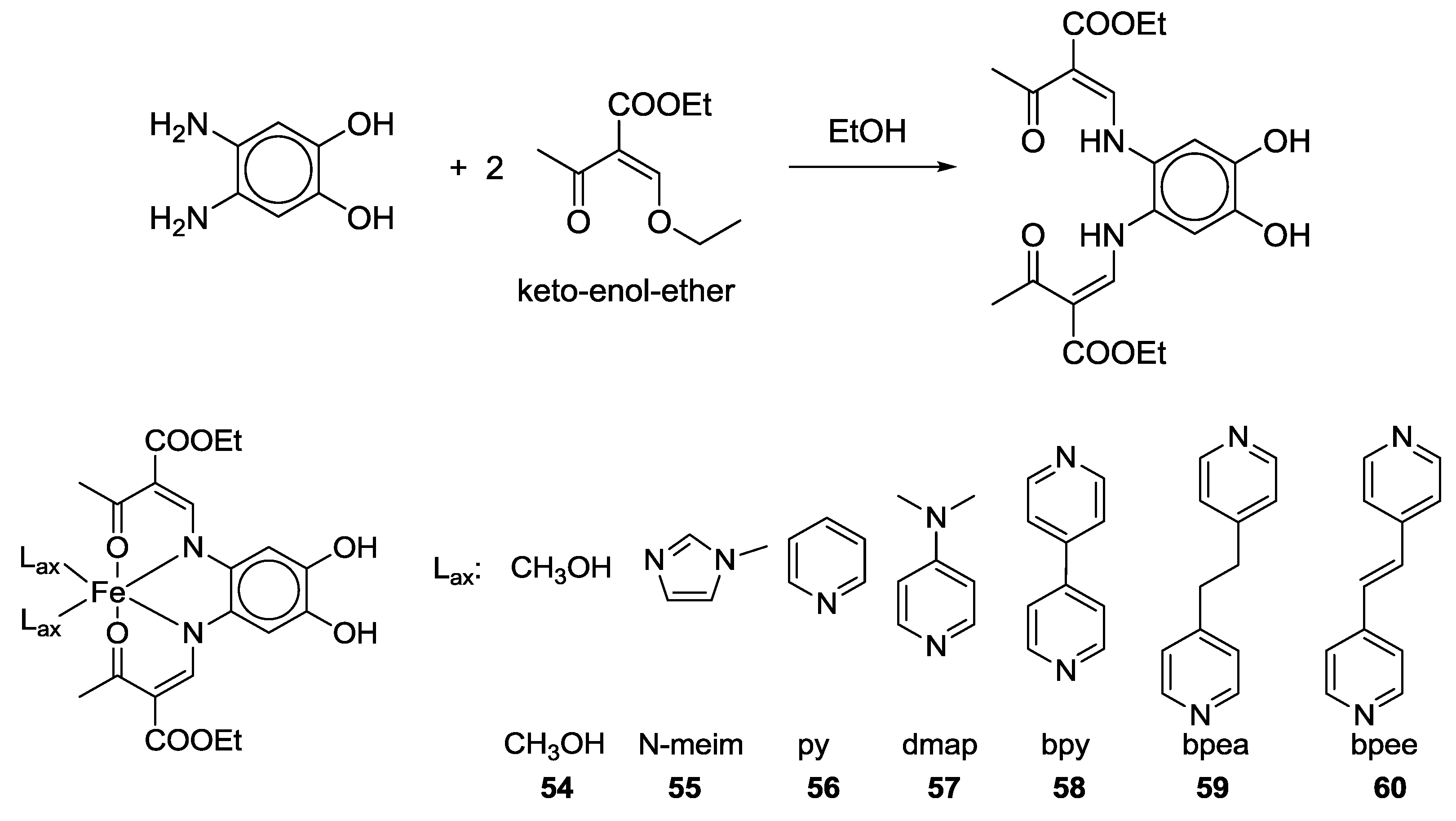
2.2. Bifunctional Catechols, Containing Schiff-Bases
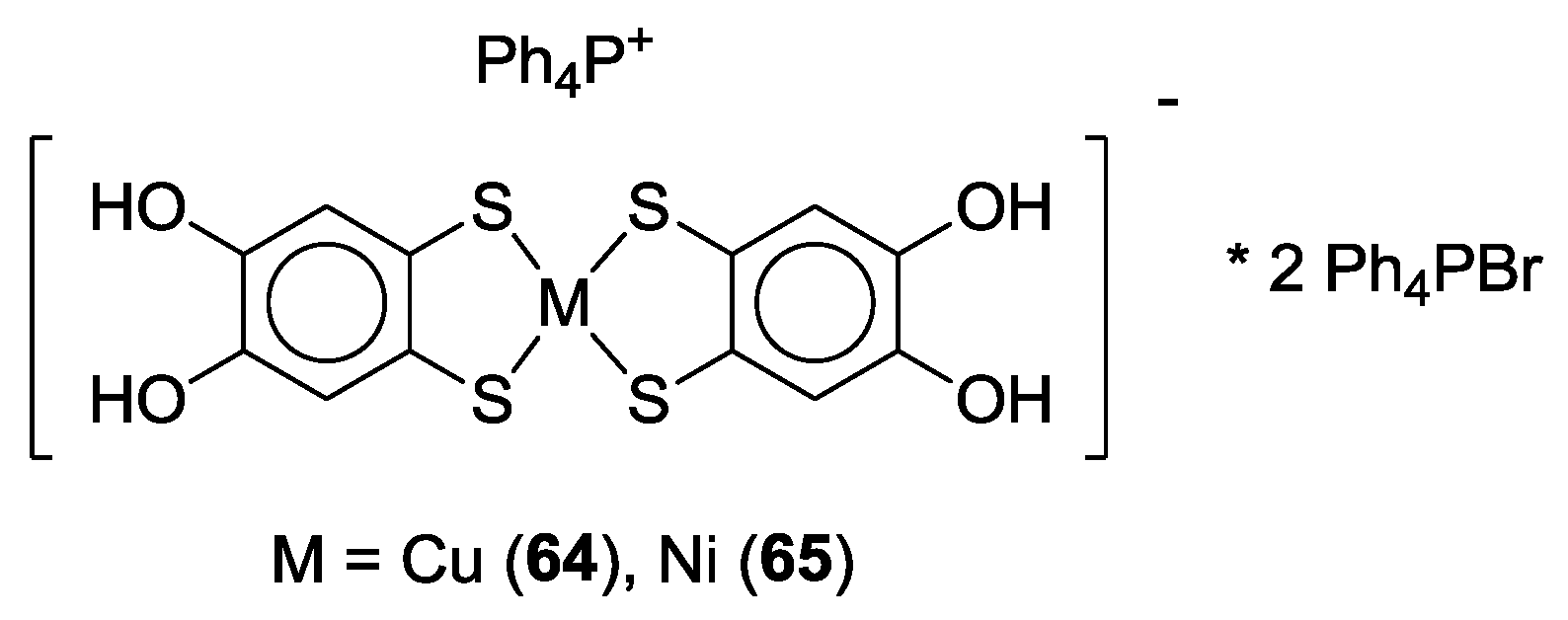
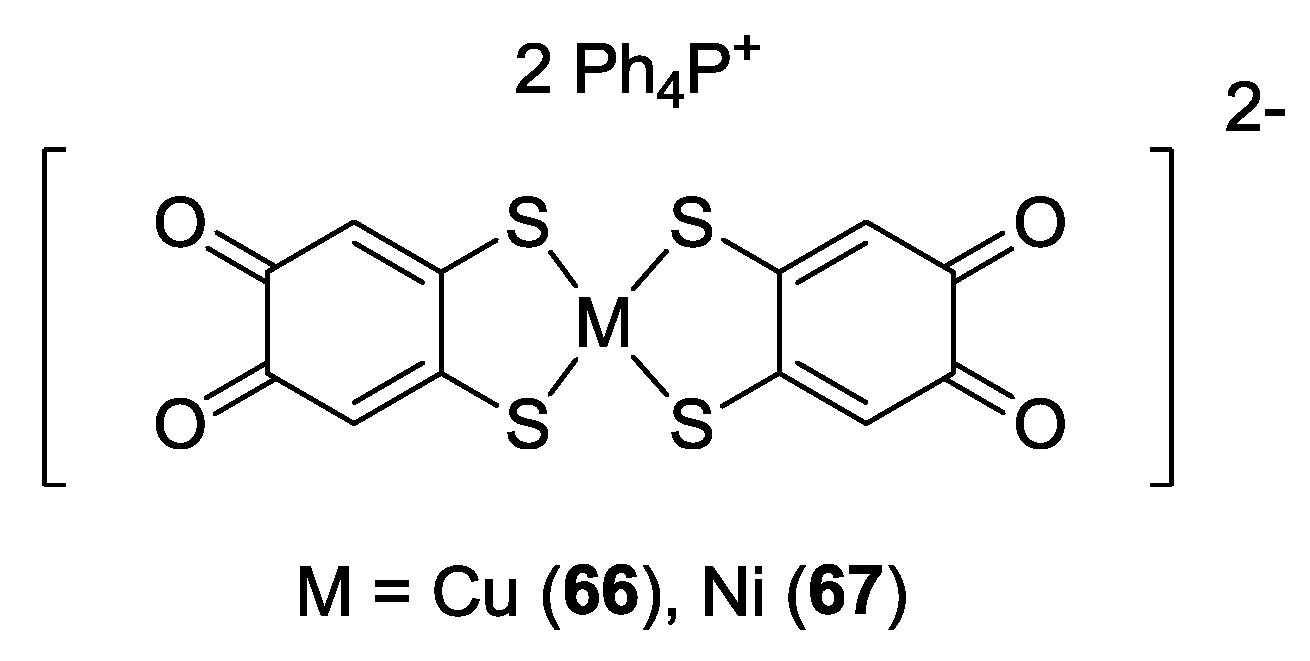
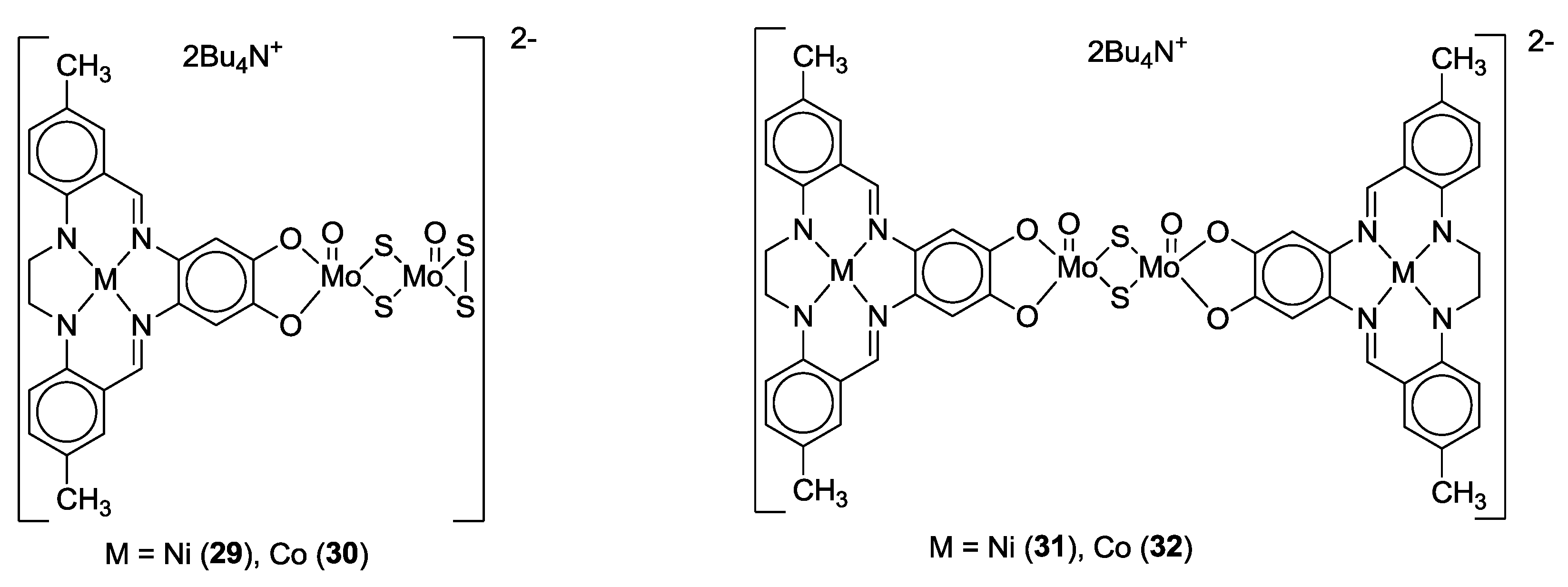
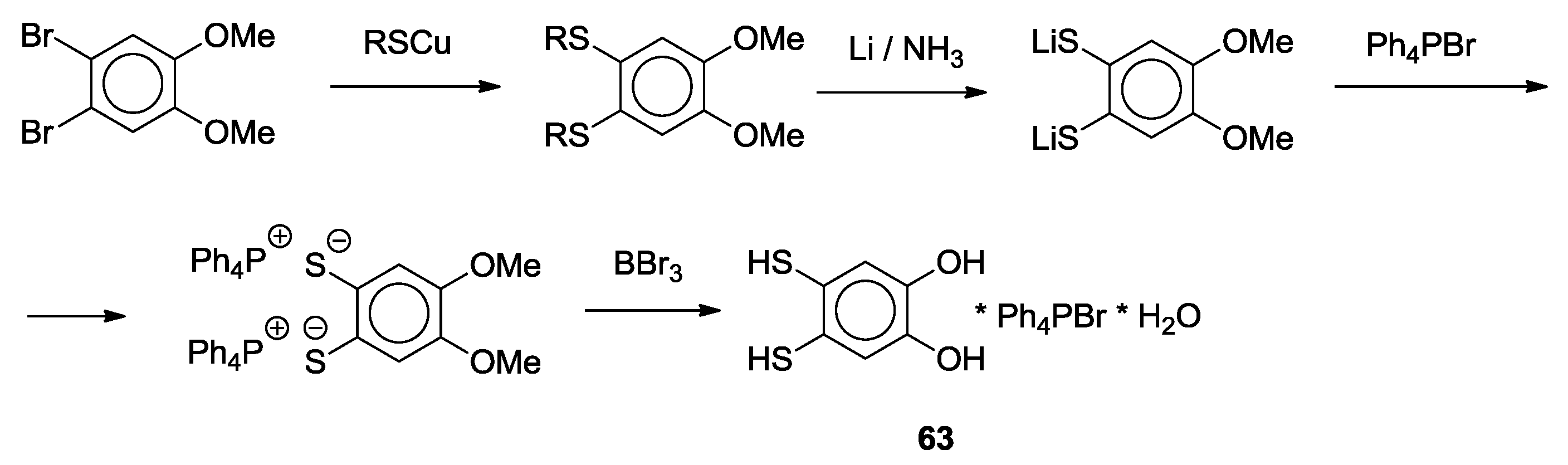
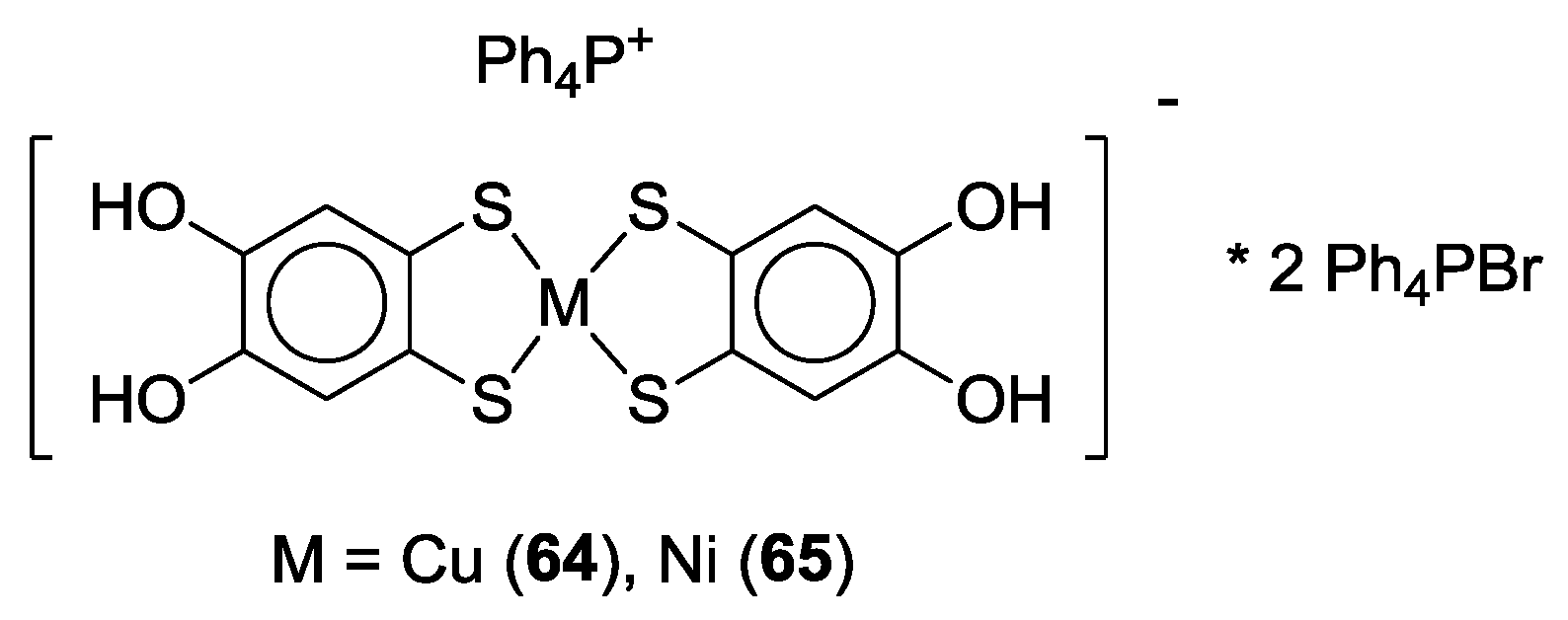
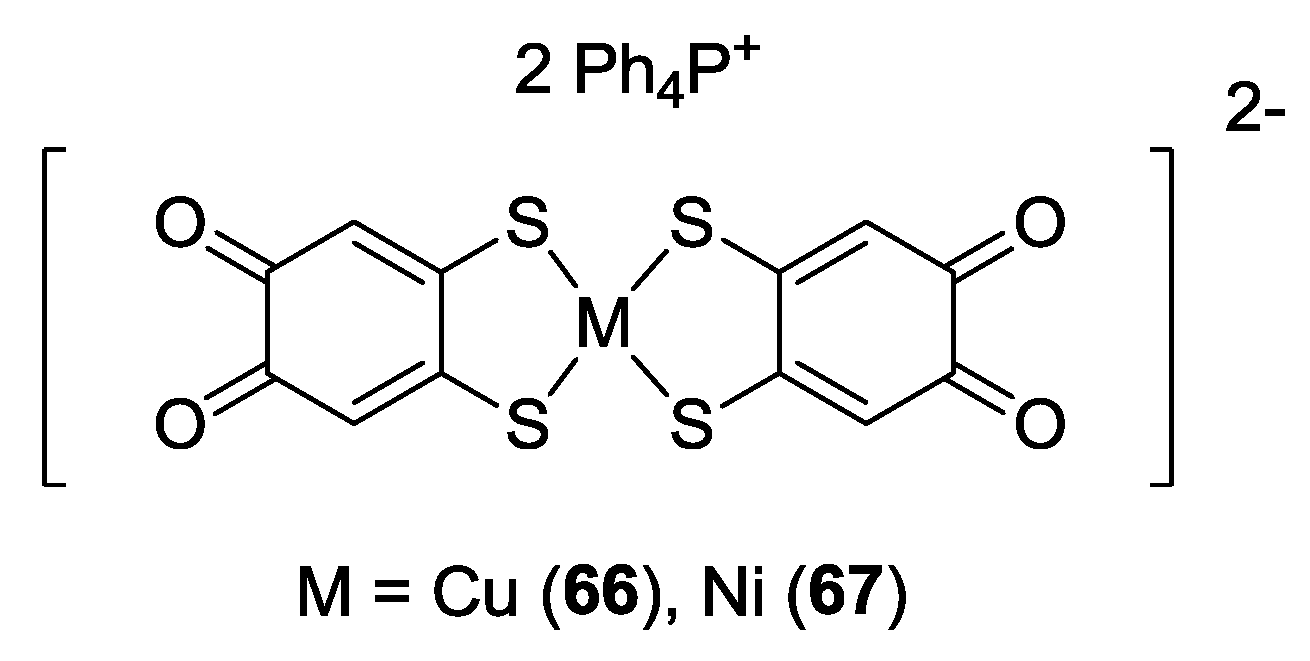
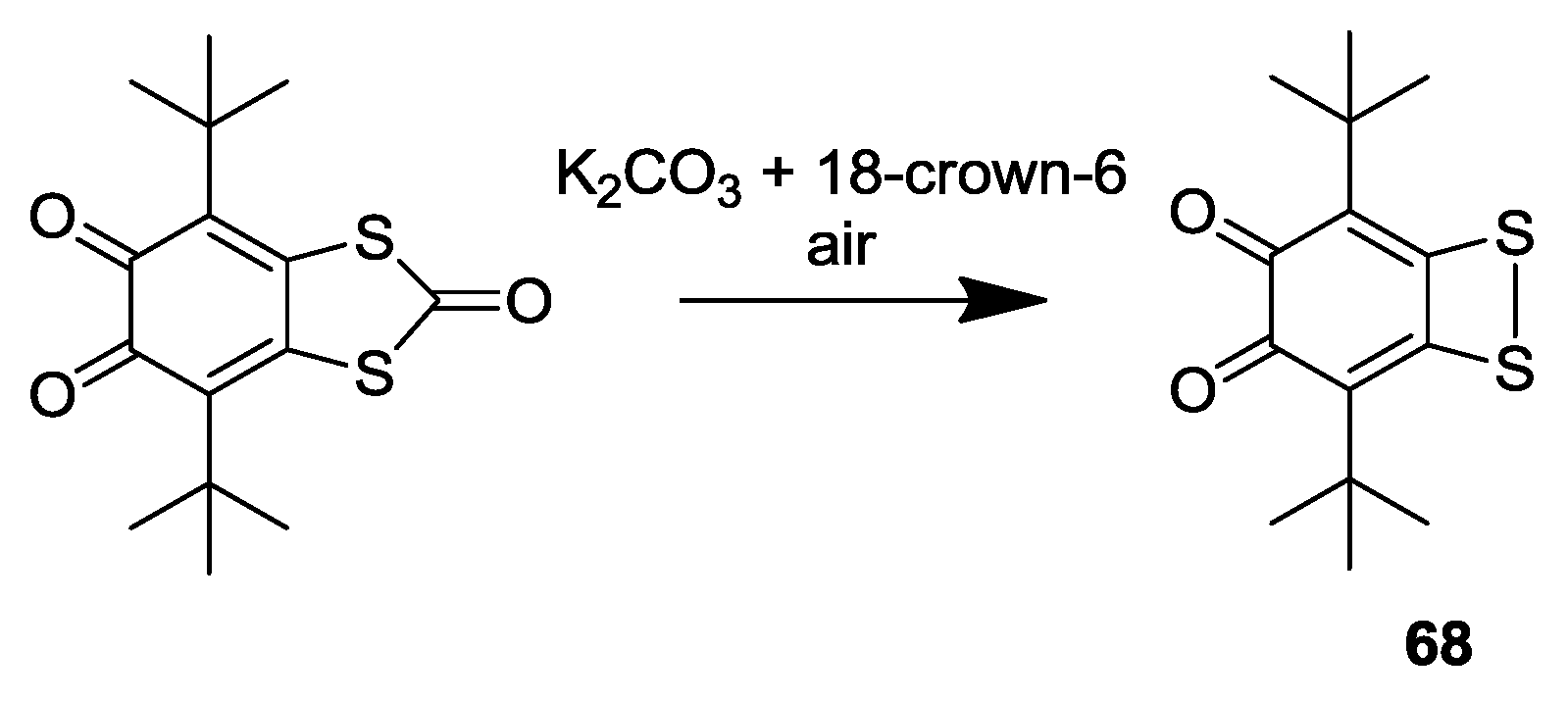
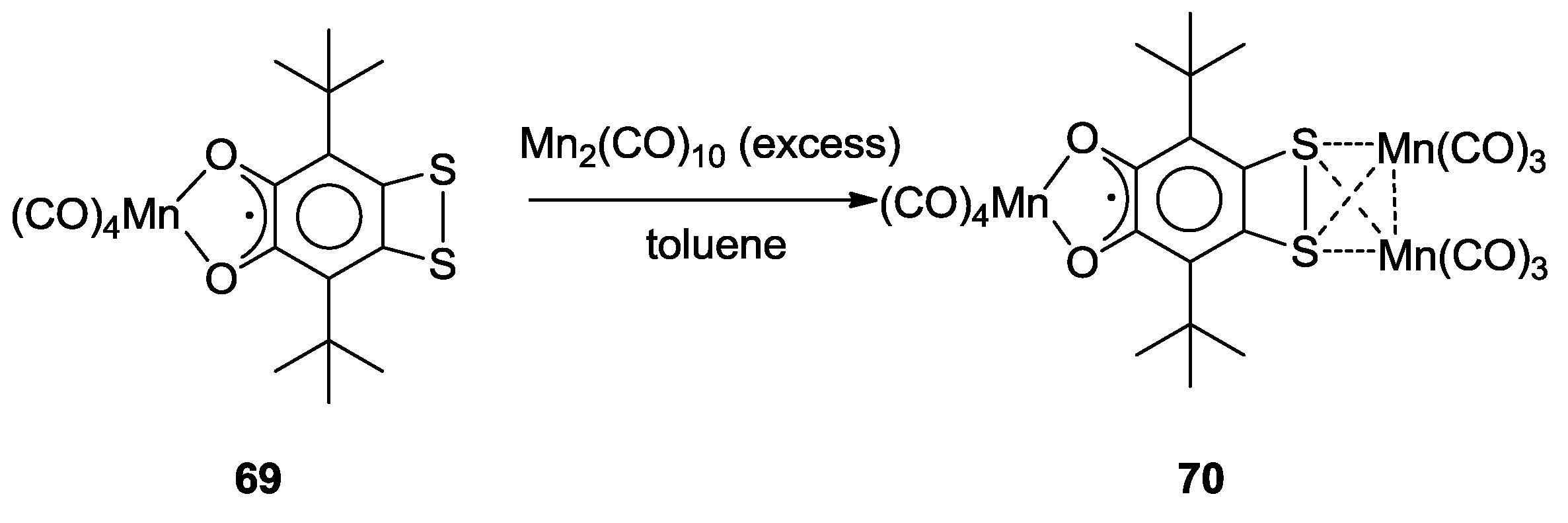
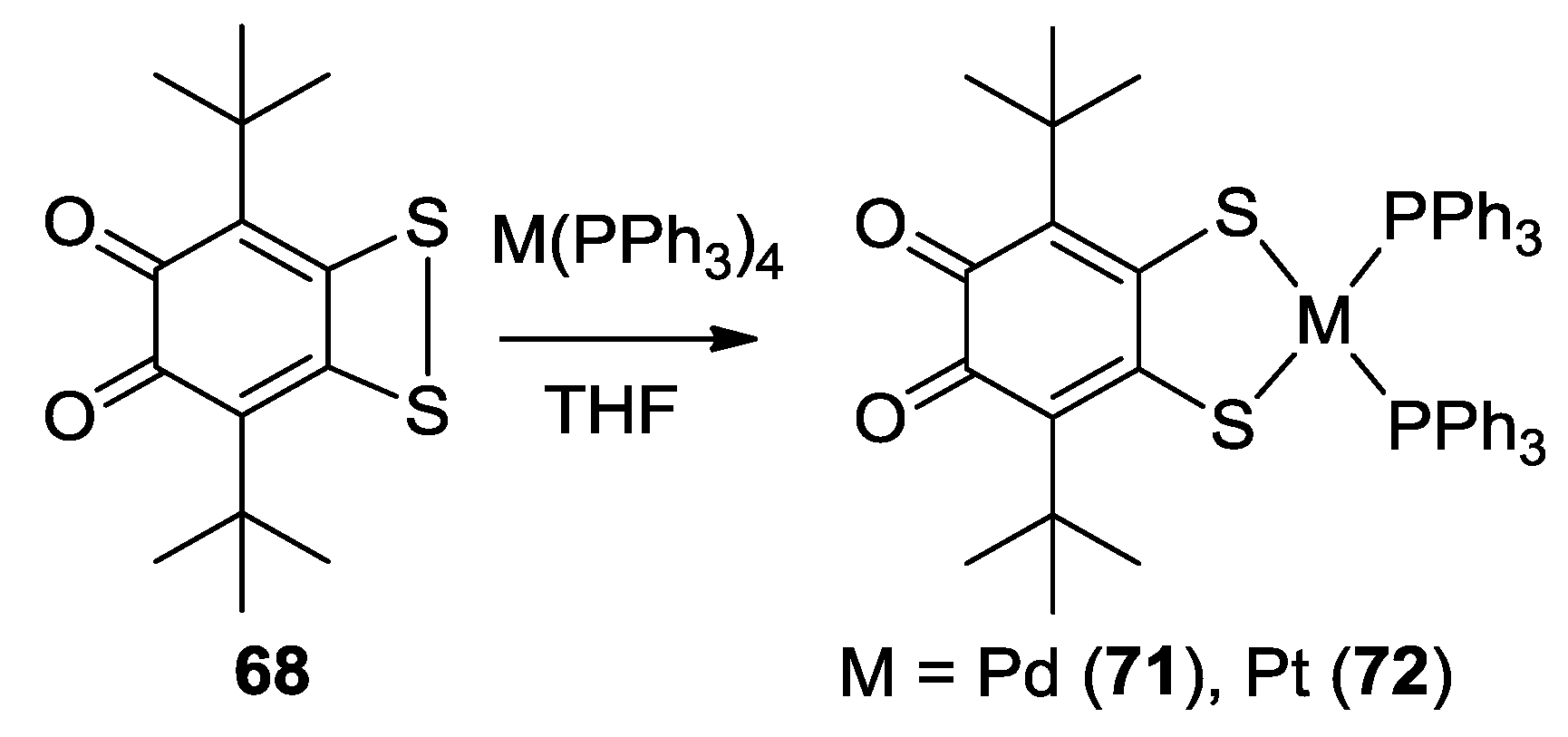
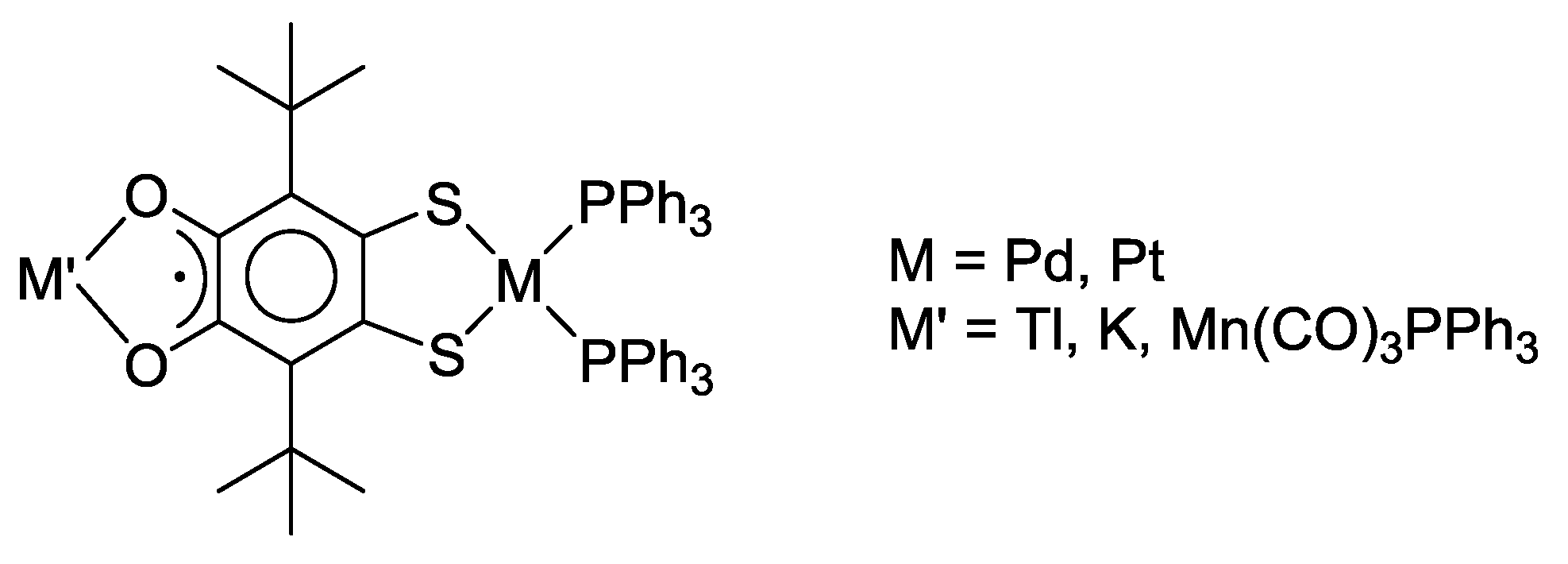
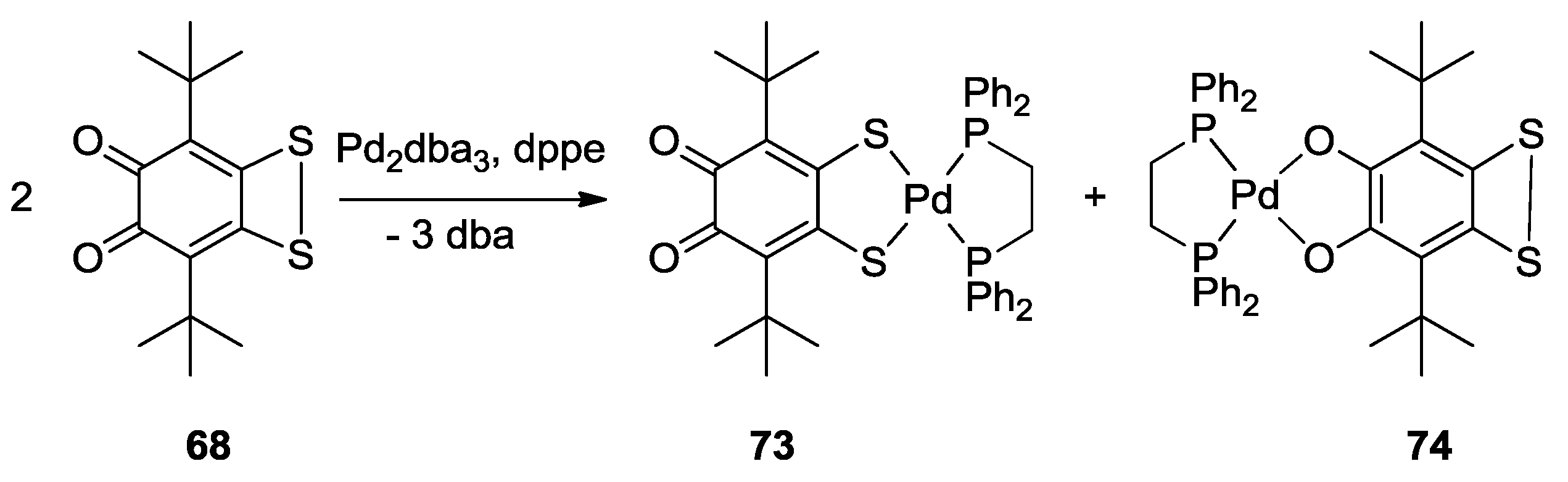
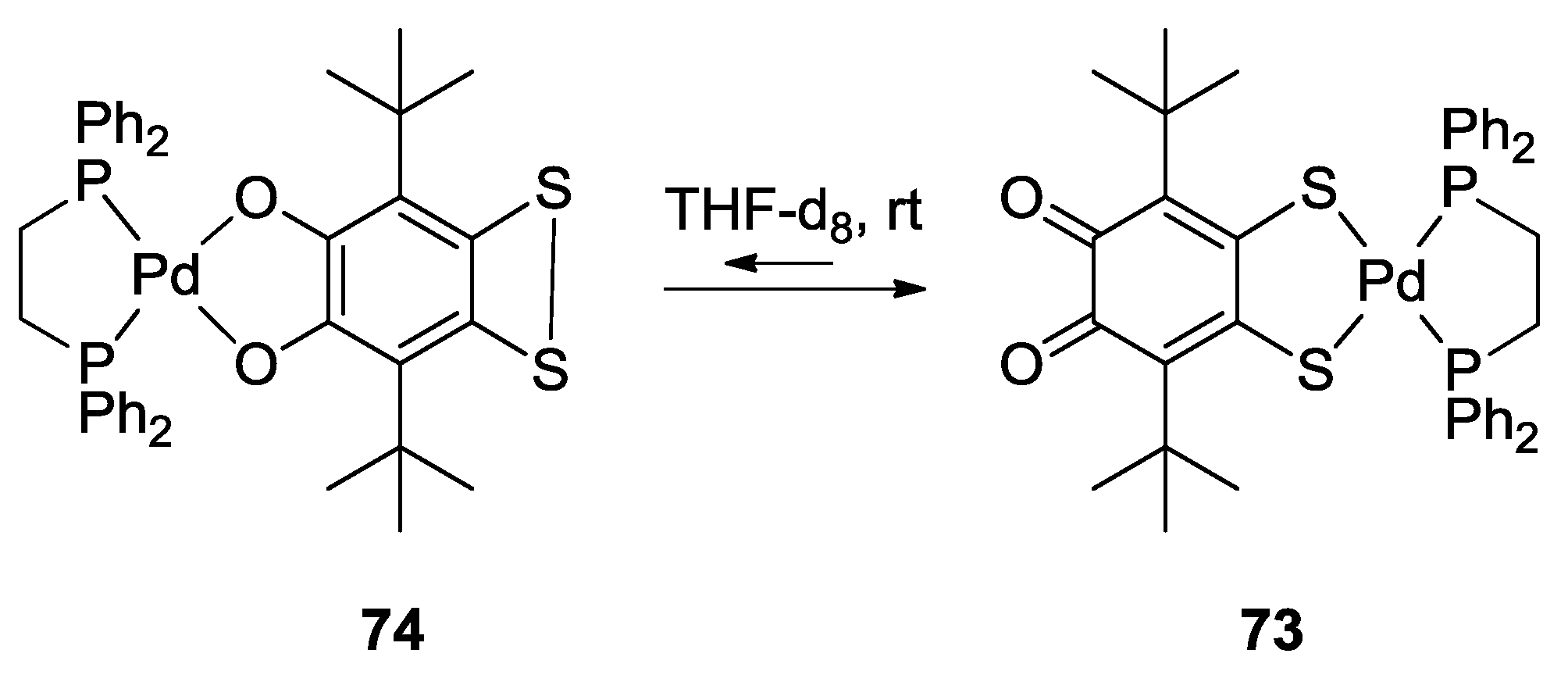
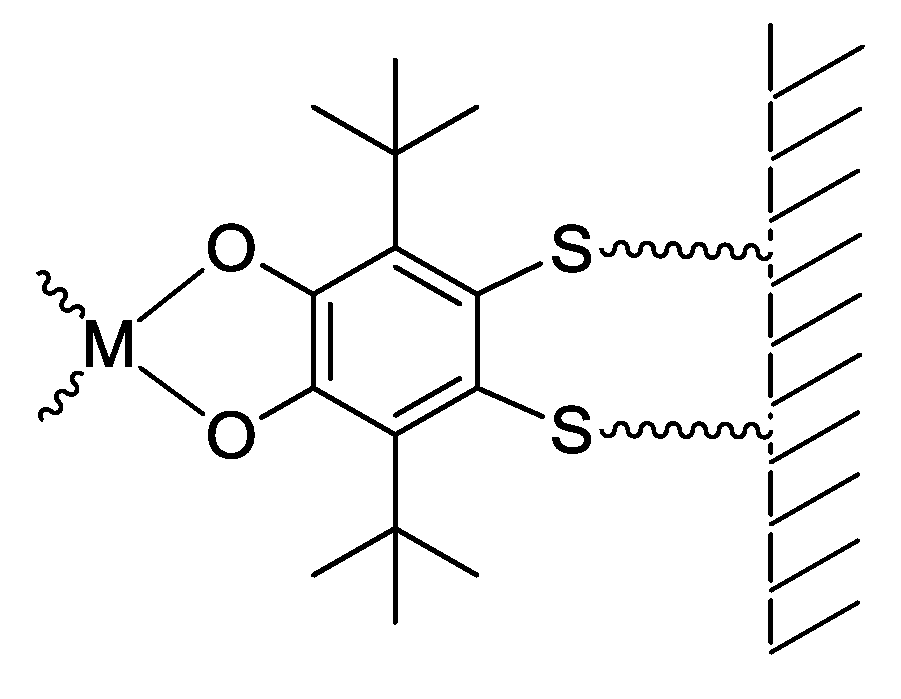
3. SS~OO Systems
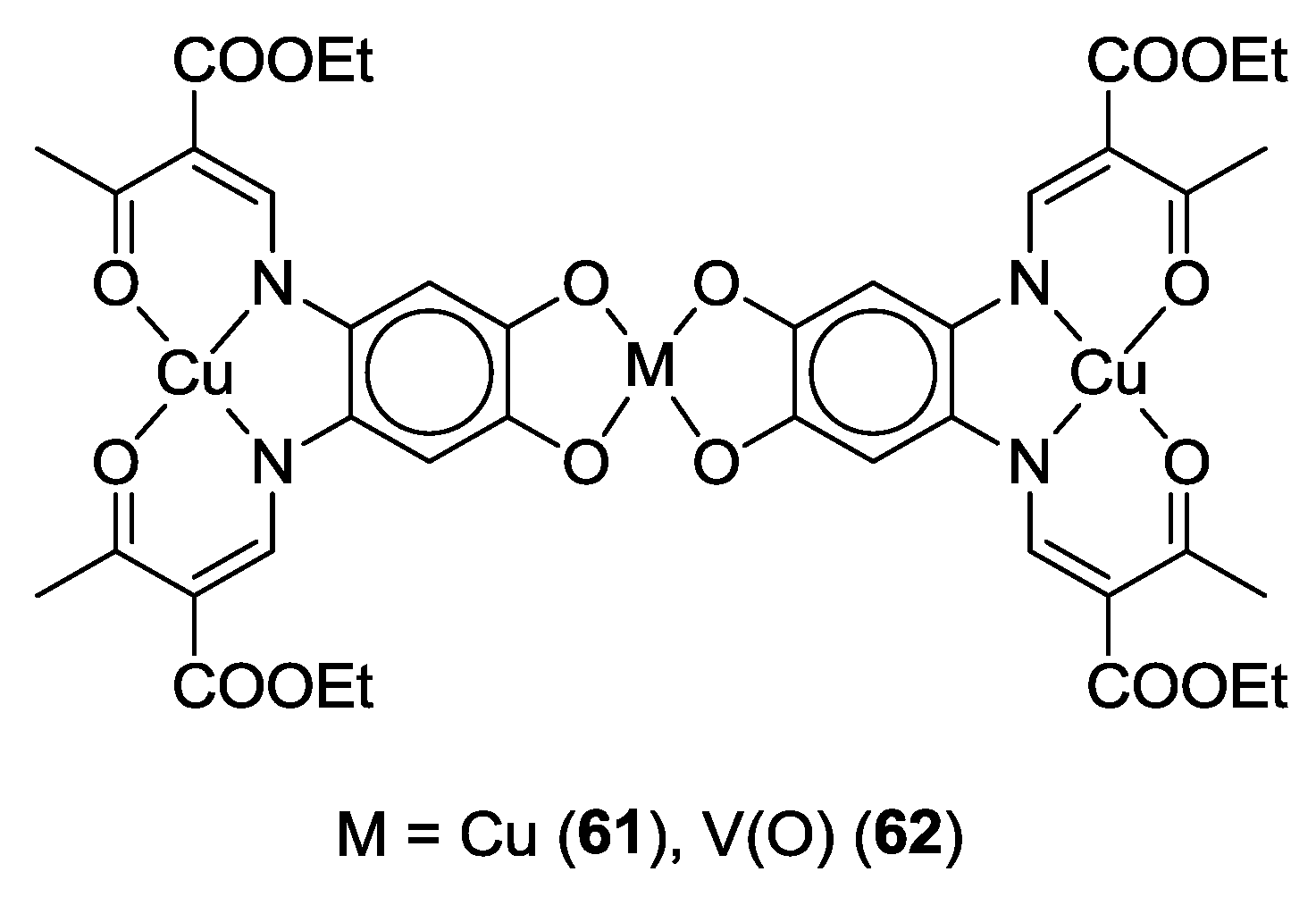
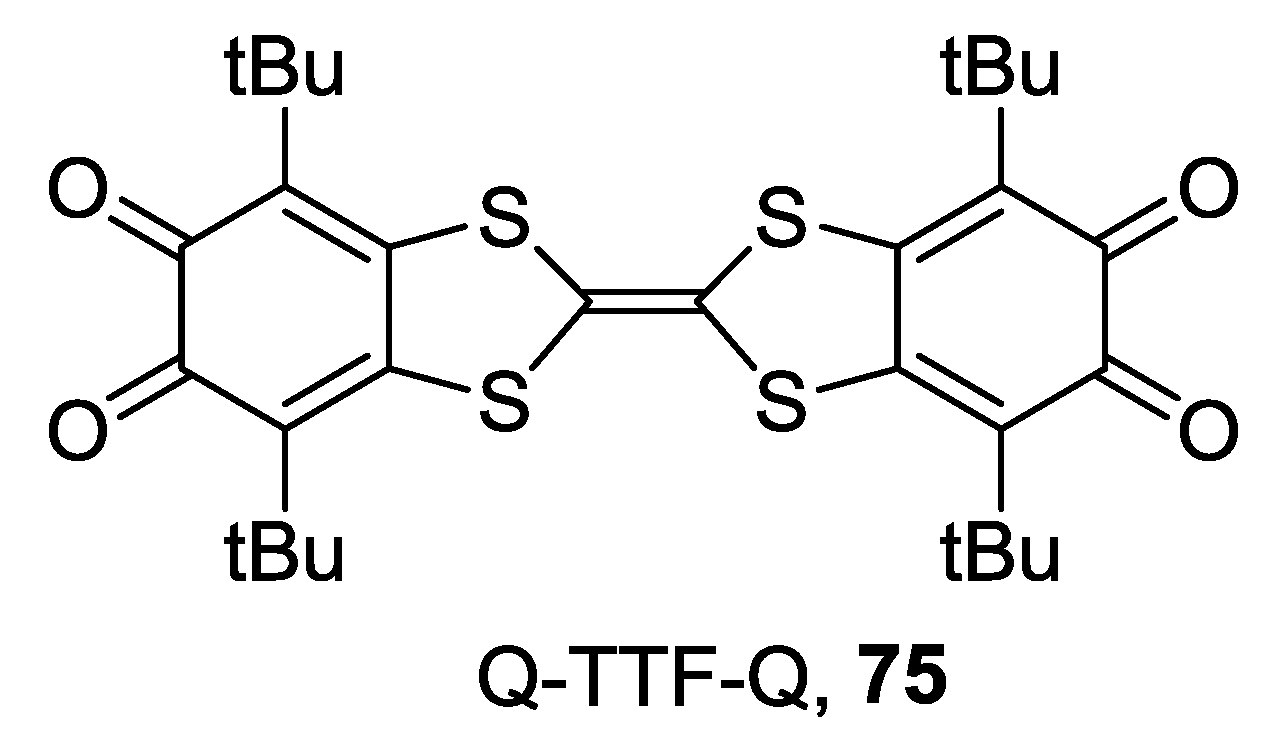
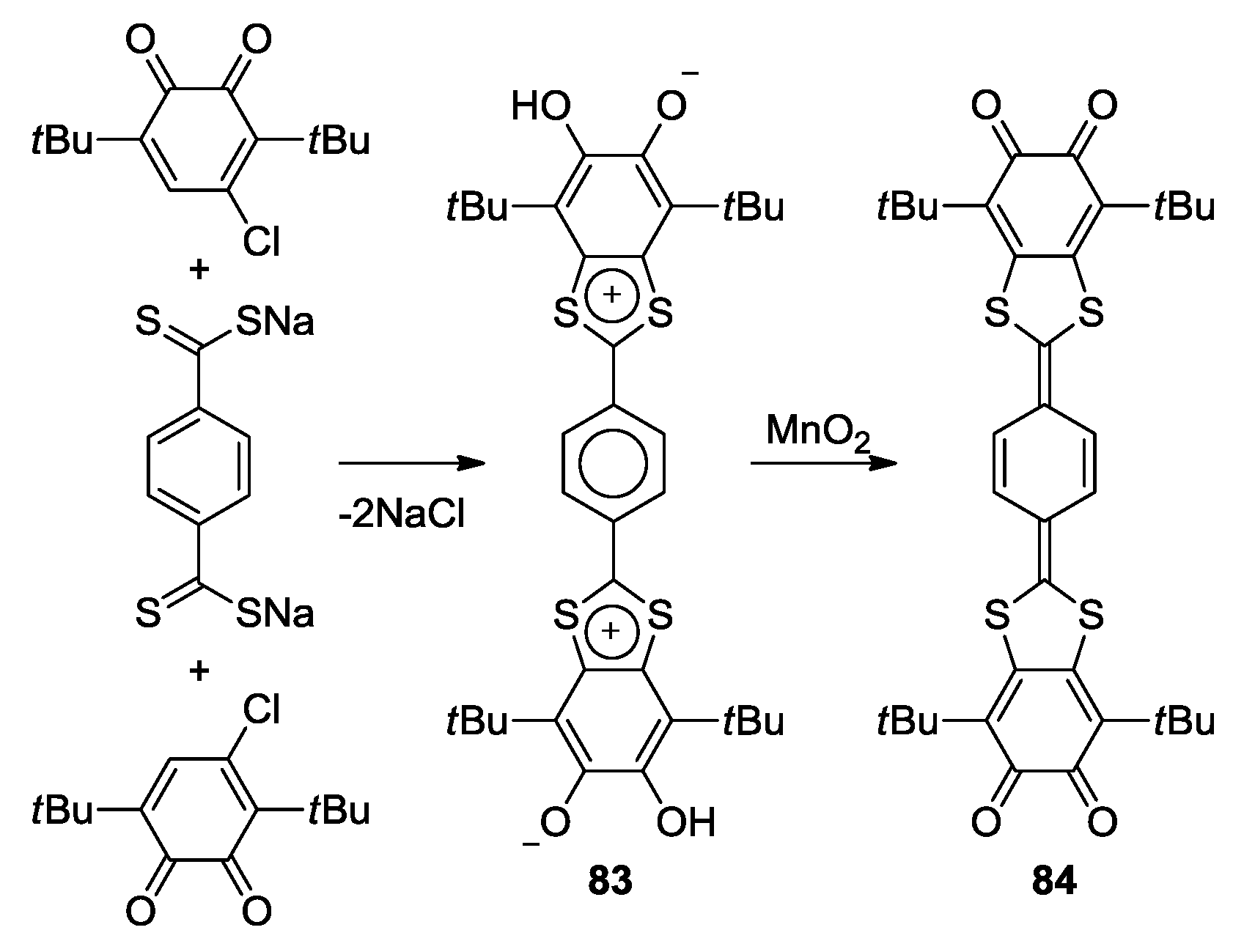
4. Di-o-Quinones Containing Acceptor-Donor-Acceptor Systems
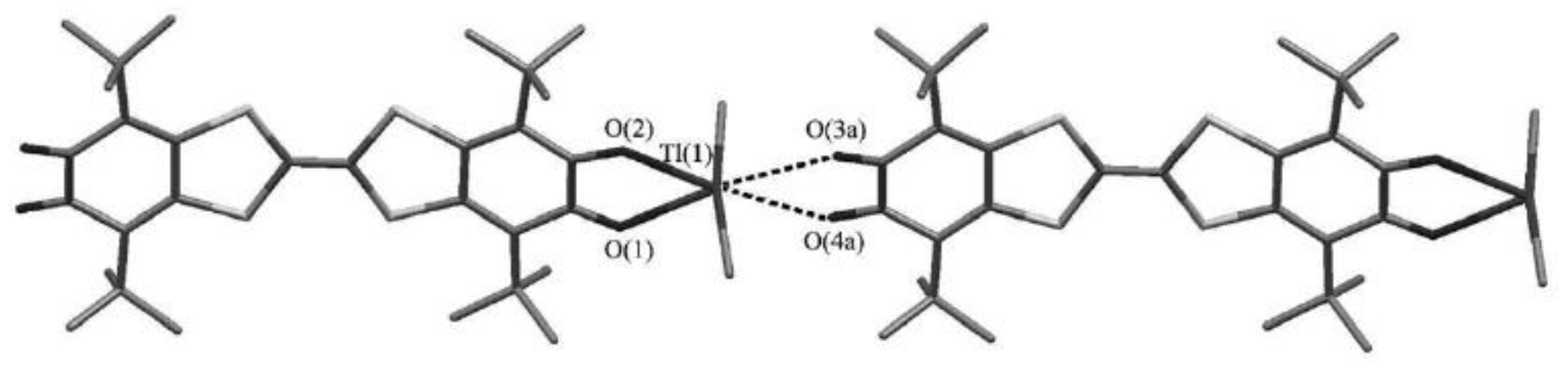
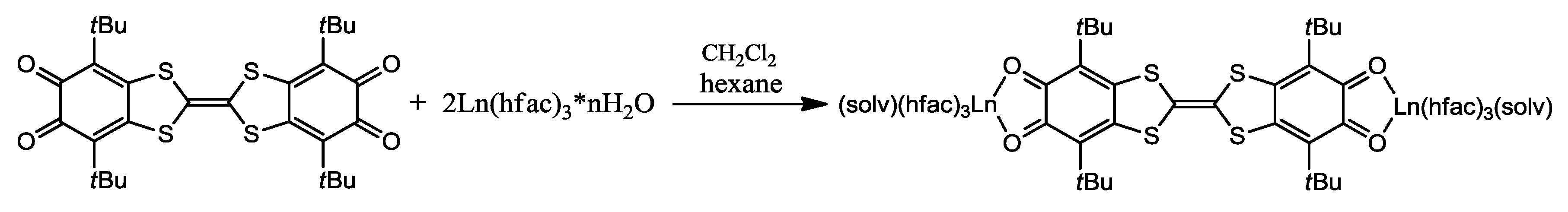
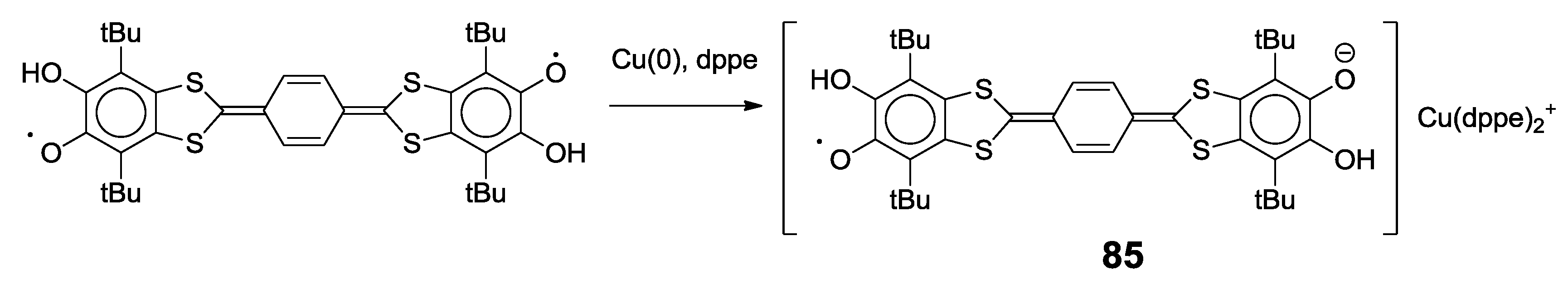
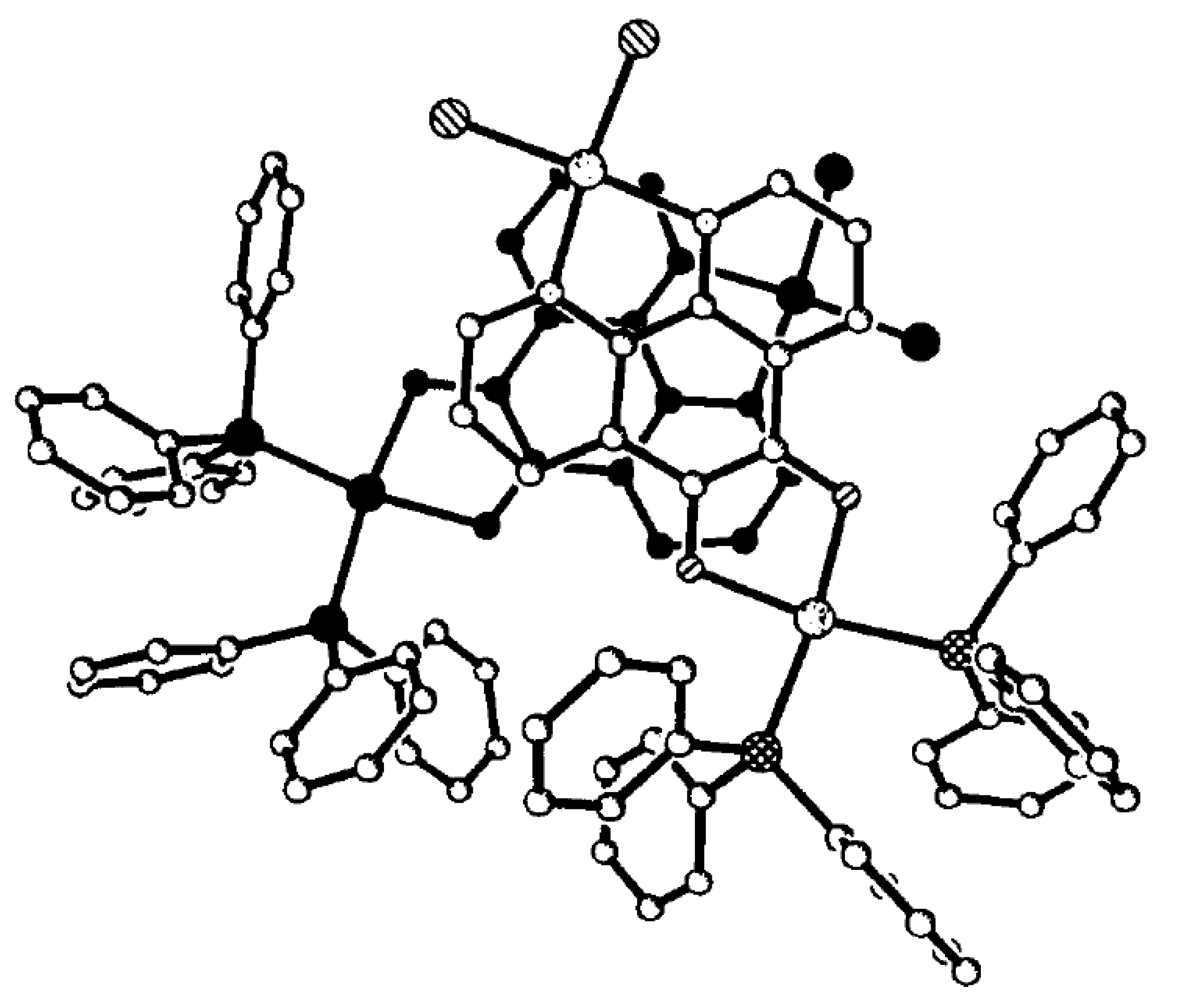
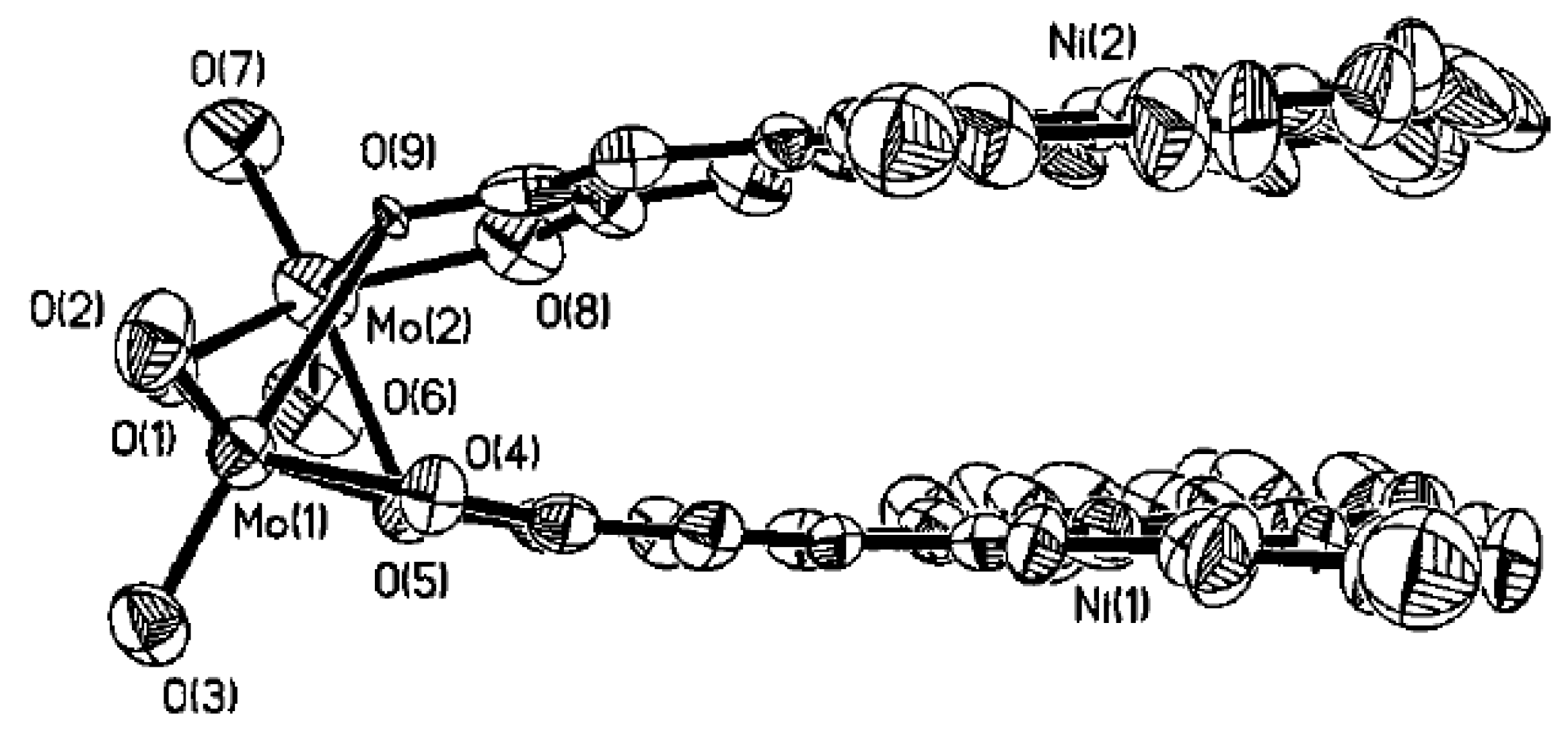
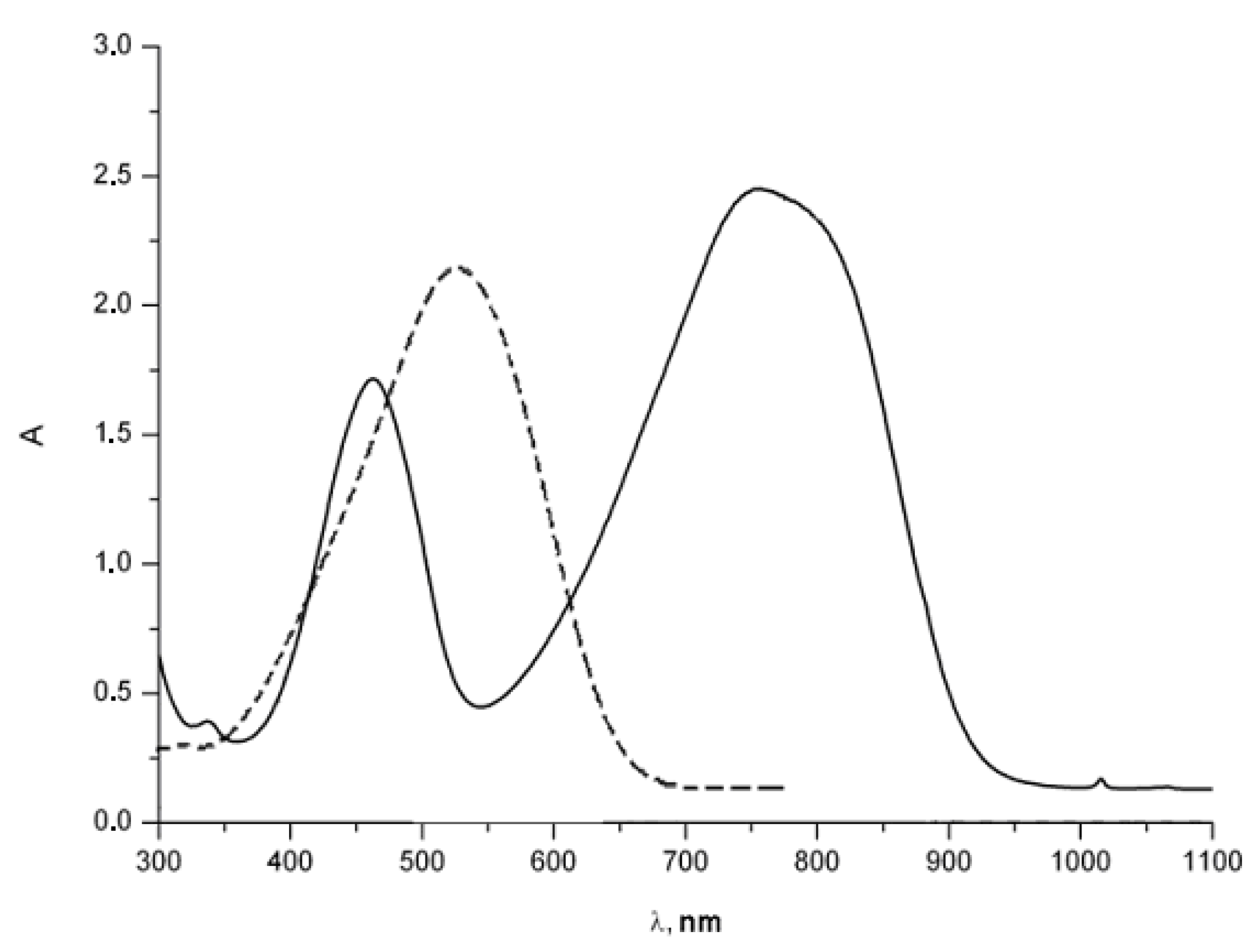
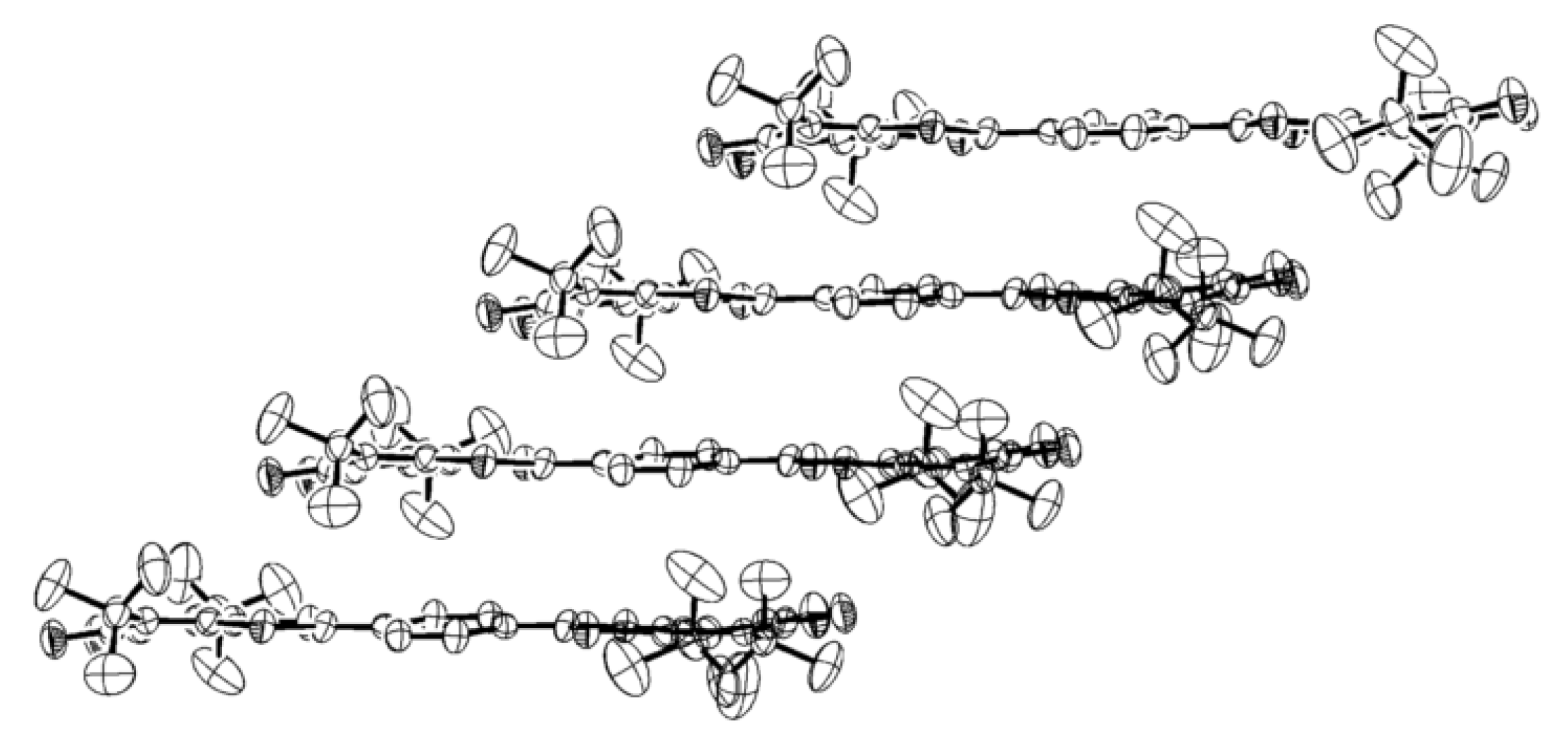
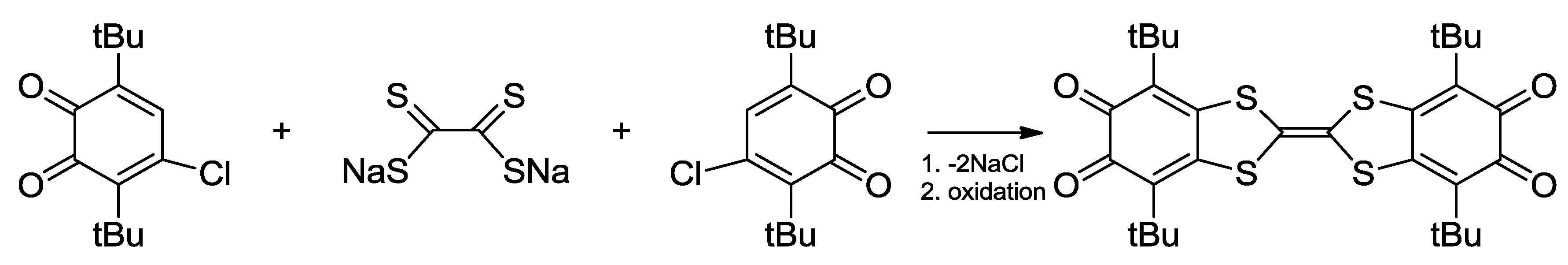
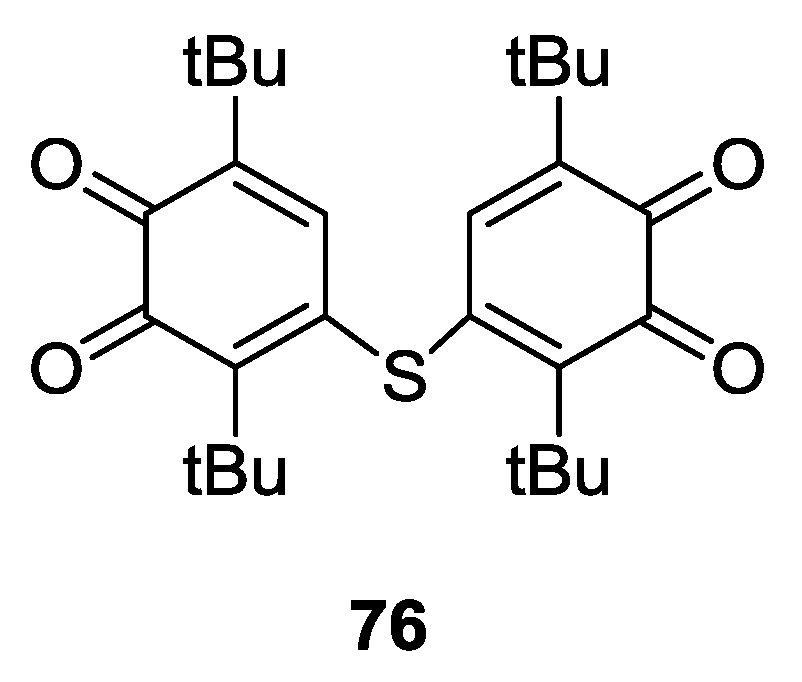
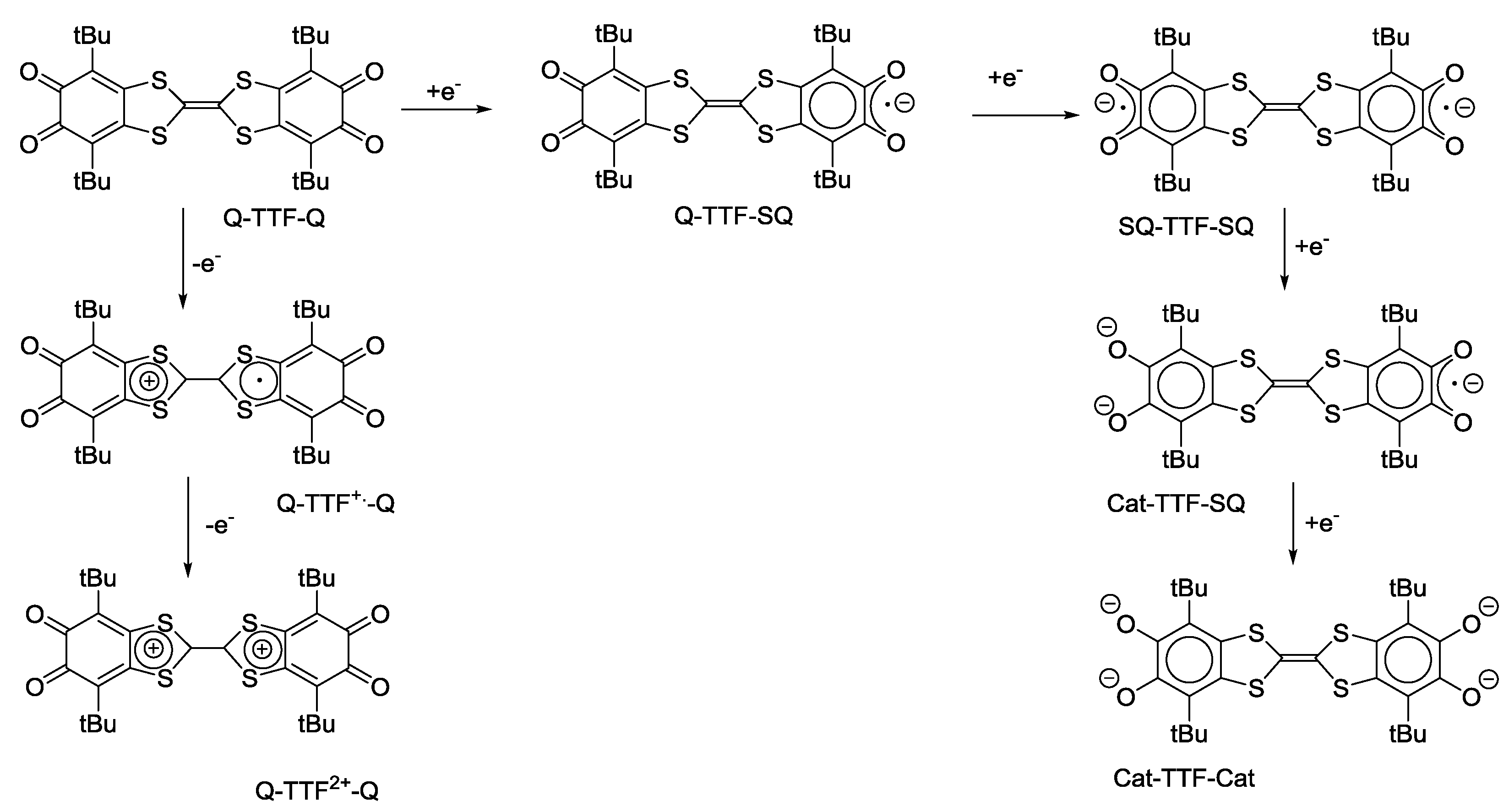
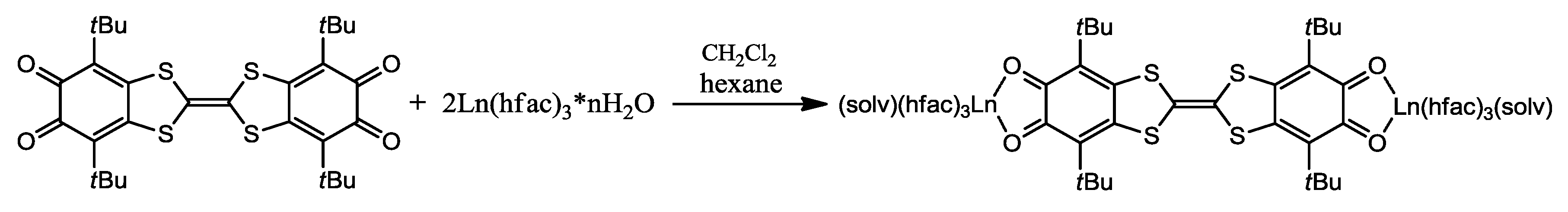
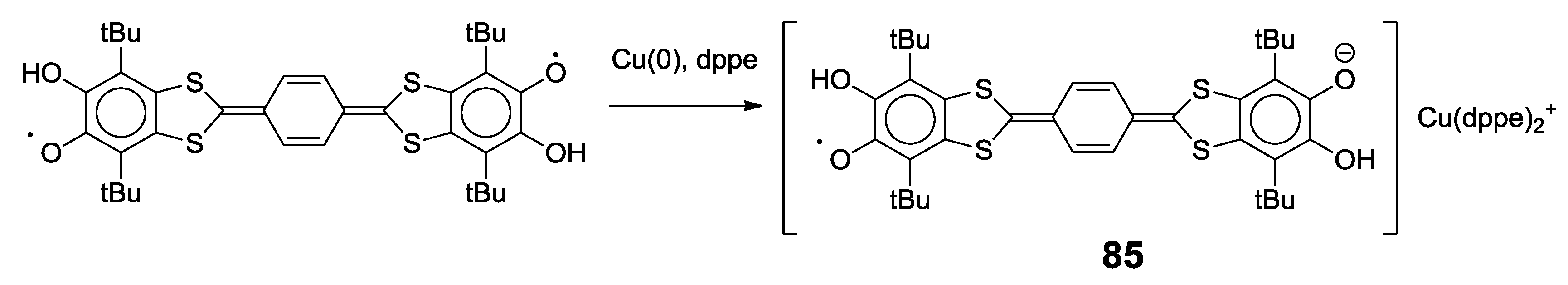
4.1. Amphoteric Redox Triad Consisting of Two o-Quinone Units Linked by TTF Bridge
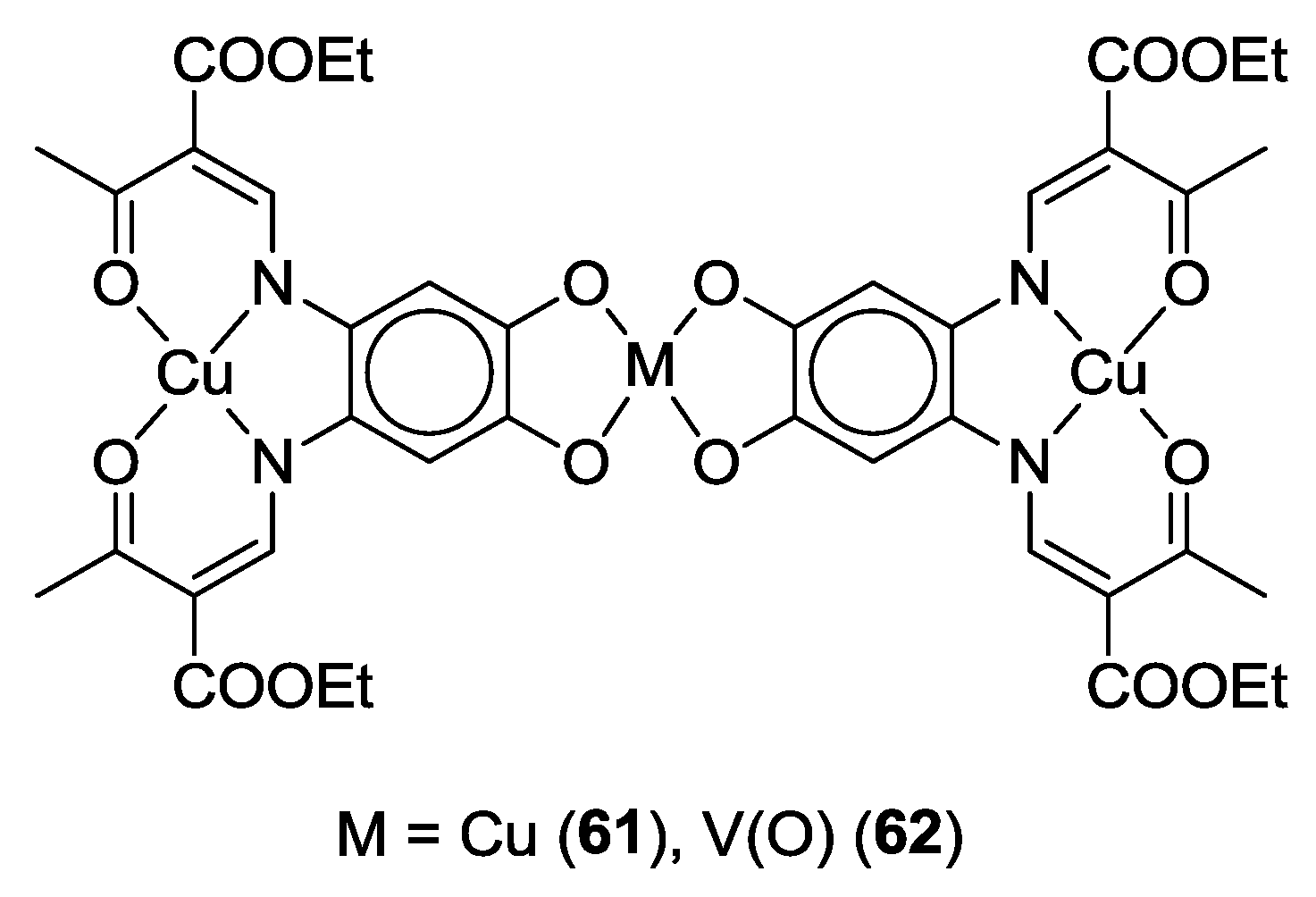
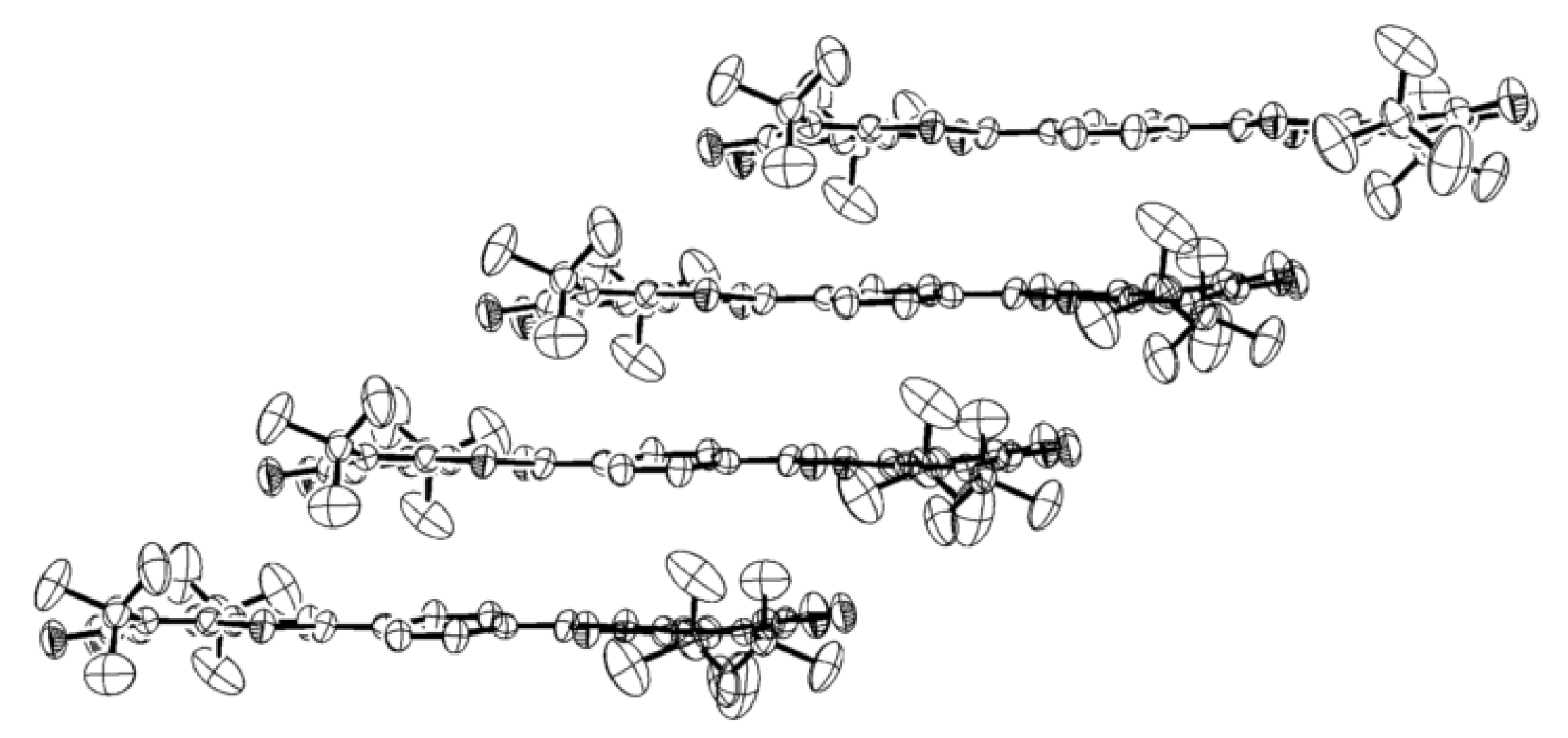
4.2. p-Phenylene Extended Amphoteric Redox Triad
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Abakumov, G.A.; Nevodchikov, V.I. Thermo- and photomechanical effect on the crystals of free-radical complex. Dokl. Akad. Nauk SSSR 1982, 266, 1407–1410. [Google Scholar]
- Jung, O.; Pierpont, C.G. Bistability and Low-Energy Electron Transfer in Cobalt Complexes Containing Catecholate and Semiquinone Ligands. Inorg. Chem. 1994, 33, 2227–2235. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Cherkasov, V.K.; Bubnov, M.P.; Ellert, O.G.; Dobrokhotova, Z.V.; Zakharov, L.N.; Struchkov, Y.T. Investigation of BpyCo(SQ)2 by X-ray diffraction, magnetochemistry, and precision calorimetry. Dokl. Akad. Nauk 1993, 328, 332–335. [Google Scholar]
