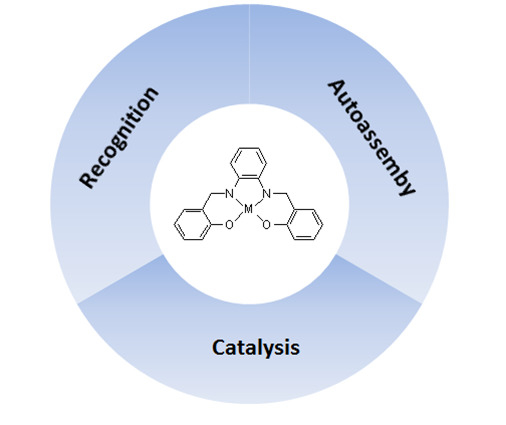
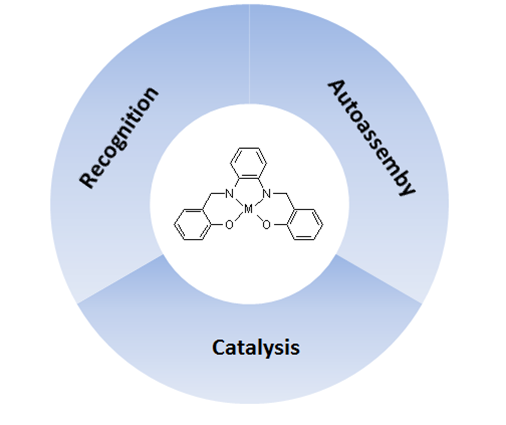
The Supramolecular Attitude