- Broere, D.L.J.; Plessius, R.; van der Vlugt, J.I. New avenues for ligand-mediated processes—Expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. [Google Scholar] [CrossRef] [PubMed]
- Moussa, J.; Rager, M.N.; Chamoreau, L.M.; Ricard, L.; Amouri, H. Unprecedented π-Bonded Rhodio- and Iridio-o-Benzoquinones as Organometallic Linkers for the Design of Chiral Octahedral Bimetallic Assemblies. Organometallics 2009, 28, 397–404. [Google Scholar] [CrossRef]
- Damas, A.; Gullo, M.P.; Rager, M.N.; Jutand, A.; Barbieri, A.; Amouri, H. Near-infrared room temperature emission from a novel class of Ru(II) heteroleptic complexes with quinonoid organometallic linker. Chem. Commun. 2013, 49, 3796–3798. [Google Scholar] [CrossRef] [PubMed]
- Damas, A.; Ventura, B.; Axet, M.R.; Degli Esposti, A.; Chamoreau, L.-M.; Barbieri, A.; Amouri, H. Organometallic Quinonoid Linkers: A Versatile Tether for the Design of Panchromatic Ruthenium(II) Heteroleptic Complexes. Inorg. Chem. 2010, 49, 10762–10764. [Google Scholar] [CrossRef] [PubMed]
- Damas, A.; Ventura, B.; Moussa, J.; Degli Esposti, A.; Chamoreau, L.-M.; Barbieri, A.; Amouri, H. Turning on Red and Near-Infrared Phosphorescence in Octahedral Complexes with Metalated Quinones. Inorg. Chem. 2012, 51, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Moussa, J.; Chamoreau, L.-M.; Degli Esposti, A.; Gullo, M.P.; Barbieri, A.; Amouri, H. Tuning Excited States of Bipyridyl Platinum(II) Chromophores with π-Bonded Catecholate Organometallic Ligands: Synthesis, Structures, TD-DFT Calculations, and Photophysical Properties. Inorg. Chem. 2014, 53, 6624–6633. [Google Scholar] [CrossRef] [PubMed]
- Moussa, J.; Loch, A.; Chamoreau, L.-M.; Degli Esposti, A.; Bandini, E.; Barbieri, A.; Amouri, H. Luminescent Cyclometalated Platinum Complexes with π-Bonded Catecholate Organometallic Ligands. Inorg. Chem. 2017, 56, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Broere, D.L.J.; Plessius, R.; Tory, J.; Demeshko, S.; de Bruin, B.; Siegler, M.A.; Hartl, F.; van der Vlugt, J.I. Localized Mixed-Valence and Redox Activity within a Triazole-Bridged Dinucleating Ligand upon Coordination to Palladium. Chem. Eur. J. 2016, 22, 13965–13975. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, B.; Schweinfurth, D.; Deibel, N.; Weisser, F. Functional metal complexes based on bridging “imino”-quinonoid ligands. Coord. Chem. Rev. 2015, 293–294, 250–262. [Google Scholar] [CrossRef]
- Pascal, S.; Siri, O. Benzoquinonediimine ligands: Synthesis, coordination chemistry and properties. Coord. Chem. Rev. 2017, 350, 178–195. [Google Scholar] [CrossRef]
- Smith, G.F.; Cagle, F.W. The improved synthesis of 5-nitro-1,10-phenanthroline. J. Org. Chem. 1947, 12, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Masaki, Y.; Yoshihito, T.; Yasuyuki, Y.; Shigeyasu, K.; Ichiro, S. Synthesis and Properties of Diamino-Substituted Dipyrido [3,2-a: 2′,3′-c]phenazine. Bull. Chem. Soc. Jpn. 1992, 65, 1006–1011. [Google Scholar]
- Ghosh, S.; Barve, A.C.; Kumbhar, A.A.; Kumbhar, A.S.; Puranik, V.G.; Datar, P.A.; Sonawane, U.B.; Joshi, R.R. Synthesis, characterization, X-ray structure and DNA photocleavage by cis-dichloro bis(diimine) Co(III) complexes. J. Inorg. Biochem. 2006, 100, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Boghaei, D.M.; Behzadian-Asl, F. Synthesis, characterization and fluorescence spectra of mononuclear Zn(II), Cd(II) and Hg(II) complexes with 1,10-phenanthroline-5,6-dione ligand. J. Coord. Chem. 2007, 60, 347–353. [Google Scholar] [CrossRef]
- Conrad, R.C.; Rund, J.V. Effect of basicity of nonreacting ligands on the rate of reaction of dithiooxamide wth dichloro (phenanthroline) platinum(II) derivatives. Inorg. Chem. 1972, 11, 129–134. [Google Scholar] [CrossRef]
- Girgis, A.Y.; Sohn, Y.S.; Balch, A.L. Preparation and oxidation of some quinone adducts of transition metal complexes. Inorg. Chem. 1975, 14, 2327–2331. [Google Scholar] [CrossRef]
- Fox, G.A.; Bhattacharya, S.; Pierpont, C.G. Structural and electrochemical properties of binuclear complexes containing 1,10-phenanthroline-5,6-diolate as a bridging ligand. Inorg. Chem. 1991, 30, 2895–2899. [Google Scholar] [CrossRef]
- Goss, C.A.; Abruna, H.D. Spectral, electrochemical and electrocatalytic properties of 1,10-phenanthroline-5,6-dione complexes of transition metals. Inorg. Chem. 1985, 24, 4263–4267. [Google Scholar] [CrossRef]
- Murphy, D.M.; McNamara, K.; Richardson, P.; Sanchez-Romaguera, V.; Winpenny, R.E.P.; Yellowlees, L.J. Electrochemical and spectroelectrochemical studies of complexes of 1,10-phenanthroline-5,6-dione. Inorg. Chim. Acta 2011, 374, 435–441. [Google Scholar] [CrossRef]