of Metal–Salophen and Metal–Salen Complexes



Abstract
:
1. Introduction
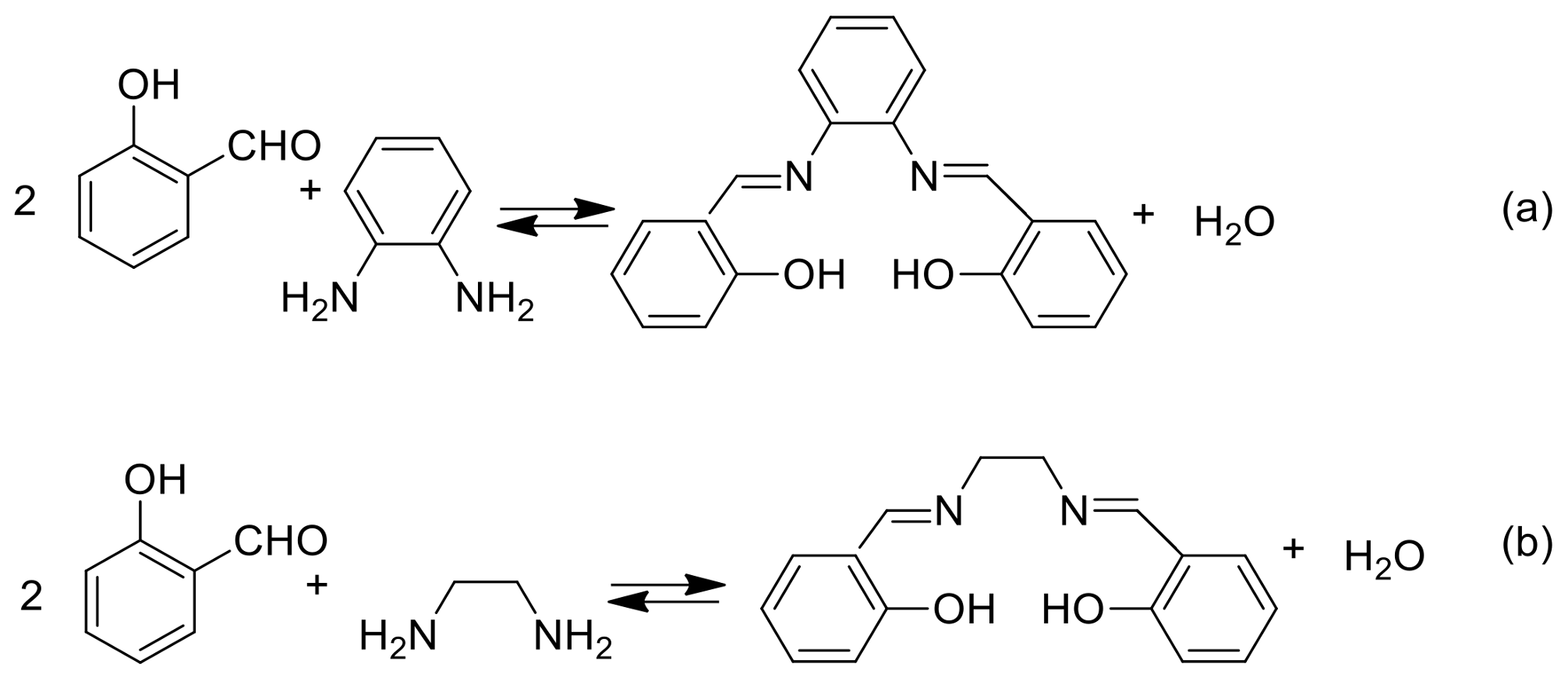