- Calderazzo, F.; Marchetti, F.; Pampaloni, G.; Passarelli, V. Co-ordination properties of 1,10-phenanthroline-5,6-dione towards group 4 and 5 metals in low and high oxidation states. J. Chem. Soc. Dalton Trans. 1999, 24, 4389–4396. [Google Scholar] [CrossRef]
- Rei, O.; Tetsuaki, F.; Tohru, W.; Koji, T. Comparison of Basicity of the Diimine and Quinoid Group of 1,10-Phenanthroline-5,6-dione Ligated on Pt(II). Bull. Chem. Soc. Jpn. 2006, 79, 106–112. [Google Scholar]
- Paw, W.; Eisenberg, R. Synthesis, Characterization, and Spectroscopy of Dipyridocatecholate Complexes of Platinum. Inorg. Chem. 1997, 36, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Calderazzo, F.; Pampaloni, G.; Passarelli, V. 1,10-Phenanthroline-5,6-dione as a building block for the synthesis of homo- and heterometallic complexes. Inorg. Chim. Acta 2002, 330, 136–142. [Google Scholar] [CrossRef]
- Calucci, L.; Pampaloni, G.; Pinzino, C.; Prescimone, A. Transition metal derivatives of 1,10-phenanthroline-5,6-dione: Controlled growth of coordination polynuclear derivatives. Inorg. Chim. Acta 2006, 359, 3911–3920. [Google Scholar] [CrossRef]
- Brechin, E.K.; Calucci, L.; Englert, U.; Margheriti, L.; Pampaloni, G.; Pinzino, C.; Prescimone, A. 1,10-Phenanthroline-5,6-dione complexes of middle transition elements: Mono- and dinuclear derivatives. Inorg. Chim. Acta 2008, 361, 2375–2384. [Google Scholar] [CrossRef]
- Shavaleev, N.M.; Moorcraft, L.P.; Pope, S.J.A.; Bell, Z.R.; Faulkner, S.; Ward, M.D. Sensitized Near-Infrared Emission from Complexes of YbIII, NdIII and ErIII by Energy-Transfer from Covalently Attached PtII-Based Antenna Units. Chem. Eur. J. 2003, 9, 5283–5291. [Google Scholar] [CrossRef] [PubMed]
- Shavaleev, N.M.; Moorcraft, L.P.; Pope, S.J.A.; Bell, Z.R.; Faulkner, S.; Ward, M.D. Sensitised near-infrared emission from lanthanides using a covalently-attached Pt(II) fragment as an antenna group. Chem. Commun. 2003, 10, 1134–1135. [Google Scholar] [CrossRef]
- Coucouvanis, D.; Jonasdottir, S.G.; Christodoulou, D.; Kim, C.G.; Kampf, J.W. Multifunctional macrocyclic ligands. Synthesis and characterization of nickel(II) and cobalt(II) macrocyclic-catechol complexes and their dimeric, antiferromagnetically coupled, semiquinone derivatives. Inorg. Chem. 1993, 32, 2987–2988. [Google Scholar] [CrossRef]
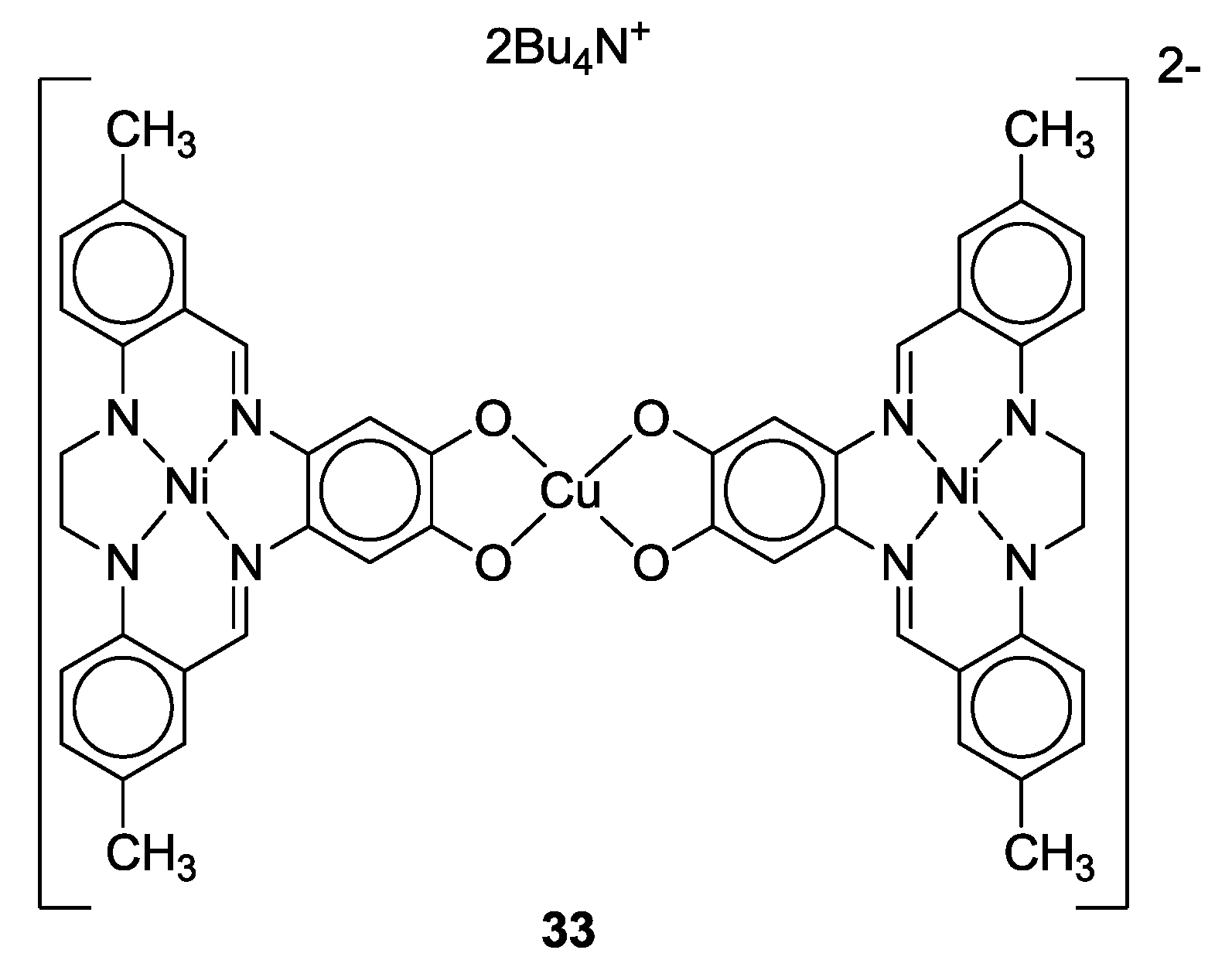
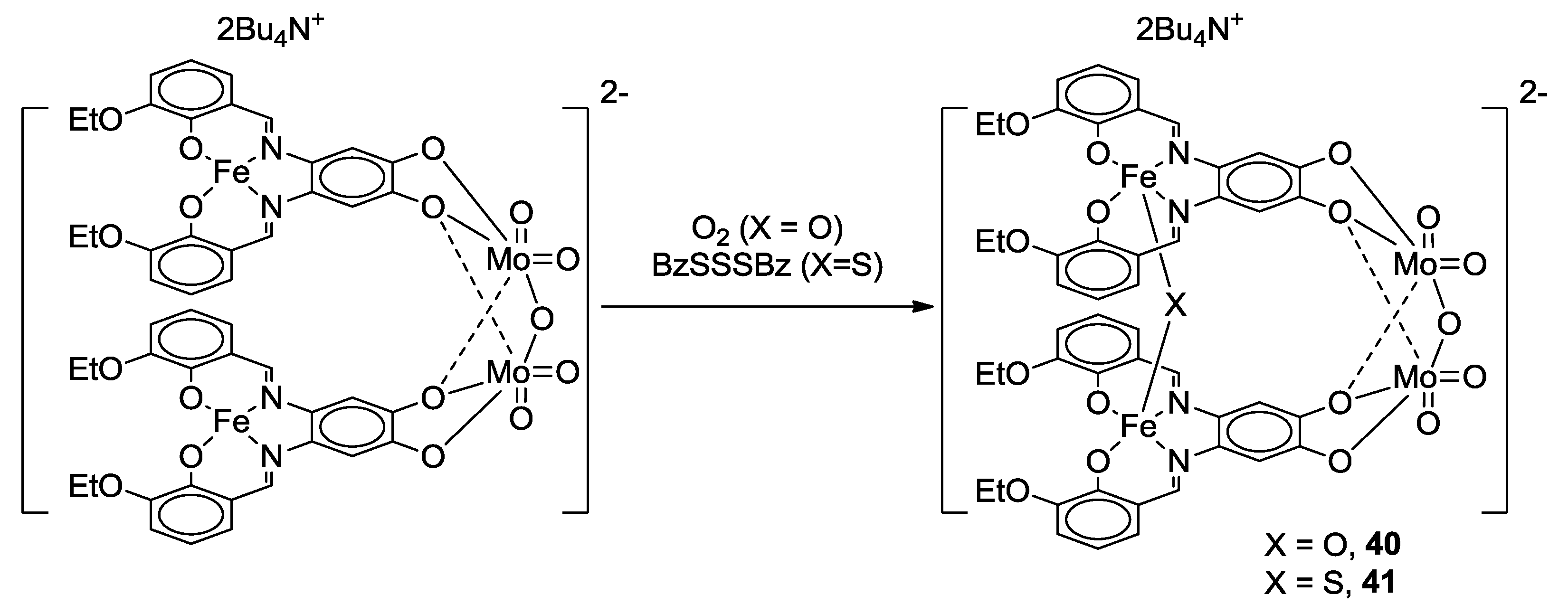
- Jonasdottir, S.G.; Kim, C.G.; Coucouvanis, D. Macrocyclic catechol complexes as ligands in the synthesis of heterometallic supramolecules. Inorg. Chem. 1993, 32, 3591–3592. [Google Scholar] [CrossRef]
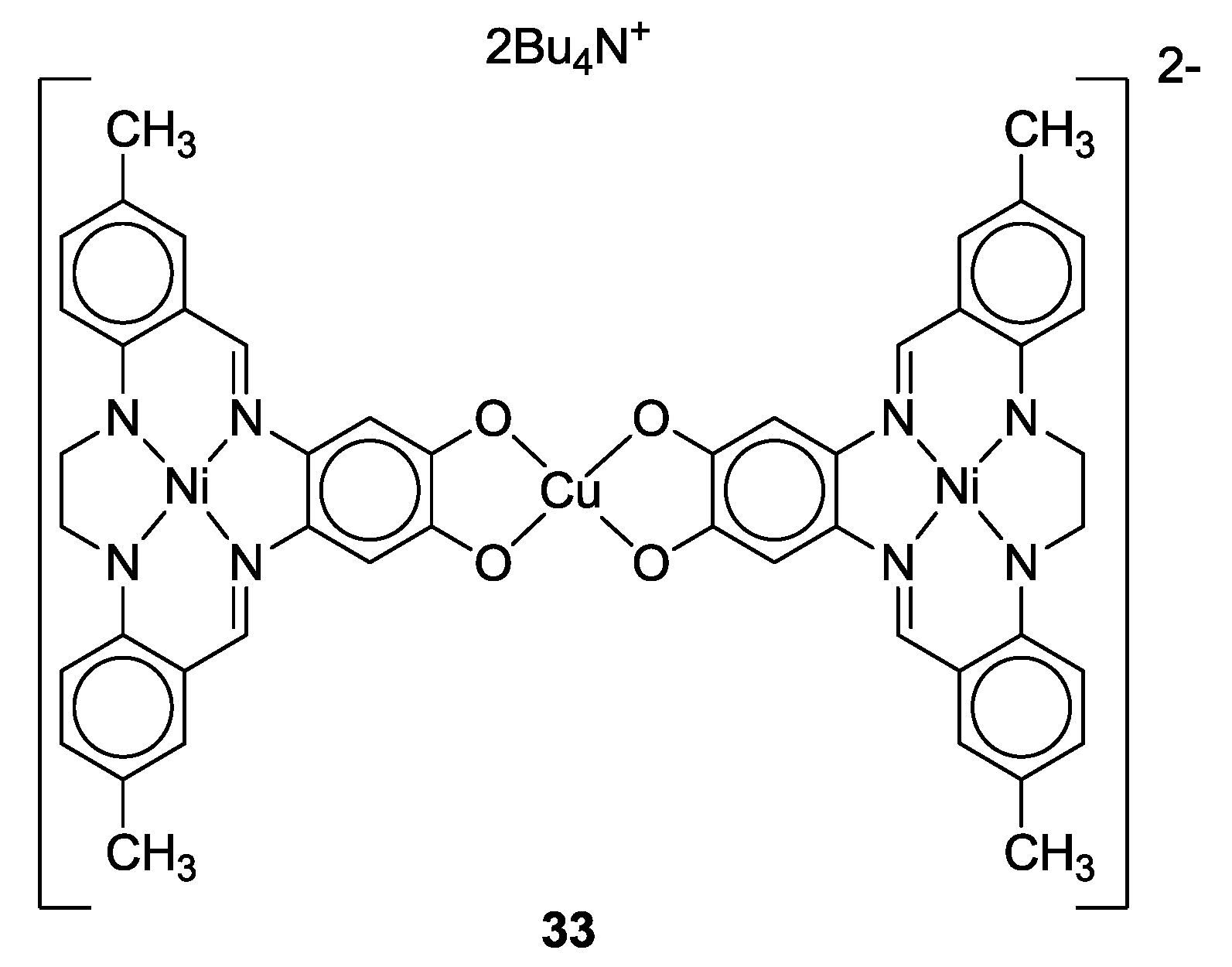
- Jonasdottir, S.; Kim, C.-G.; Kampf, J.; Coucouvanis, D. Macrocyclic catecholate complexes as ligands. Synthesis, structural characterization and properties of the [MH2ETC] and {[Ni(ETC)]Cu}2− complexes (M = Co, Ni; H4ETC = the macrocyclic catechol, 2,3-ethylene-5,6:13,14-di(5′-methylbzo)-9,10-(4′,5′-diolbzo)-[14]-1,4,8,11-[N4]-7,12-diene). Inorg. Chim. Acta 1996, 243, 255–270. [Google Scholar]
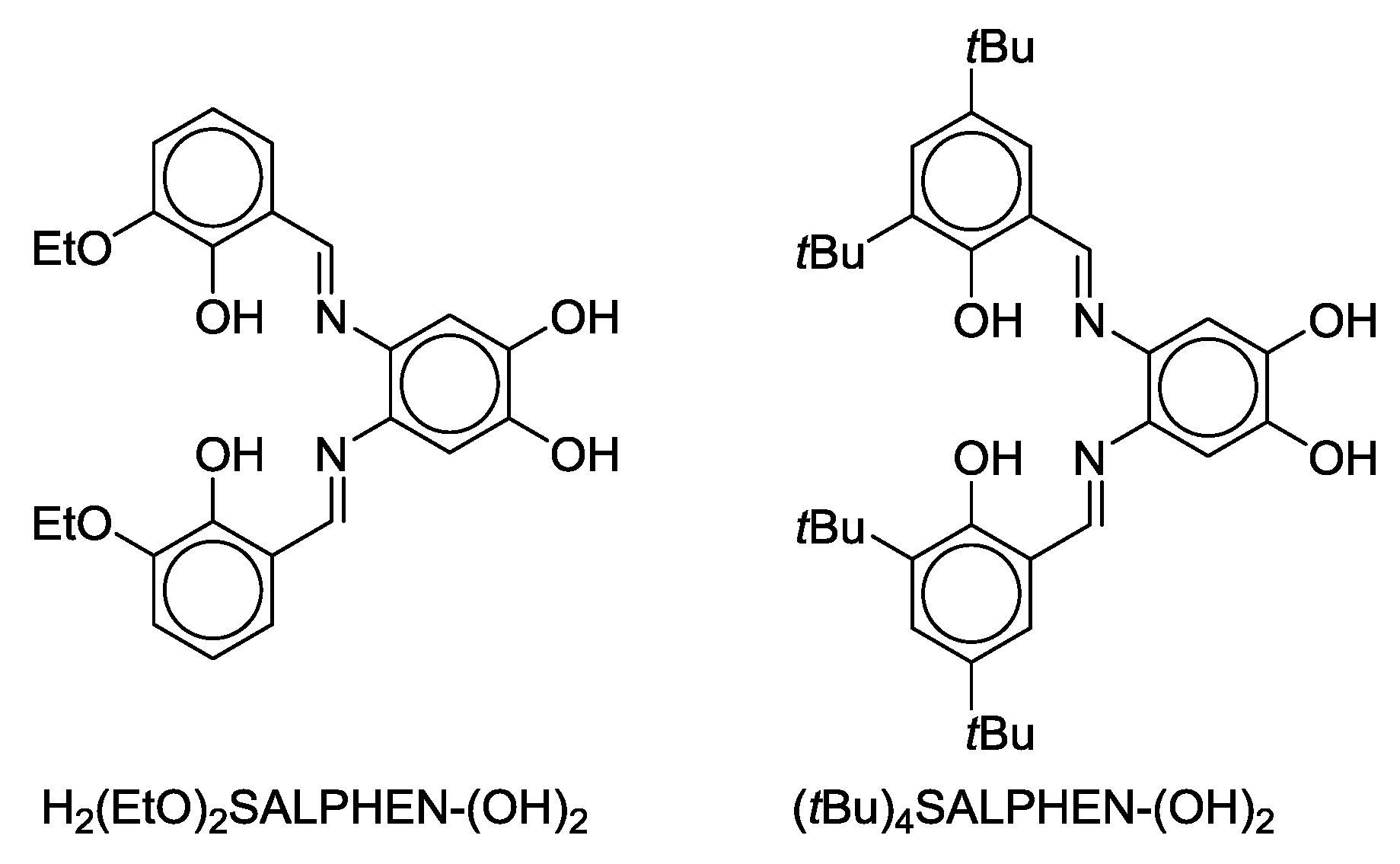
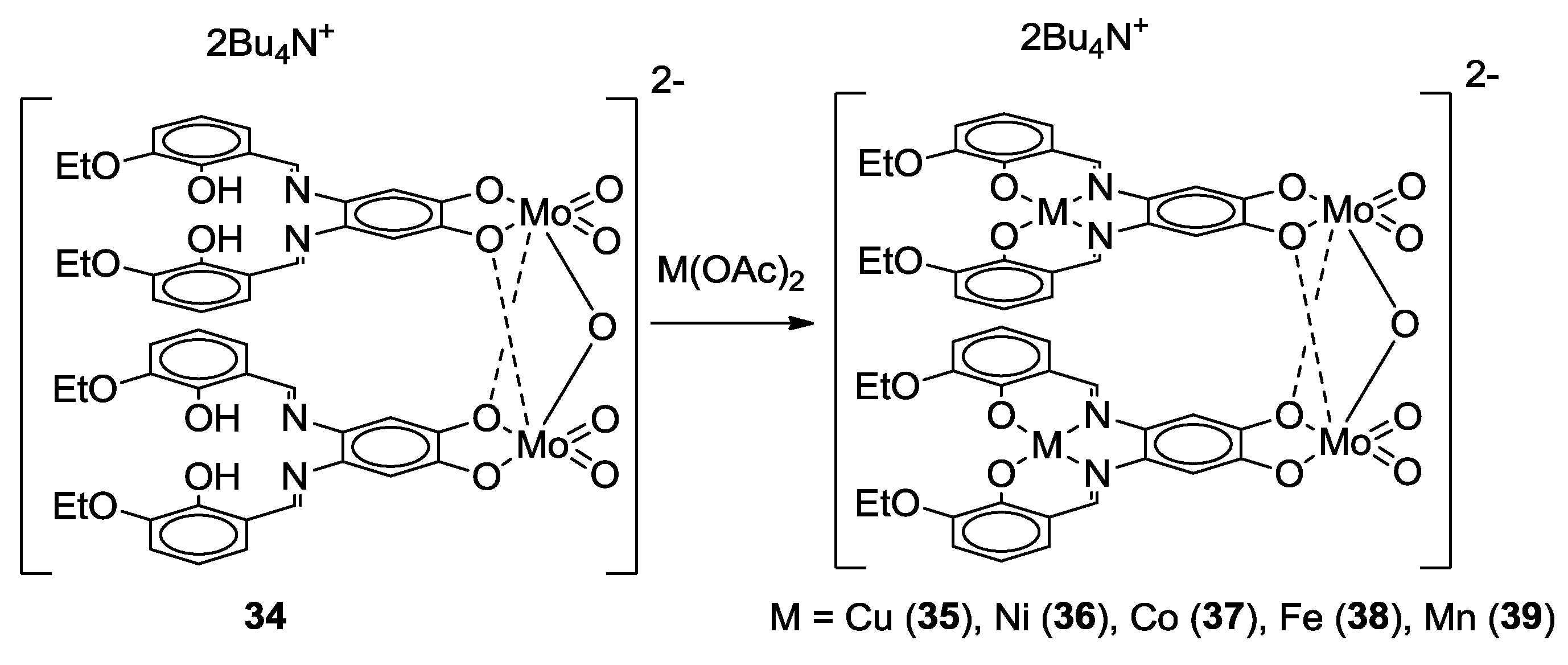
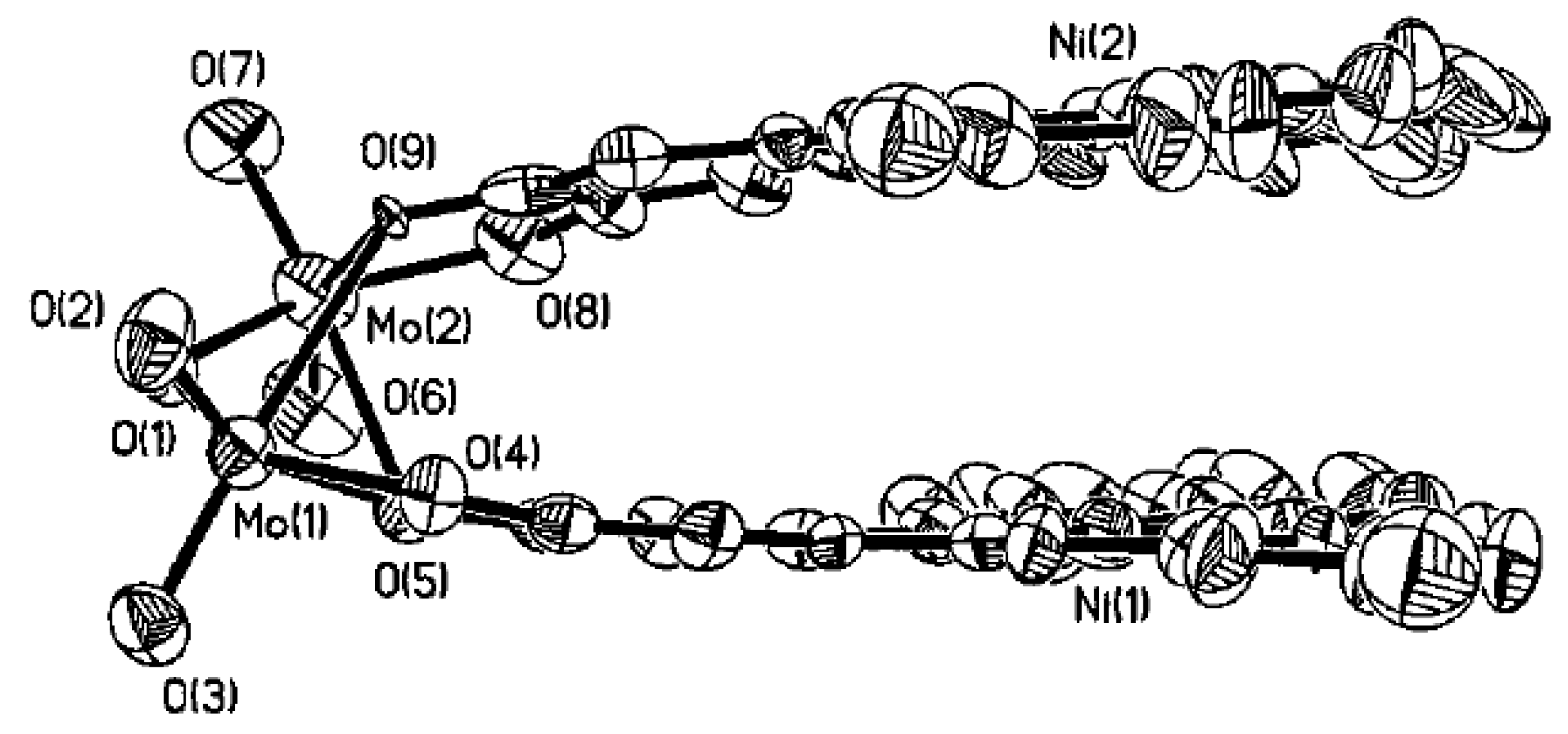
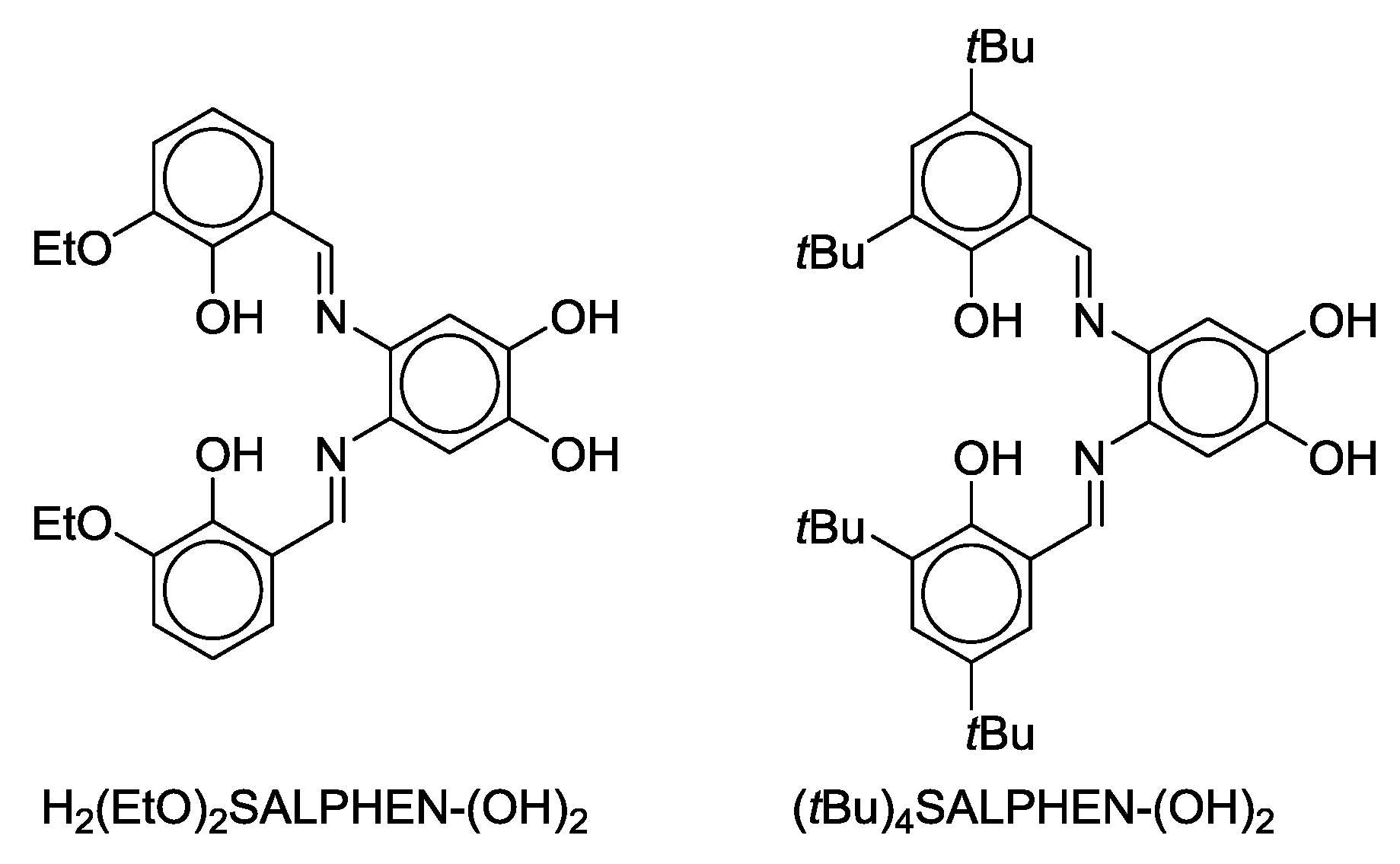
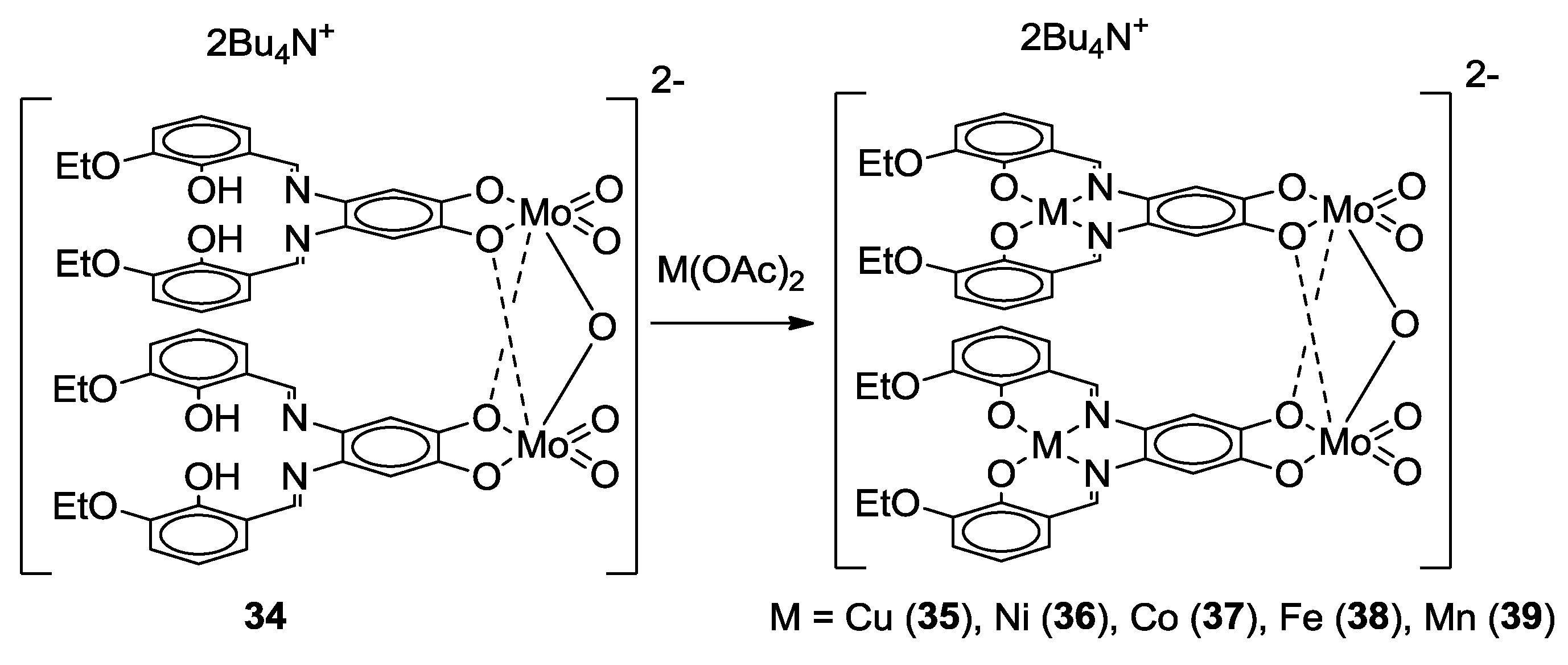
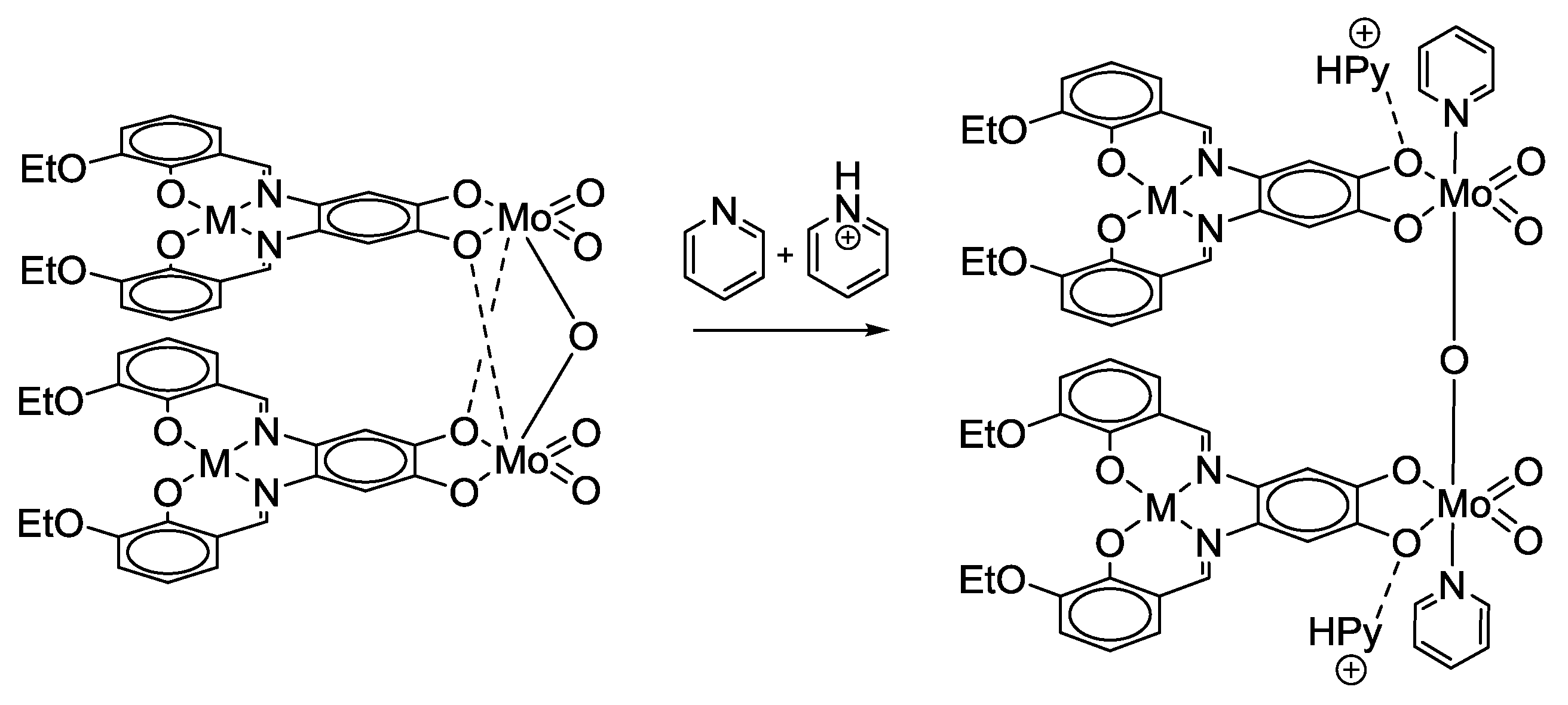
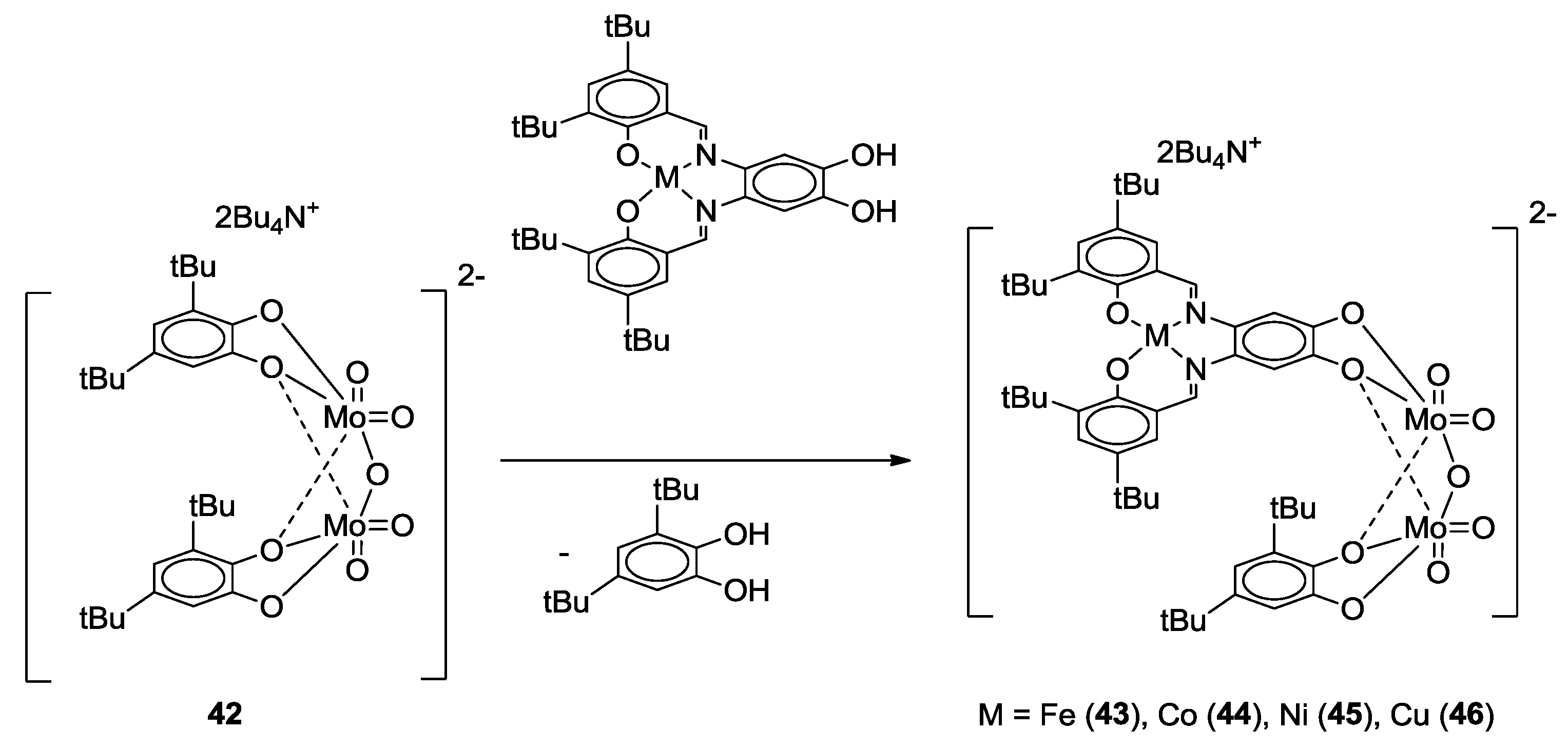
- Malinak, S.M.; Coucouvanis, D. Synthesis and Characterization of [Mo2O5]2+-Bridged Complexes Containing Cofacially-Oriented, Catechol-Functionalized Macrocyclic and SALPHEN Ligands. Inorg. Chem. 1996, 35, 4810–4811. [Google Scholar] [CrossRef] [PubMed]
- Malinak, S.M.; Rosa, D.T.; Coucouvanis, D. A New Class of Complexes Possessing Cofacially-Oriented, Planar, Metal-Containing Subunits. Synthesis, Characterization, and Reactivity of [(MoO2)2(μ-O)]2+-Linked, Catechol-Functionalized, Tetraazamacrocyclic and Salicylideneamine Complexes. Inorg. Chem. 1998, 37, 1175–1190. [Google Scholar] [CrossRef] [PubMed]
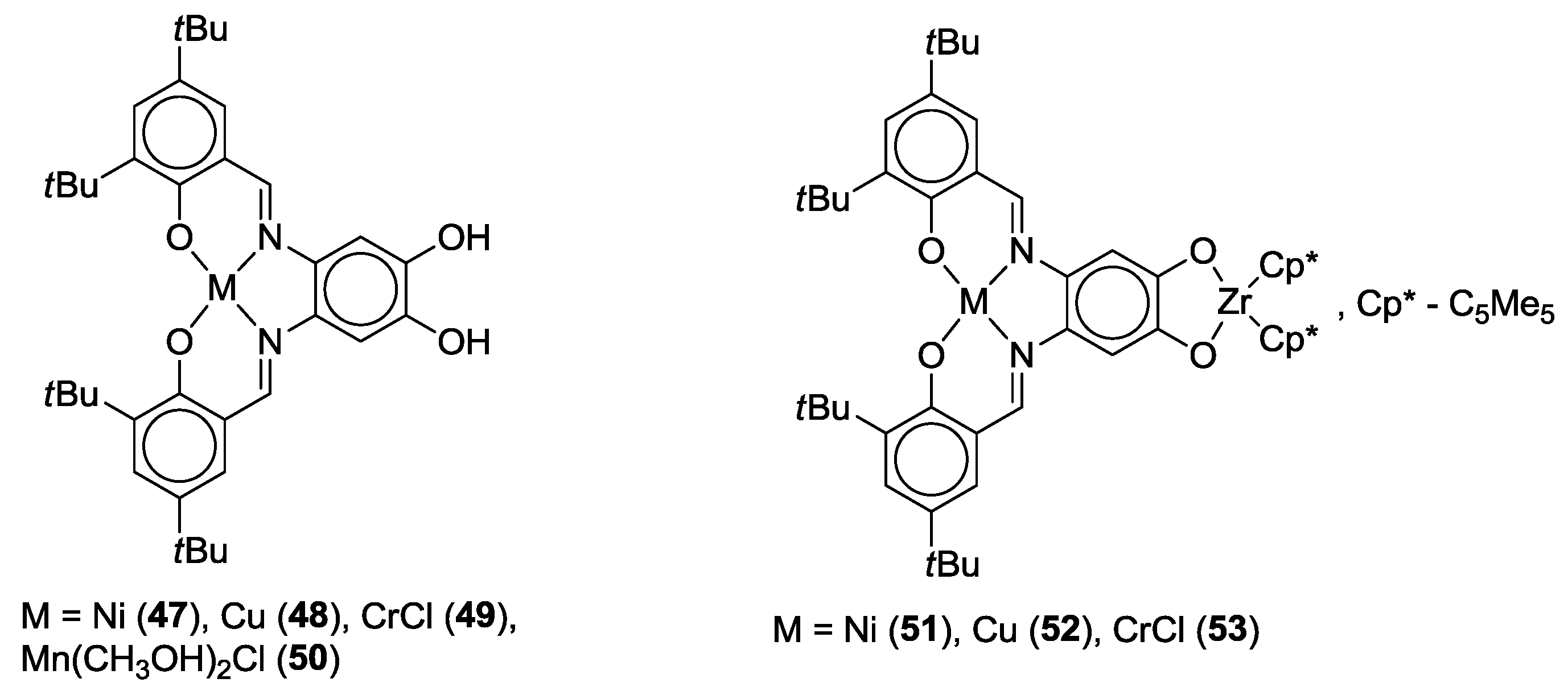
- Schley, M.; Lönnecke, P.; Hey-Hawkins, E. Monometallic and heterobimetallic complexes derived from salen-type ligands. J. Organomet. Chem. 2009, 694, 2480–2487. [Google Scholar] [CrossRef]
- Weber, B.; Obel, J. Synthesis and Characterisation of a New Schiff-Base-like Ligand and its Iron(II) Complexes. Zeitschrift für Anorganische und Allgemeine Chemie 2009, 635, 2474–2479. [Google Scholar] [CrossRef]
- Weber, B.; Obel, J.; Henner-Vásquez, D.; Bauer, W. Two New Iron(II) Spin-Crossover Complexes with N4O2 Coordination Sphere and Spin Transition around Room Temperature. Eur. J. Inorg. Chem. 2009, 2009, 5527–5534. [Google Scholar] [CrossRef]