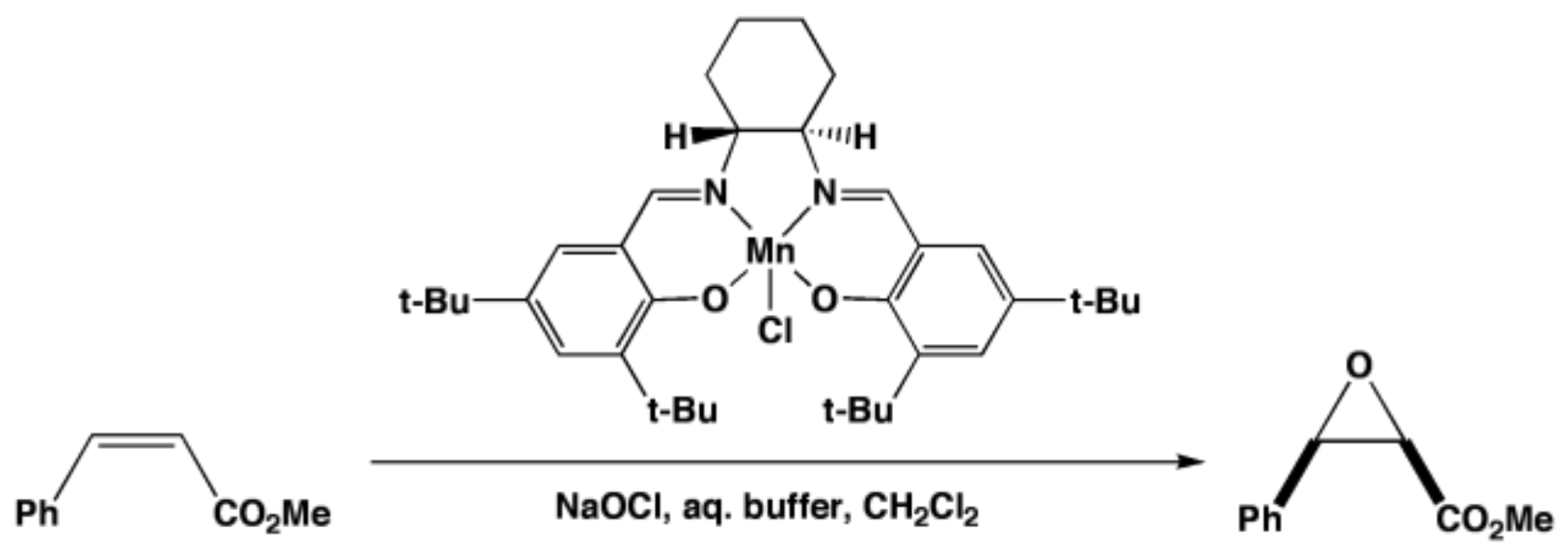
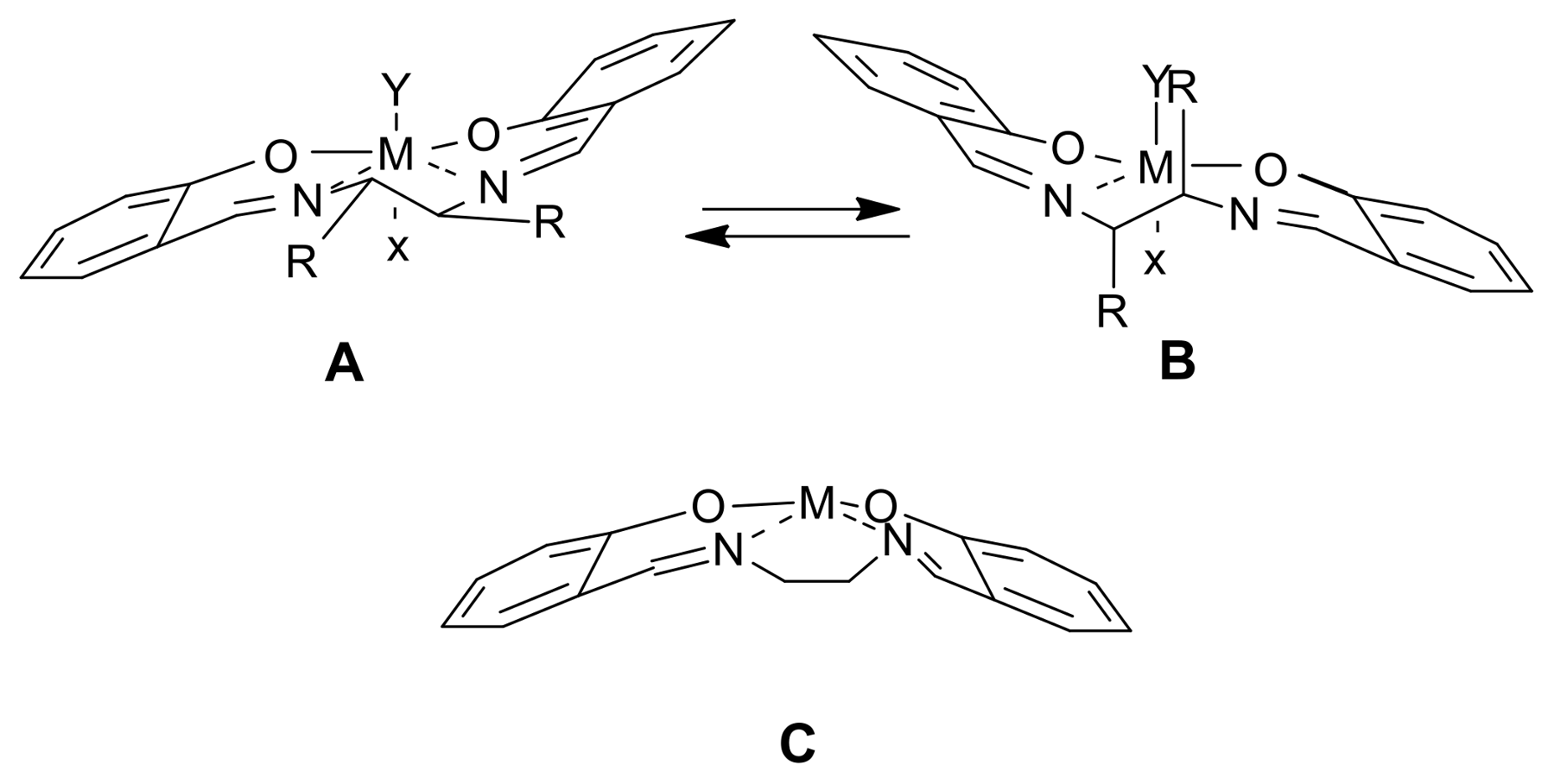