- Bauer, W.; Lochenie, C.; Weber, B. Synthesis and characterization of 1D iron(II) spin crossover coordination polymers with hysteresis. Dalton Trans. 2014, 43, 1990–1999. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Obel, J.; Lorenz, L.R.; Bauer, W.; Carrella, L.; Rentschler, E. Control of Exchange Interactions in Trinuclear Complexes Based on Orthogonal Magnetic Orbitals. Eur. J. Inorg. Chem. 2009, 2009, 5535–5540. [Google Scholar] [CrossRef]
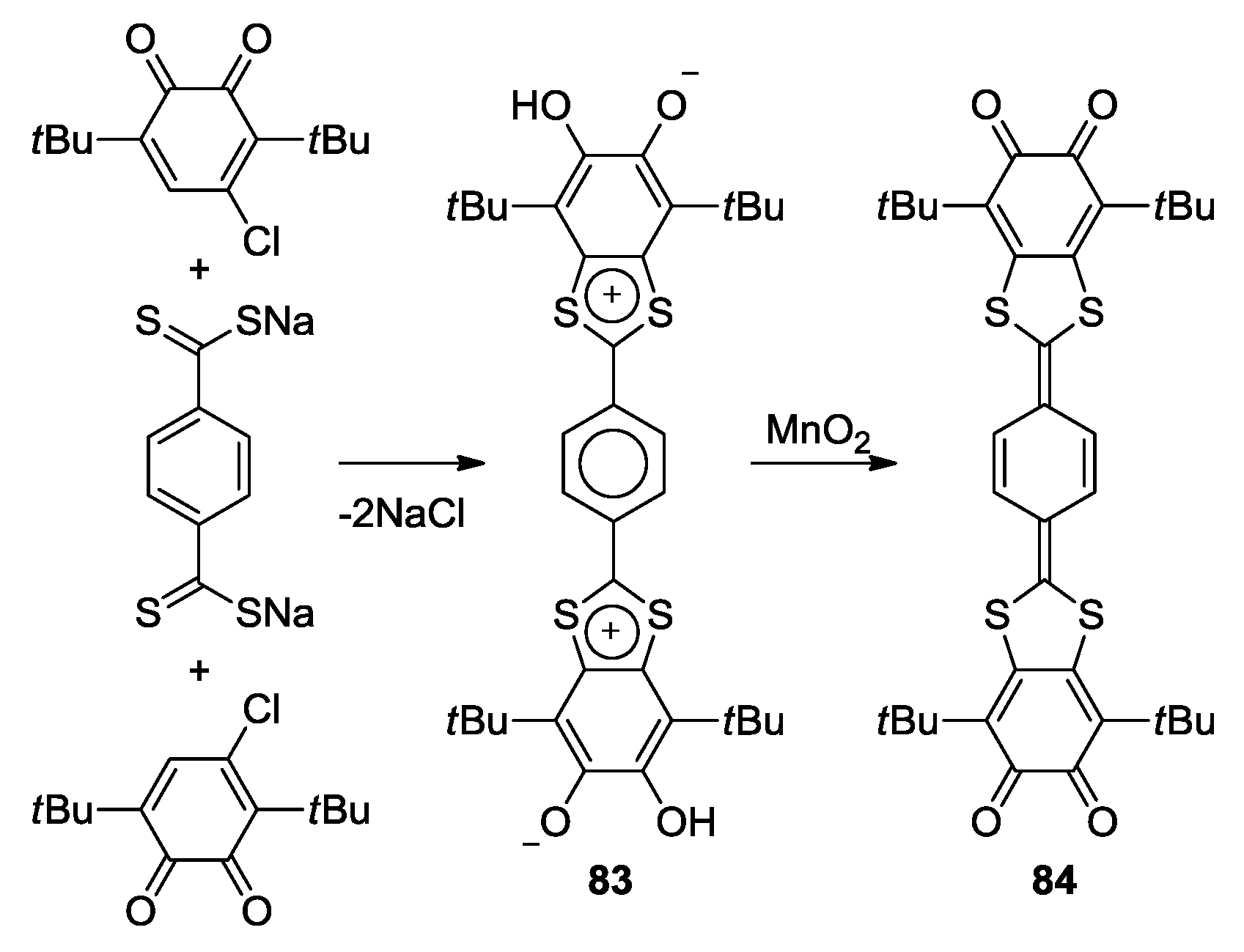
- Coucouvanis, D.; Paital, A.R.; Zhang, Q.; Lehnert, N.; Ahlrichs, R.; Fink, K.; Fenske, D.; Powell, A.K.; Lan, Y. Synthesis, electronic structure, and structural characterization of the new, “non-innocent” 4,5-dithio-catecholate ligand, its metal complexes, and their oxidized 4,5-dithio-o-quinone derivatives. Inorg. Chem. 2009, 48, 8830–8844. [Google Scholar] [CrossRef] [PubMed]
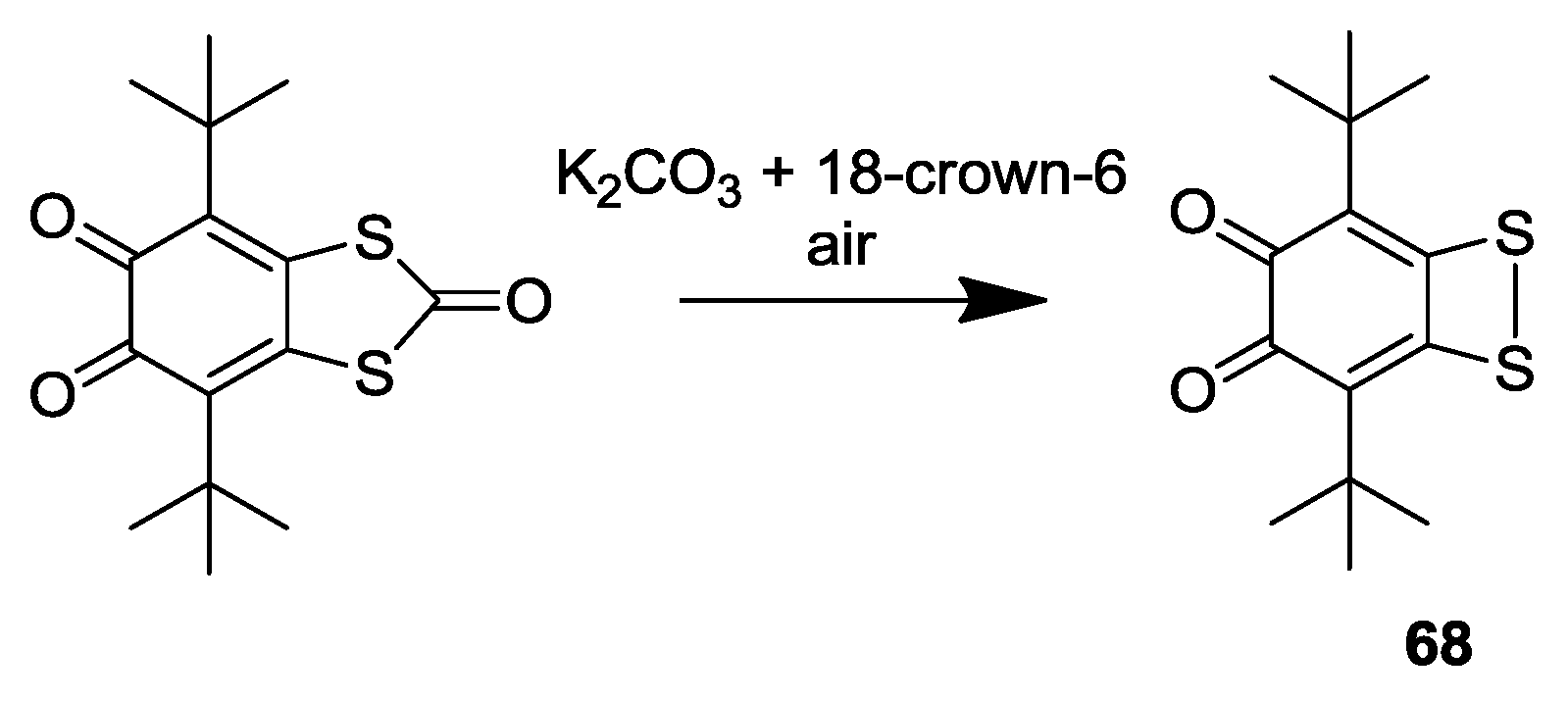
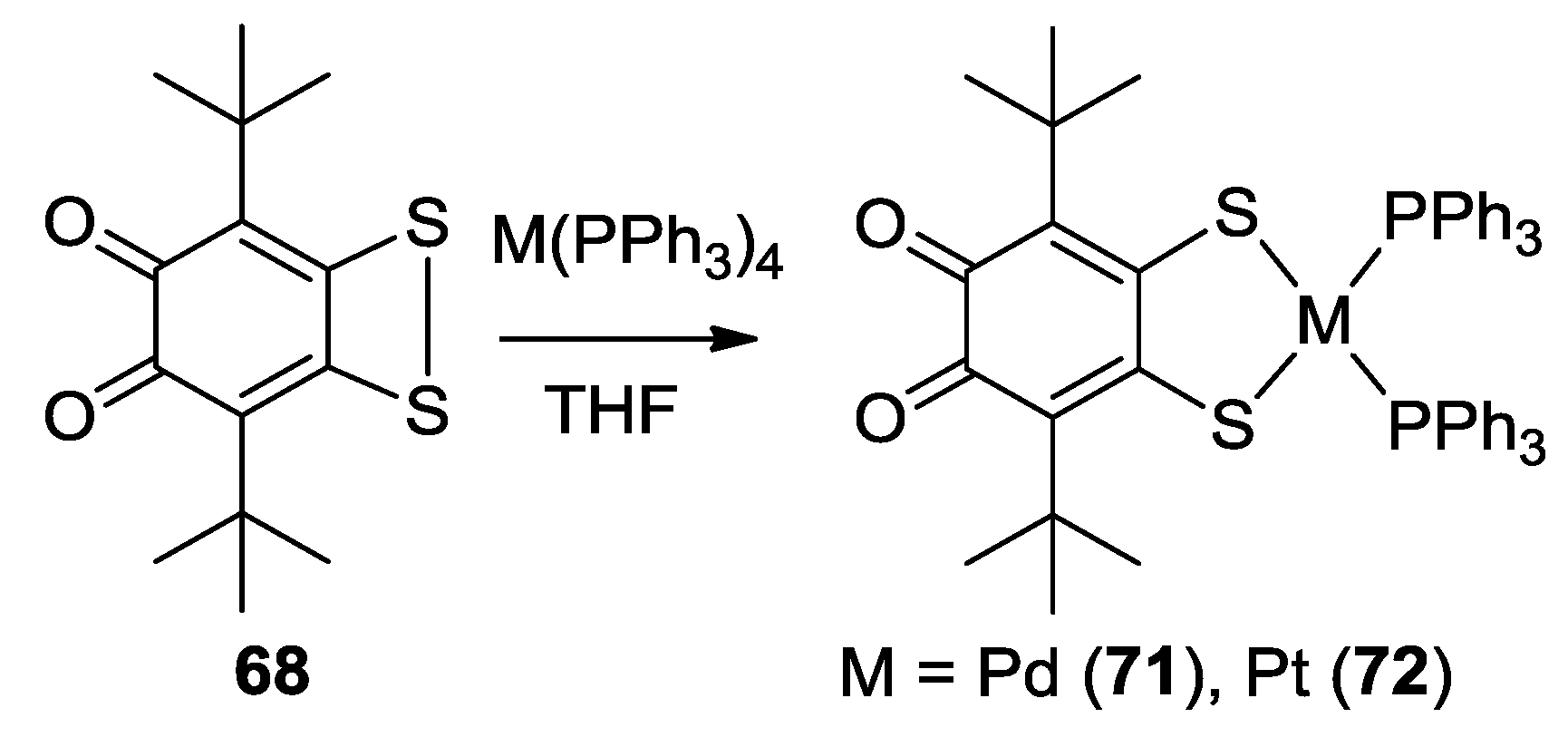
- Cherkasov, V.K.; Abakumov, G.A.; Fukin, G.K.; Klementyeva, S.V.; Kuropatov, V.A. Sterically Hindered o-Quinone Annulated with Dithiete: A Molecule Comprising Diolate and Dithiolate Coordination Sites. Chem. Eur. J. 2012, 18, 13821–13827. [Google Scholar] [CrossRef] [PubMed]
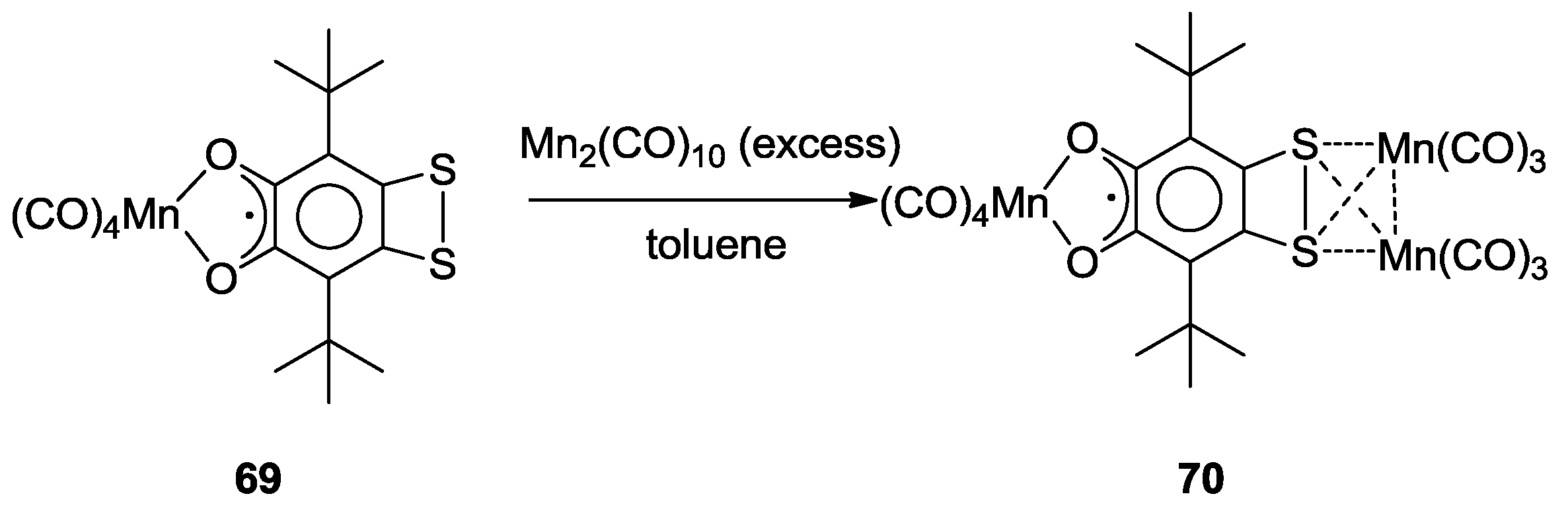
- Martyanov, K.A.; Cherkasov, V.K.; Abakumov, G.A.; Samsonov, M.A.; Khrizanforova, V.V.; Budnikova, Y.H.; Kuropatov, V.A. New sterically-hindered o-quinones annelated with metal-dithiolates: Regiospecificity in oxidative addition reactions of a bifacial ligand to the Pd and Pt complexes. Dalton Trans. 2016, 45, 7400–7405. [Google Scholar] [CrossRef] [PubMed]
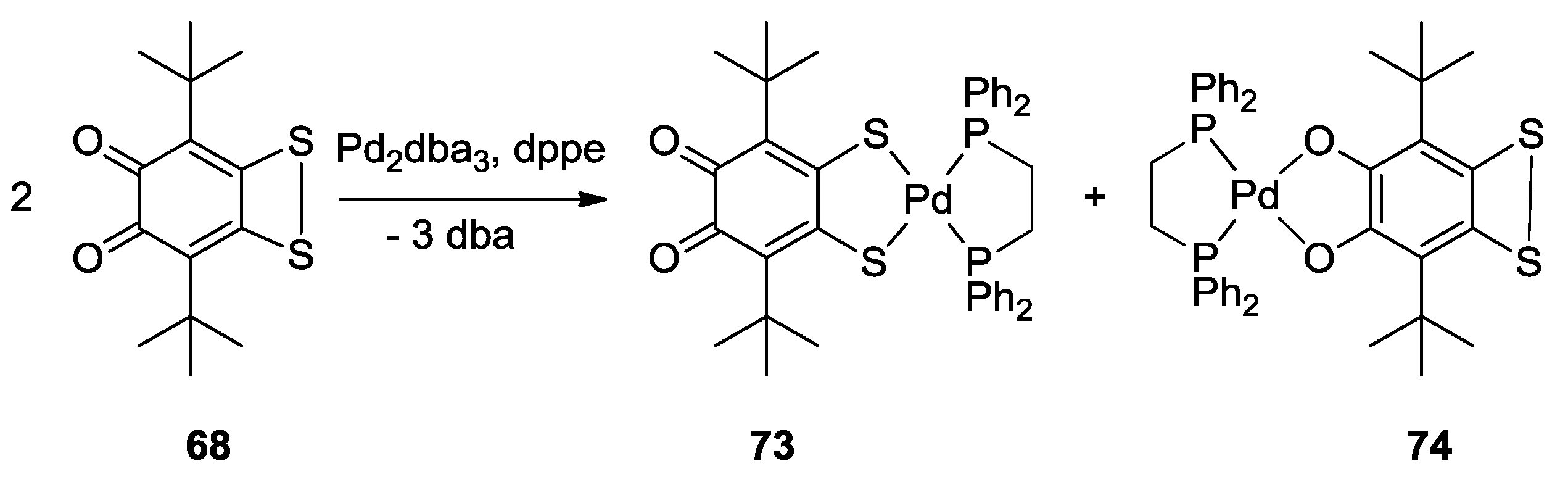
- Martyanov, K.A.; Cherkasov, V.K.; Abakumov, G.A.; Baranov, E.V.; Shavyrin, A.S.; Kuropatov, V.A. Regioisomerism in coordination chemistry: Oxidative addition of a bifunctional ligand to palladium, stabilized with 1,2-bis (diphenylphosphino)ethane. Dalton Trans. 2017, 46, 16783–16786. [Google Scholar] [CrossRef] [PubMed]
- Razuvaev, G.A.; Abakumov, G.A.; Teplova, I.A.; Shalnova, K.G.; Cherkasov, V.K. Palladium and platinum paramagnetic complexes formed by oxidation of catecholate derivatives of these elements. Inorg. Chim. Acta 1981, 53, L267-L269. [Google Scholar] [CrossRef]
- Perepichka, D.F.; Bryce, M.R.; Batsanov, A.S.; McInnes, E.J.L.; Zhao, J.P.; Farley, R.D. Engineering a remarkably low HOMO–LUMO gap by covalent linkage of a strong π-donor and a π-acceptor-tetrathiafulvalene-σ-polynitrofluorene diads: Their amphoteric redox behavior, electron transfer and spectroscopic properties. Chem. Eur. J. 2002, 8, 4656–4669. [Google Scholar] [CrossRef]
- Kuropatov, V.; Klementieva, S.; Fukin, G.; Mitin, A.; Ketkov, S.; Budnikova, Y.; Cherkasov, V.; Abakumov, G. Novel method for the synthesis of functionalized tetrathiafulvalenes, an acceptor–donor–acceptor molecule comprising of two o-quinone moieties linked by a TTF bridge. Tetrahedron 2010, 66, 7605–7611. [Google Scholar] [CrossRef]
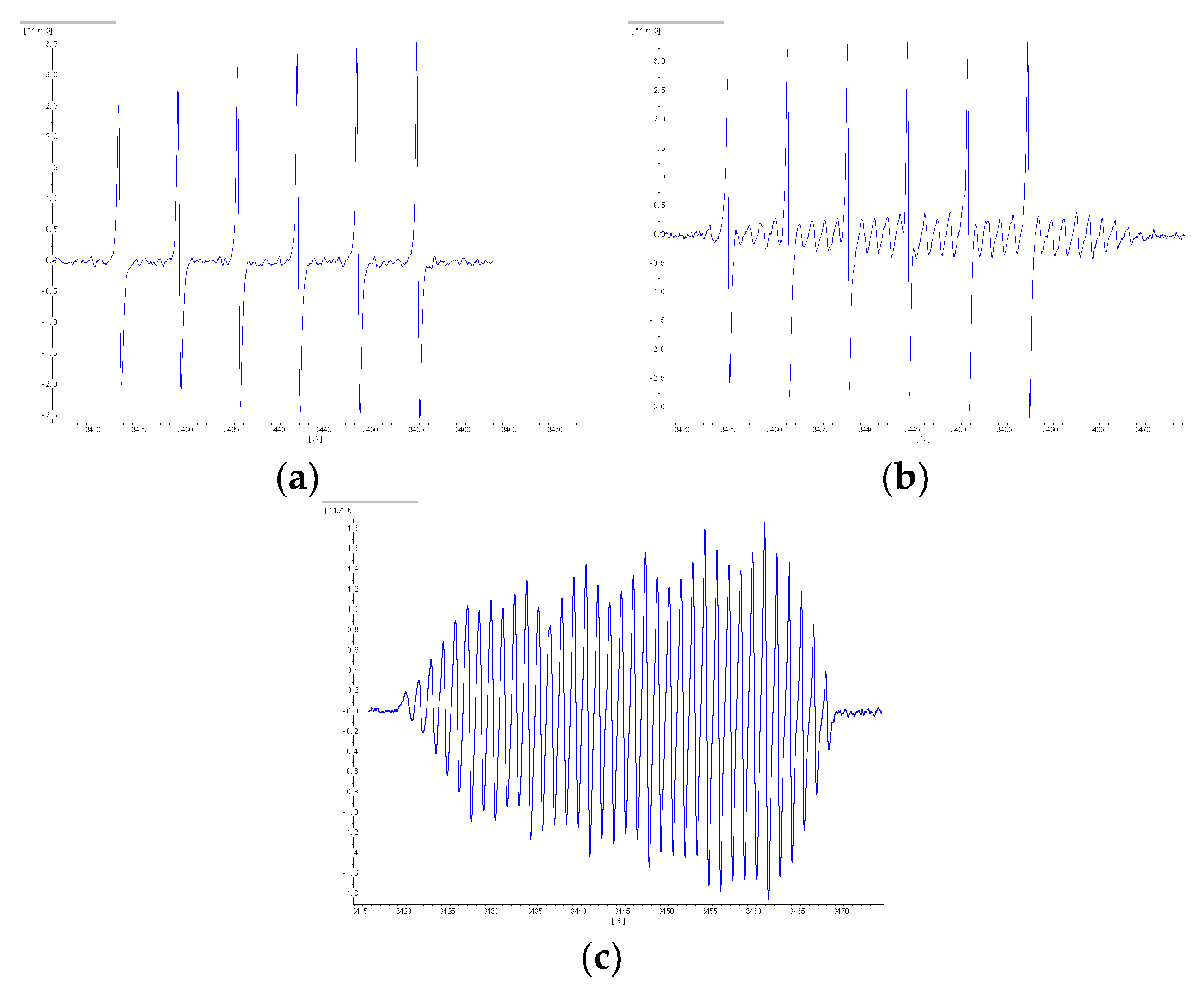
- Kuropatov, V.A.; Klementieva, S.V.; Poddel’sky, A.I.; Cherkasov, V.K.; Abakumov, G.A. ESR study of paramagnetic derivatives of sterically hindered di-o-quinone with the tetrathiafulvalene bridge. Russ. Chem. Bull. 2010, 59, 1698–1706. [Google Scholar] [CrossRef]
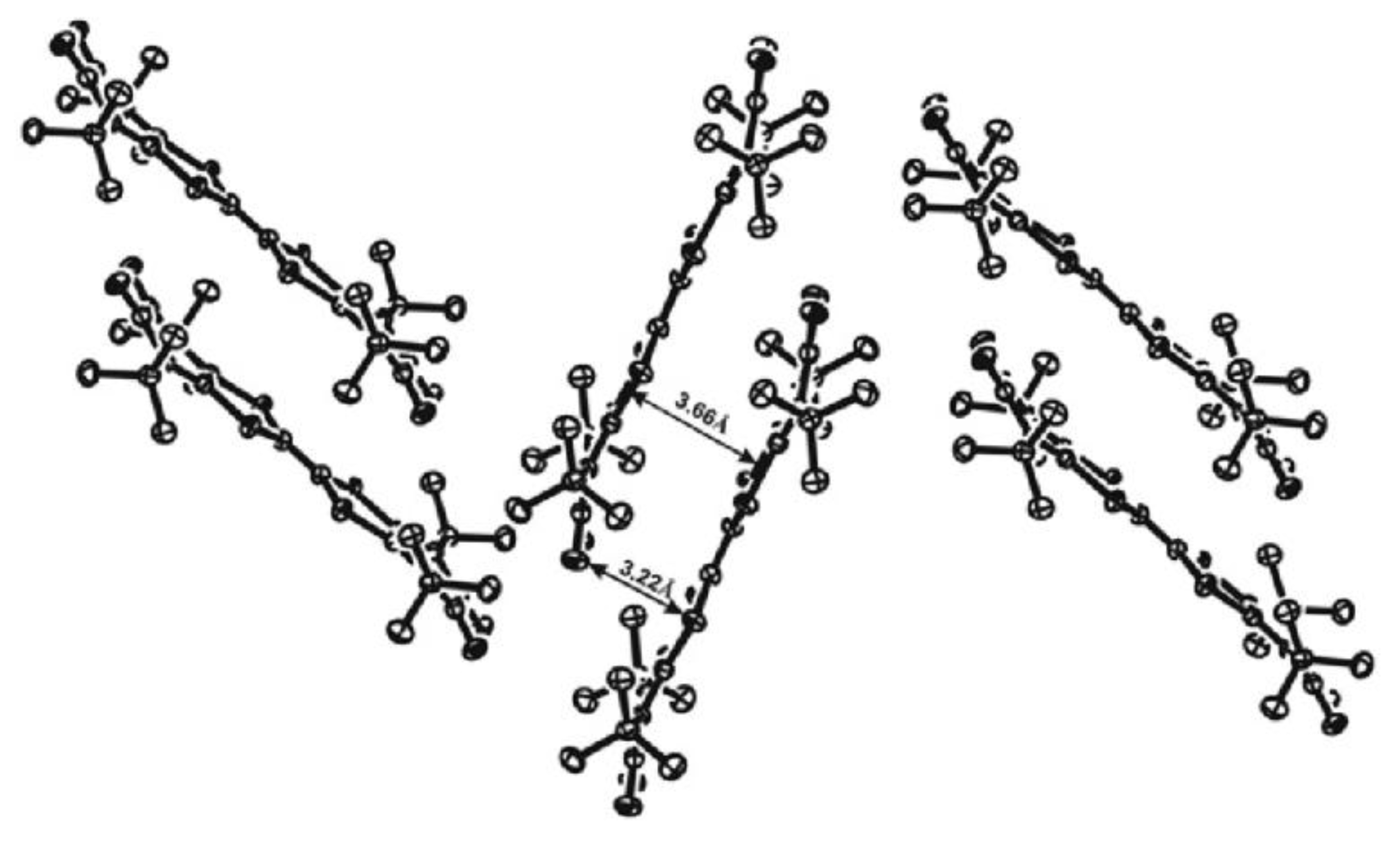
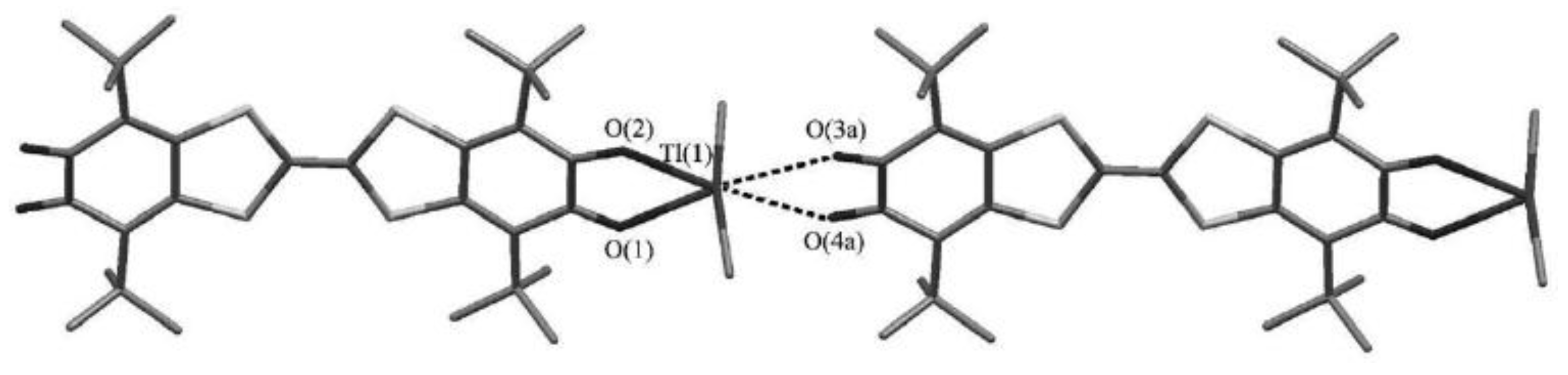
- Klementieva, S.V.; Kuropatov, V.A.; Fukin, G.K.; Romanenko, G.V.; Bogomyakov, A.S.; Cherkasov, V.K.; Abakumov, G.A. Mono- and Binuclear Dimethylthallium(III) Complexes with o-Benzoquinone-TTF-o-Benzoquinone Ligand; Synthesis, Spectroscopy and X-ray Study. Zeitschrift für Anorganische und Allgemeine Chemie 2011, 637, 232–241. [Google Scholar] [CrossRef]
- Pointillart, F.; Klementieva, S.; Kuropatov, V.; Le Gal, Y.; Golhen, S.; Cador, O.; Cherkasov, V.; Ouahab, L. A single molecule magnet behaviour in a D3h symmetry Dy(III) complex involving a quinone-tetrathiafulvalene-quinone bridge. Chem. Commun. 2012, 48, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; Kuropatov, V.; Mitin, A.; Maury, O.; Le Gal, Y.; Golhen, S.; Cador, O.; Cherkasov, V.; Ouahab, L. Lanthanide-Based Dinuclear Complexes Involving an o-Quinone–Tetrathiafulvalene–o-Quinone Bridging Ligand: X-ray Structures, Magnetic and Photophysical Properties. Eur. J. Inorg. Chem. 2012, 2012, 4708–4718. [Google Scholar] [CrossRef]
- Yamashita, Y.; Kobayashi, Y.; Miyashi, T. p-Quinodimethane Analogues of Tetrathiafulvalene. Angew. Chem. Int. Ed. Engl. 1989, 28, 1052–1053. [Google Scholar] [CrossRef]