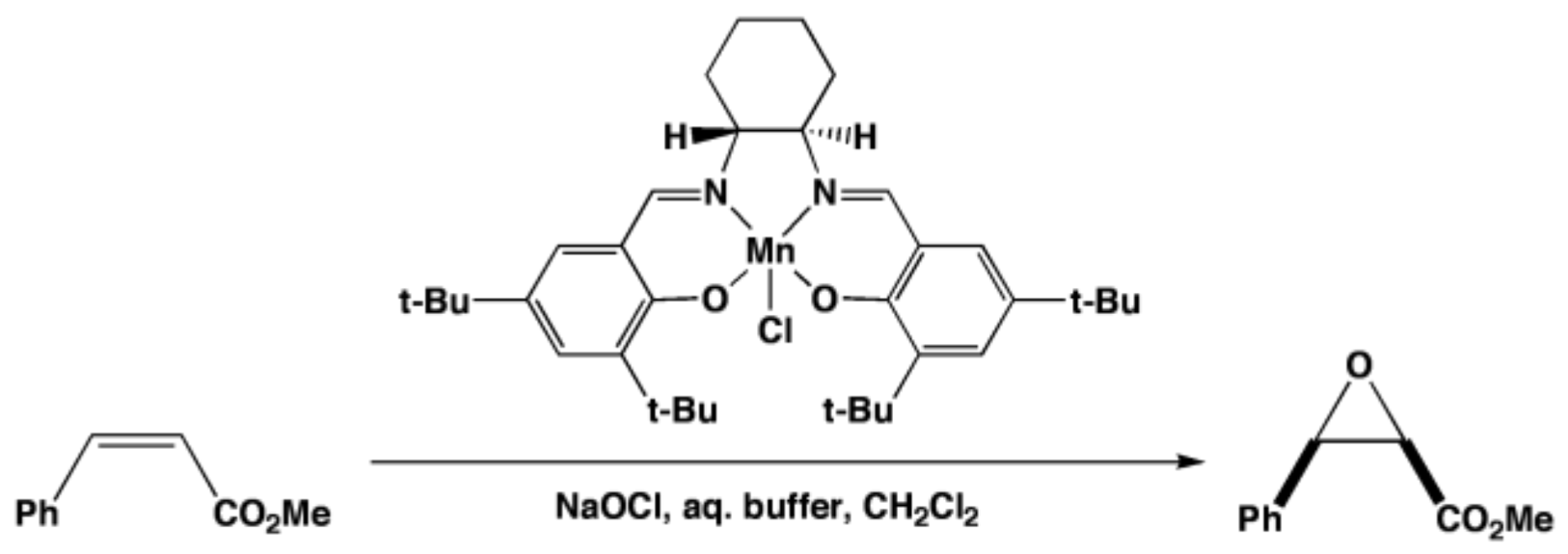
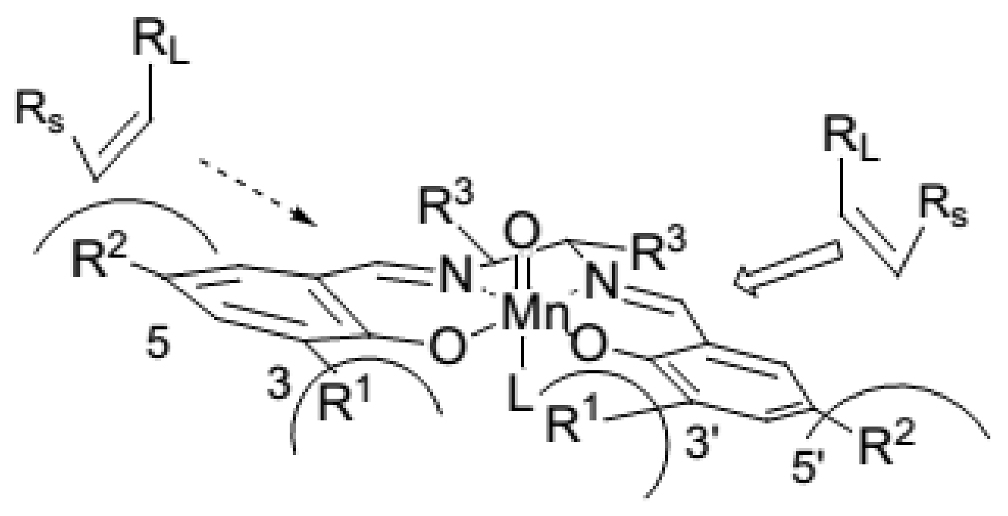
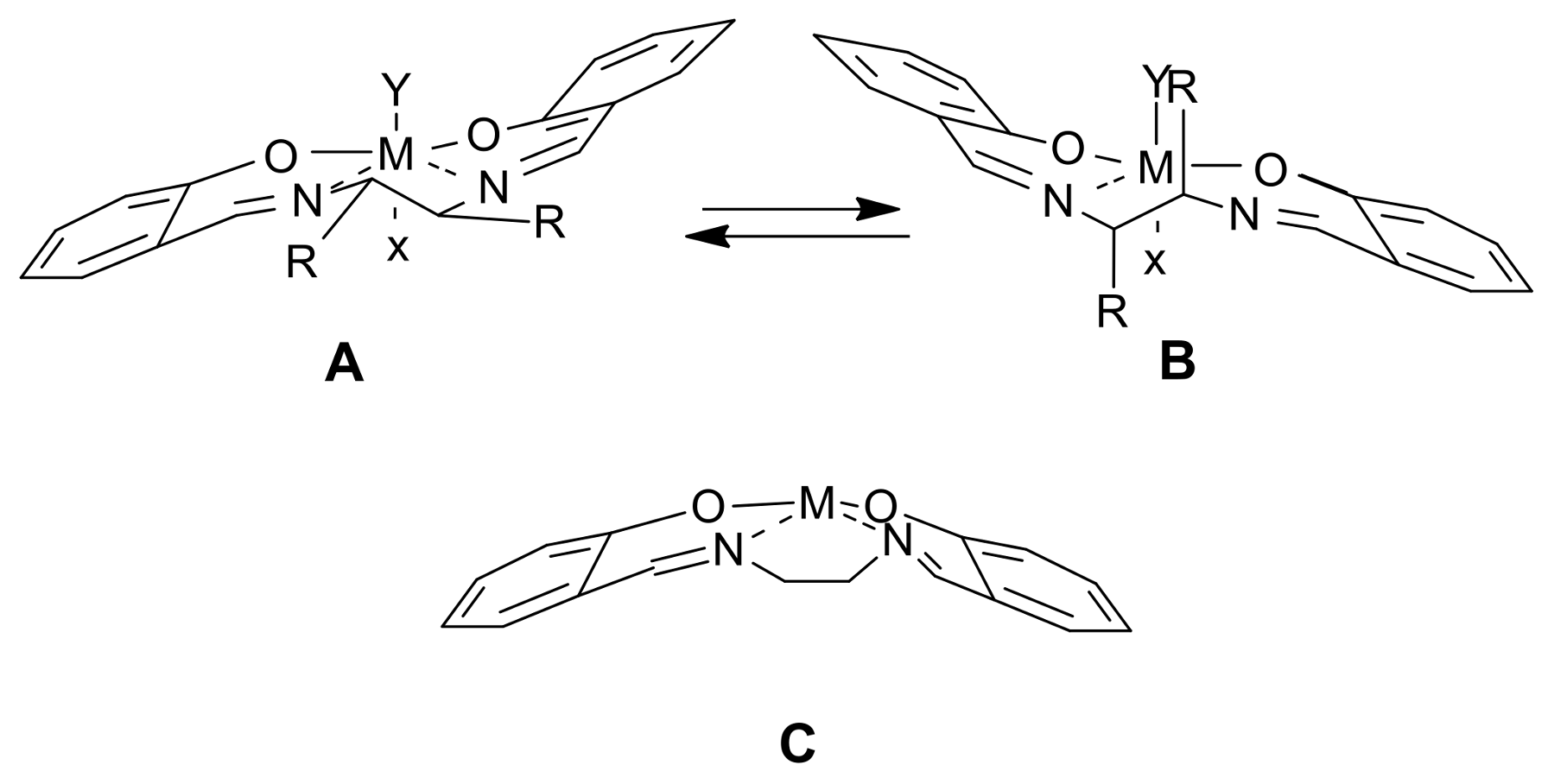