- Chalkov, N.O.; Cherkasov, V.K.; Abakumov, G.A.; Romanenko, G.V.; Ketkov, S.Y.; Smolyaninov, I.V.; Starikov, A.G.; Kuropatov, V.A. Compactly Fused o-Quinone-Extended Tetrathiafulvalene-o-Quinone Triad—A Redox-Amphoteric Ligand. Eur. J. Org. Chem. 2014, 2014, 4571–4576. [Google Scholar] [CrossRef]
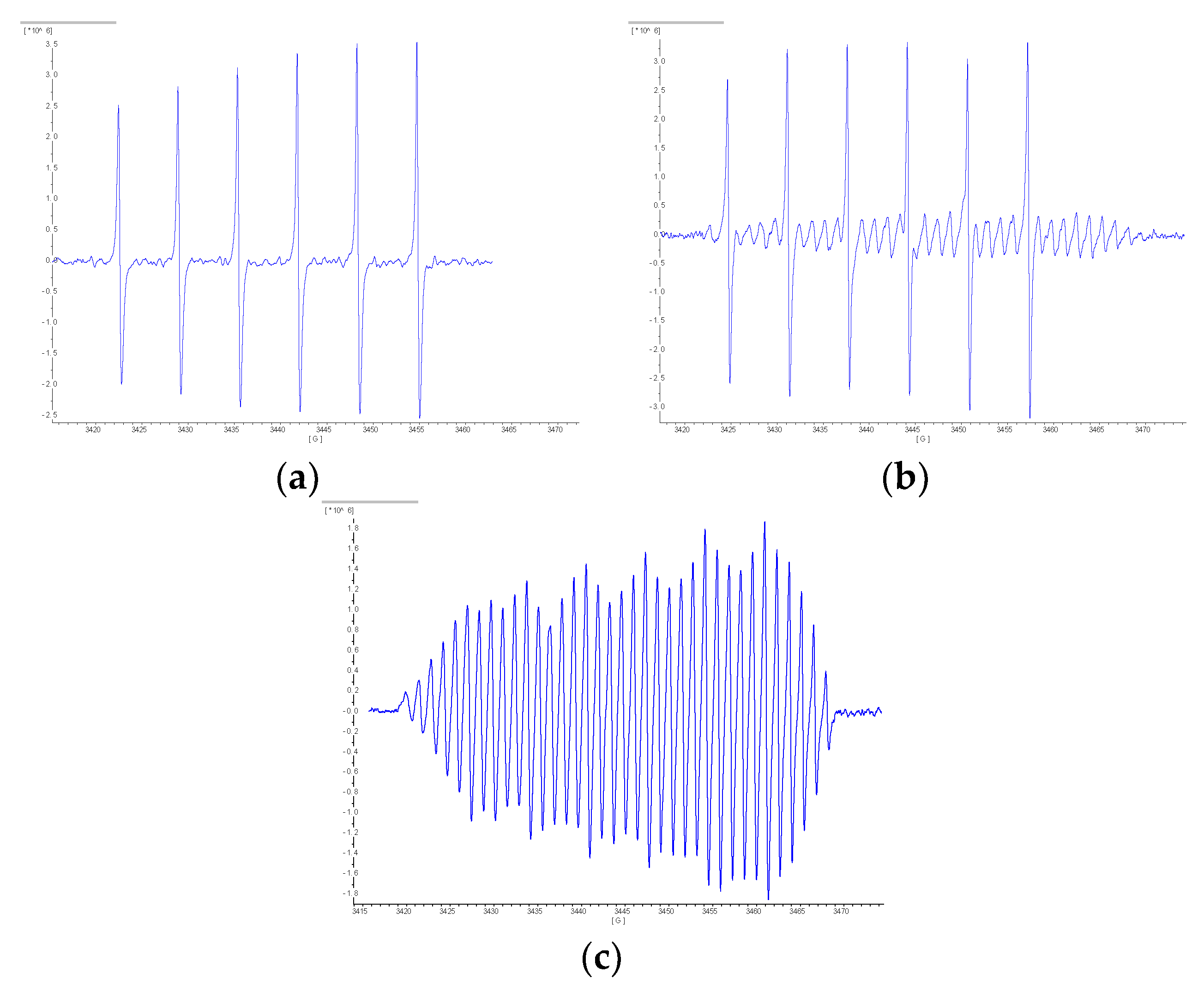
- Chalkov, N.O.; Cherkasov, V.K.; Abakumov, G.A.; Starikov, A.G.; Kuropatov, V.A. EPR spectroscopy study of di-o-quinone bridged by π-extended TTF: Redox behavior and binding modes as a ligand. New J. Chem. 2016, 40, 1244–1249. [Google Scholar] [CrossRef]
- Chalkov, N.O.; Cherkasov, V.K.; Abakumov, G.A.; Starikov, A.G.; Kuropatov, V.A. Protonated paramagnetic redox forms of di-o-quinone bridged with p-phenylene-extended TTF: A EPR spectroscopy study. Beilstein J. Org. Chem. 2016, 12, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.B.; Day, P. Mixed Valence Chemistry-A Survey and Classification. Adv. Inorg. Chem. Radiochem. 1967, 10, 247–422. [Google Scholar]






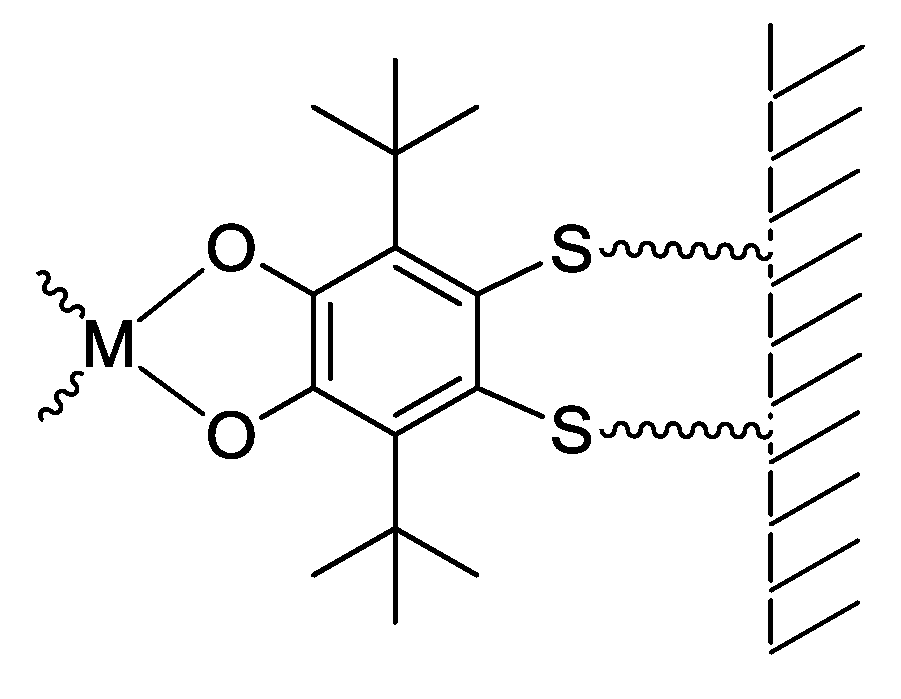
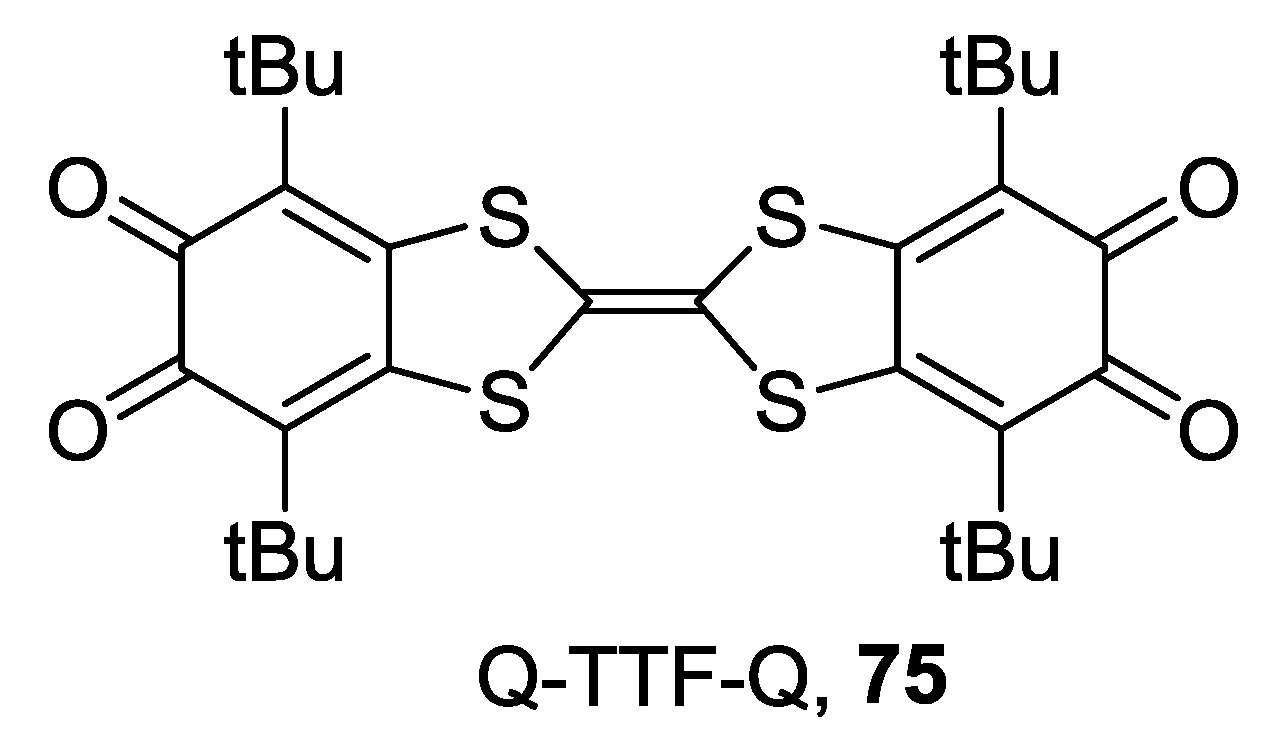

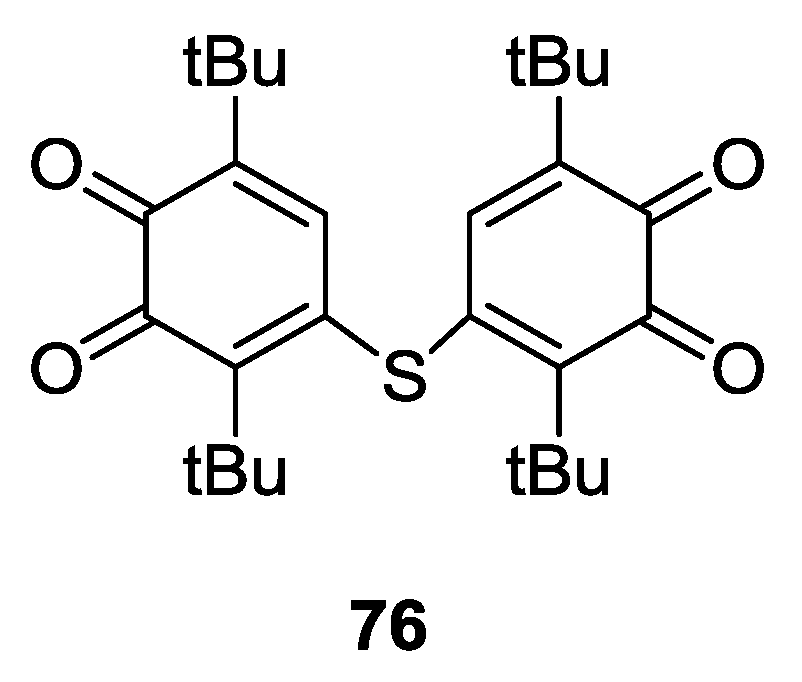


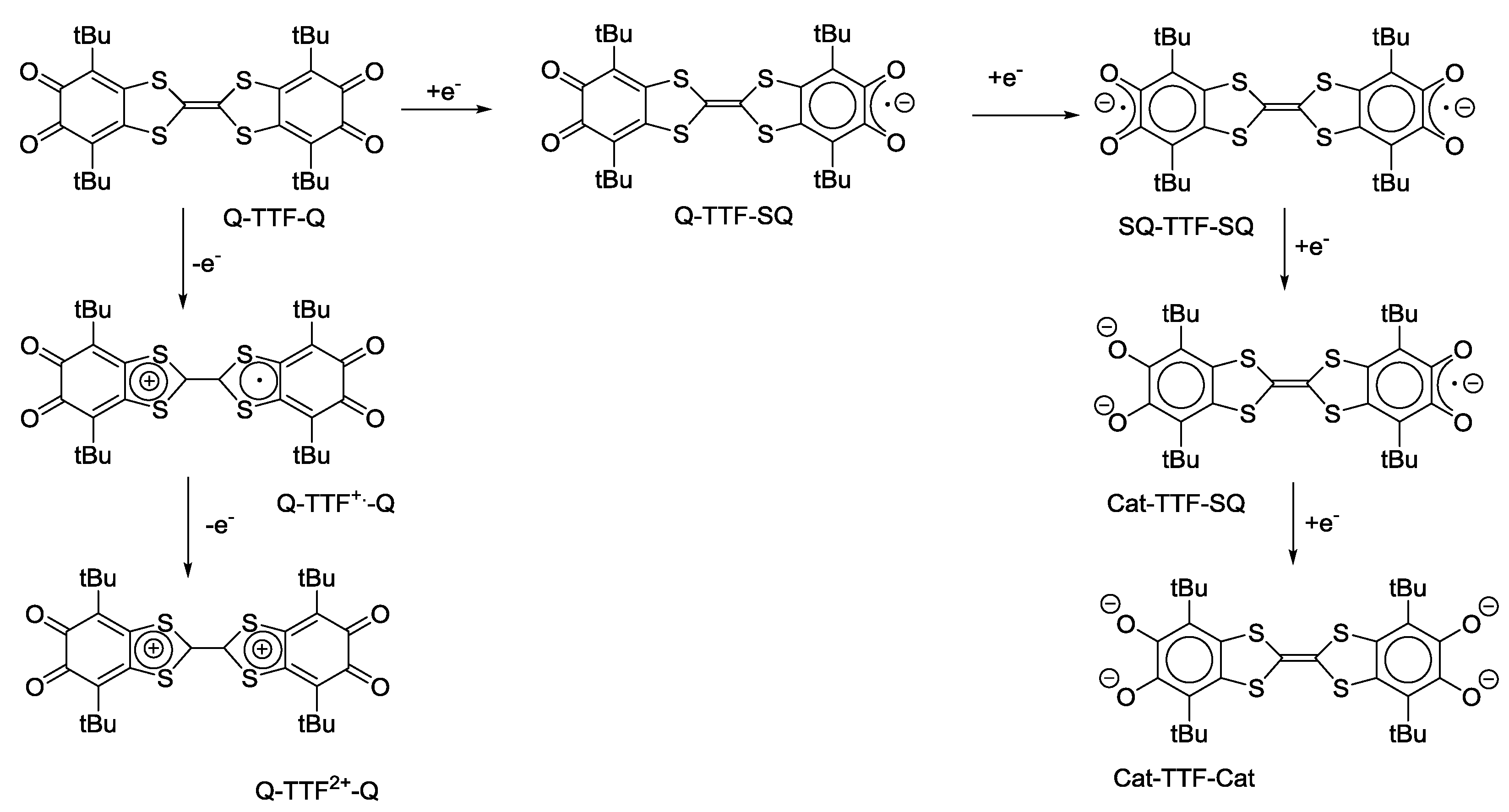

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Redox State | Coordination Mode | |
|---|---|---|
| Bridging | Terminating | |
| 0 | 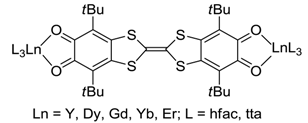 | - |
| −1 | - |  |
| −2 | 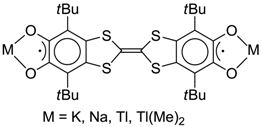 | 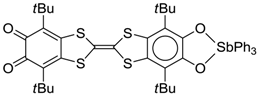 |
| −3 | 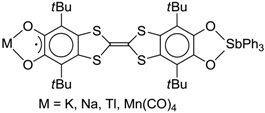 | - |
| −4 |  | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martyanov, K.; Kuropatov, V. Functionalized o-Quinones: Concepts, Achievements and Prospects. Inorganics 2018, 6, 48. https://doi.org/10.3390/inorganics6020048
Martyanov K, Kuropatov V. Functionalized o-Quinones: Concepts, Achievements and Prospects. Inorganics. 2018; 6(2):48. https://doi.org/10.3390/inorganics6020048
Chicago/Turabian StyleMartyanov, Konstantin, and Viacheslav Kuropatov. 2018. "Functionalized o-Quinones: Concepts, Achievements and Prospects" Inorganics 6, no. 2: 48. https://doi.org/10.3390/inorganics6020048
APA StyleMartyanov, K., & Kuropatov, V. (2018). Functionalized o-Quinones: Concepts, Achievements and Prospects. Inorganics, 6(2), 48. https://doi.org/10.3390/inorganics6020048