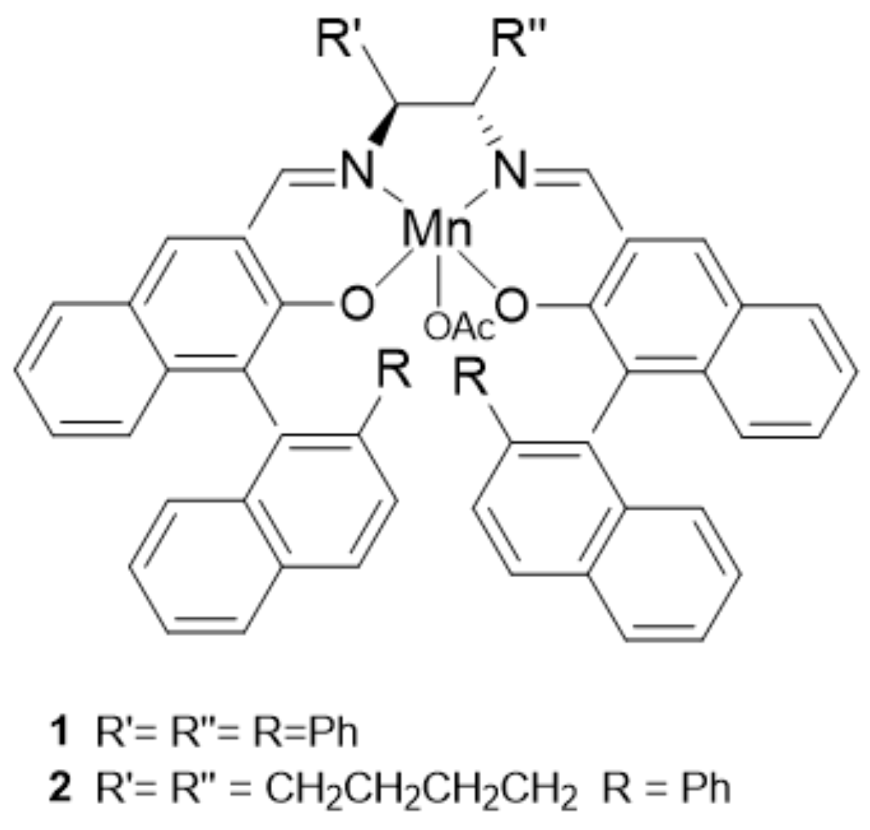
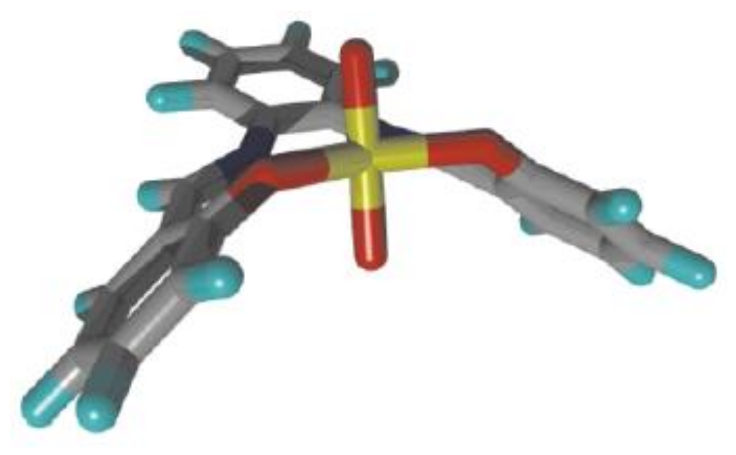
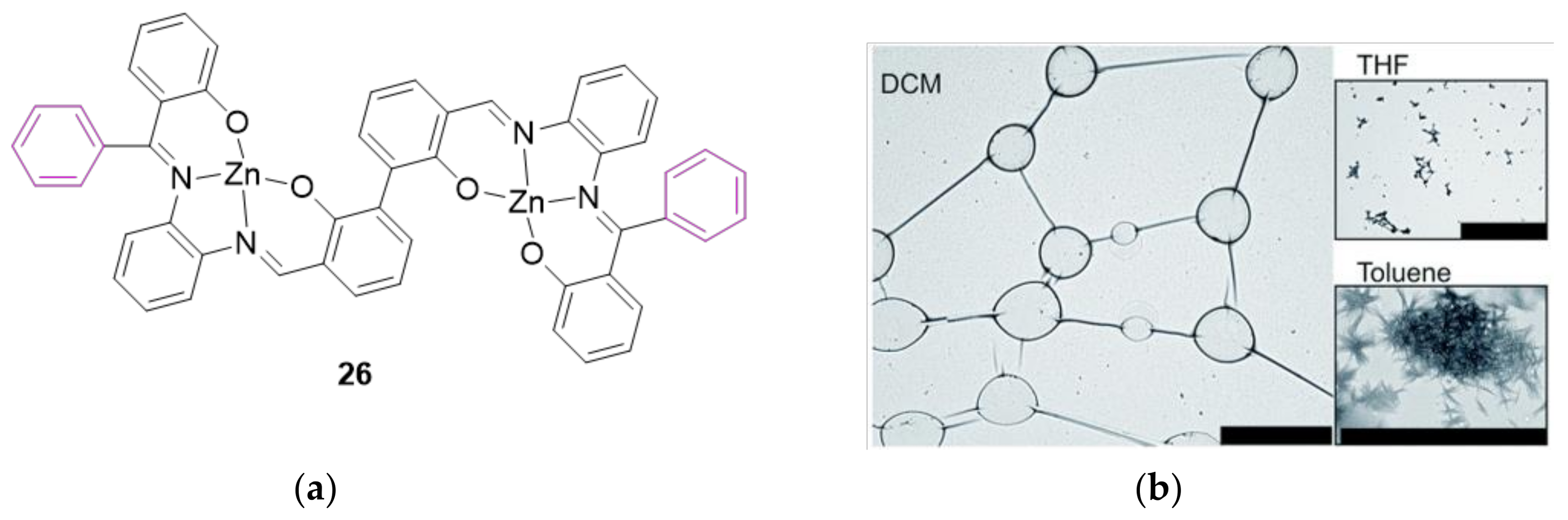
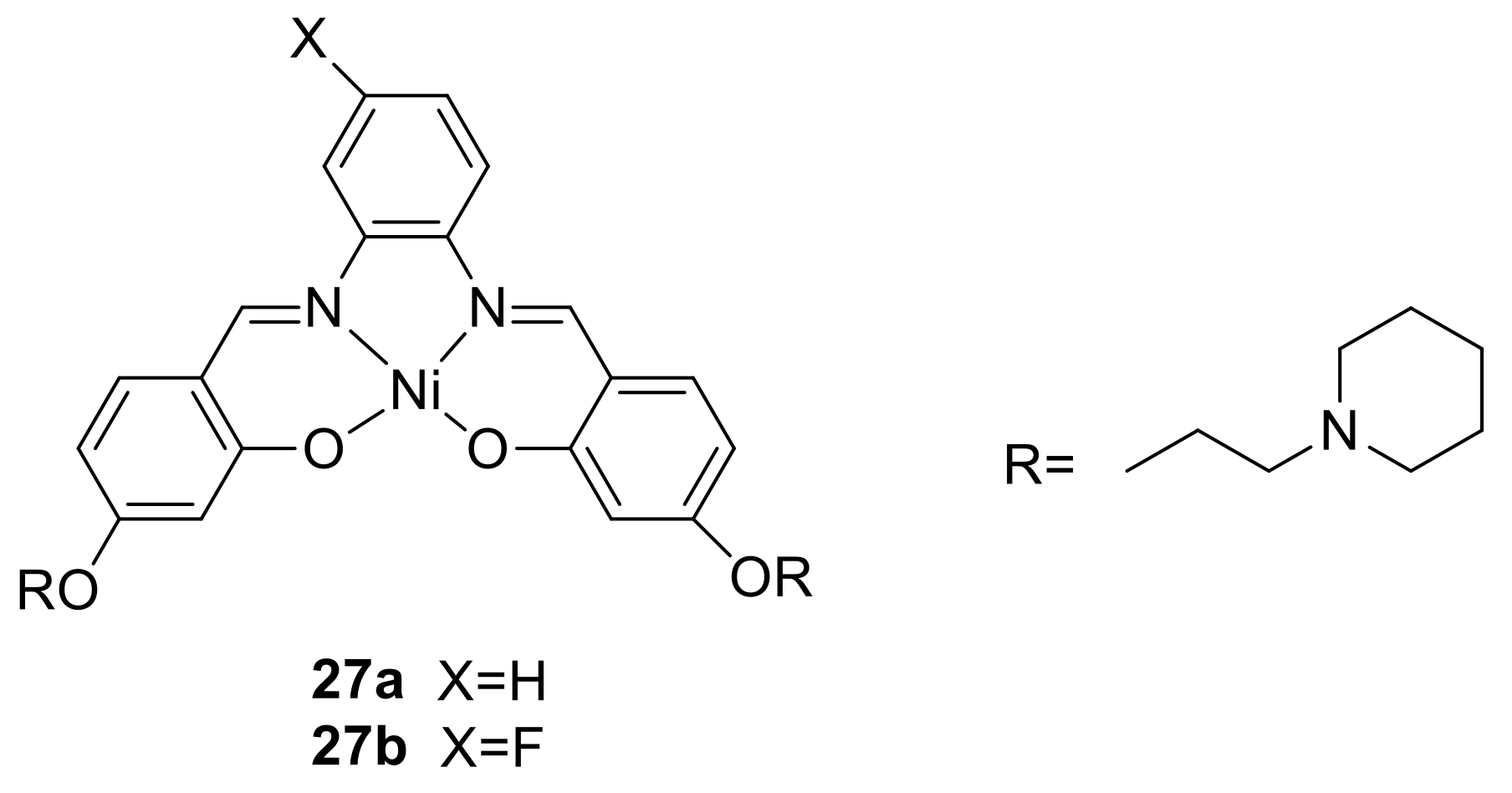
2. A Brief Survey
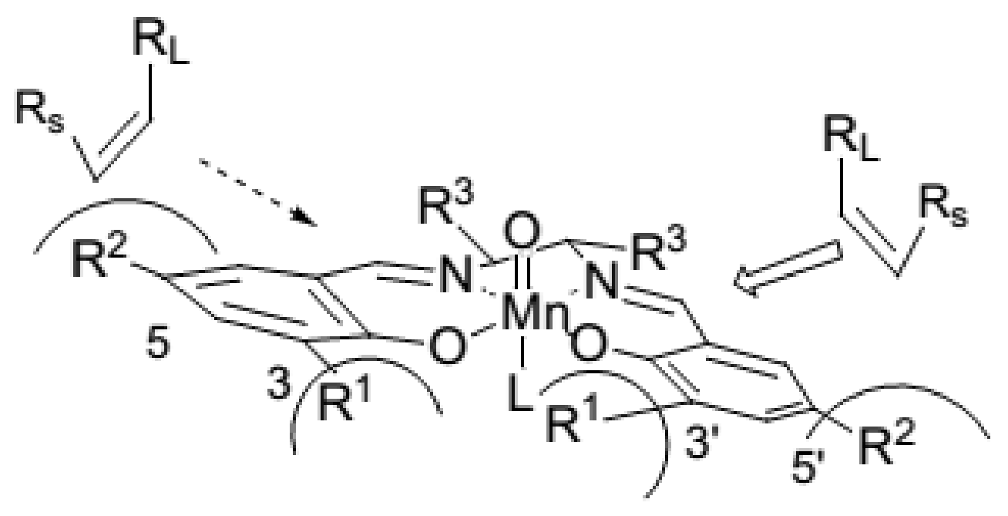
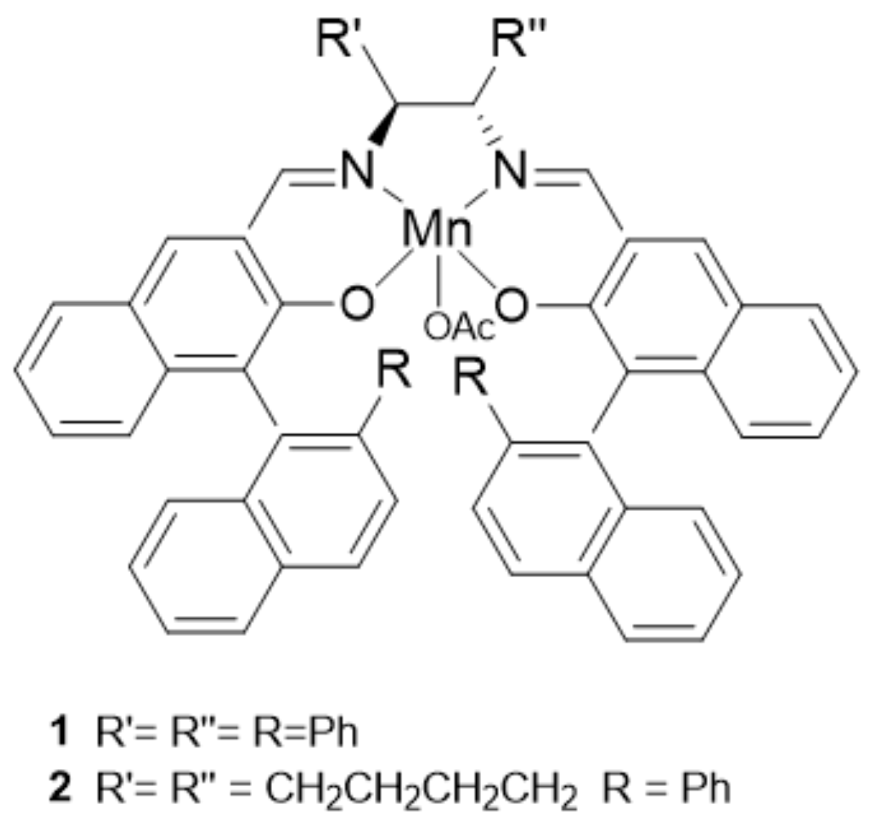
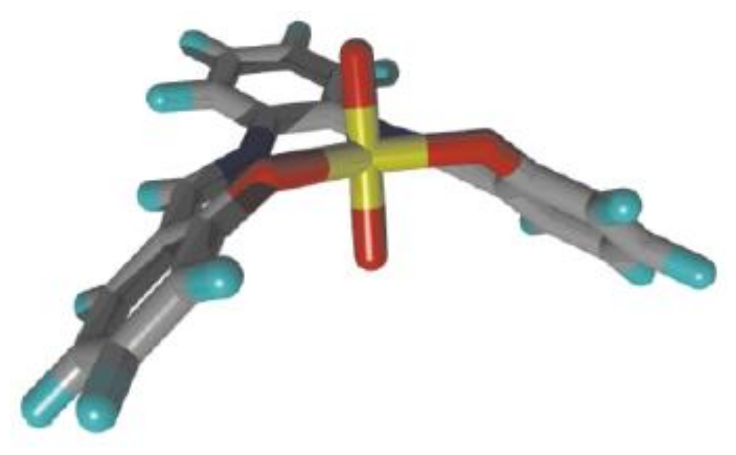
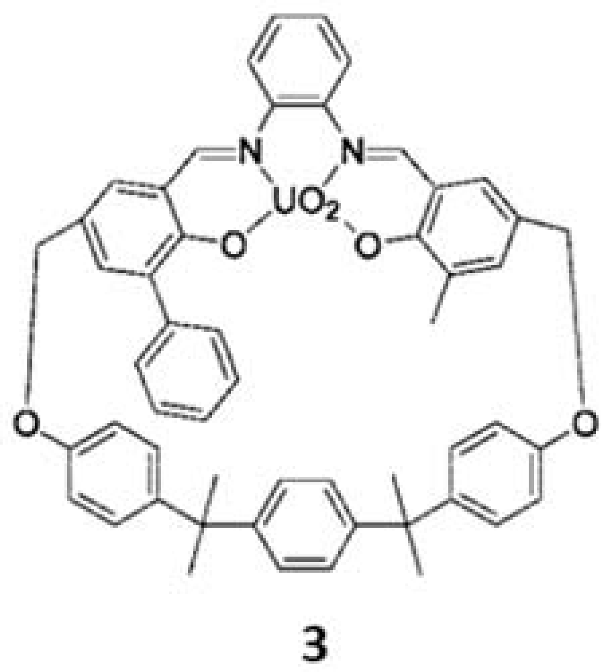
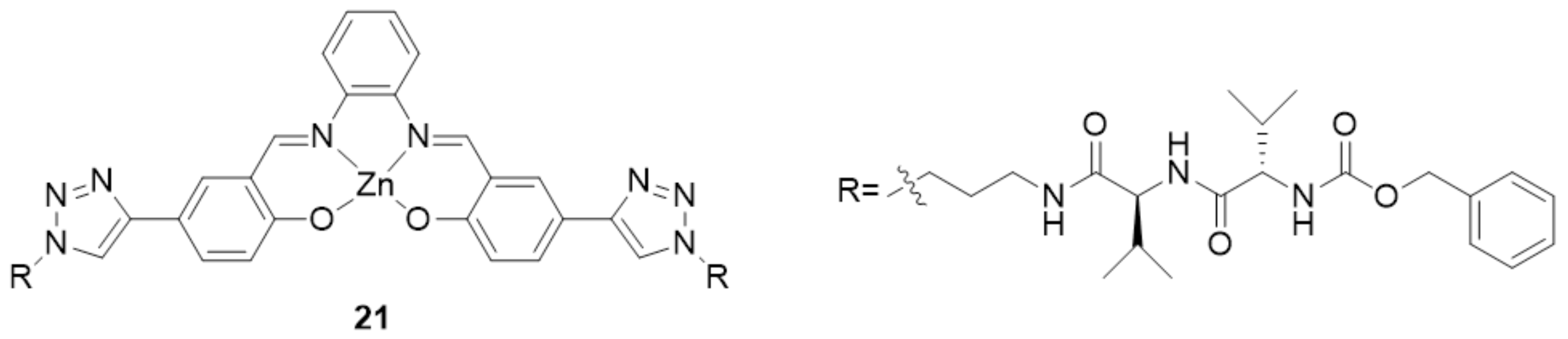
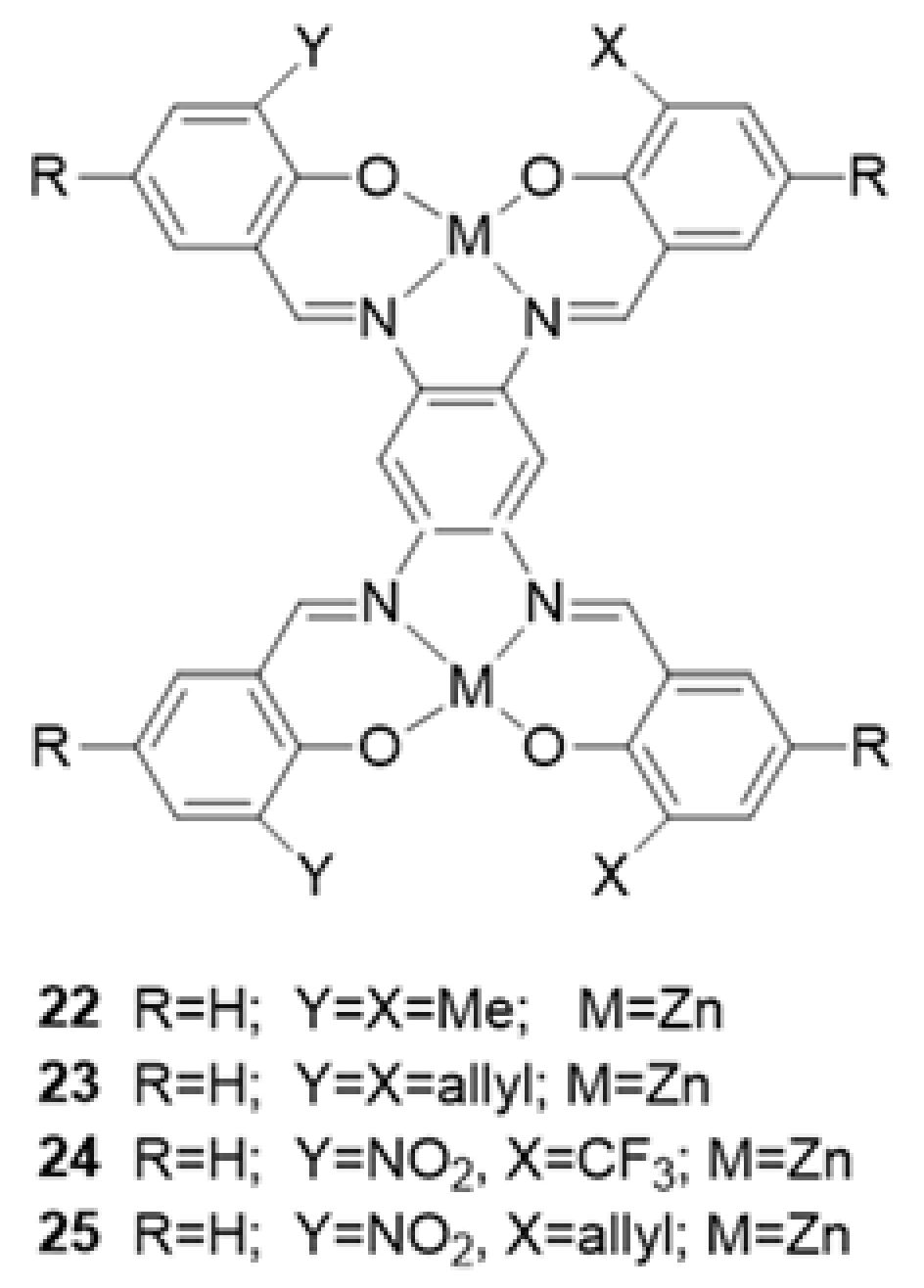
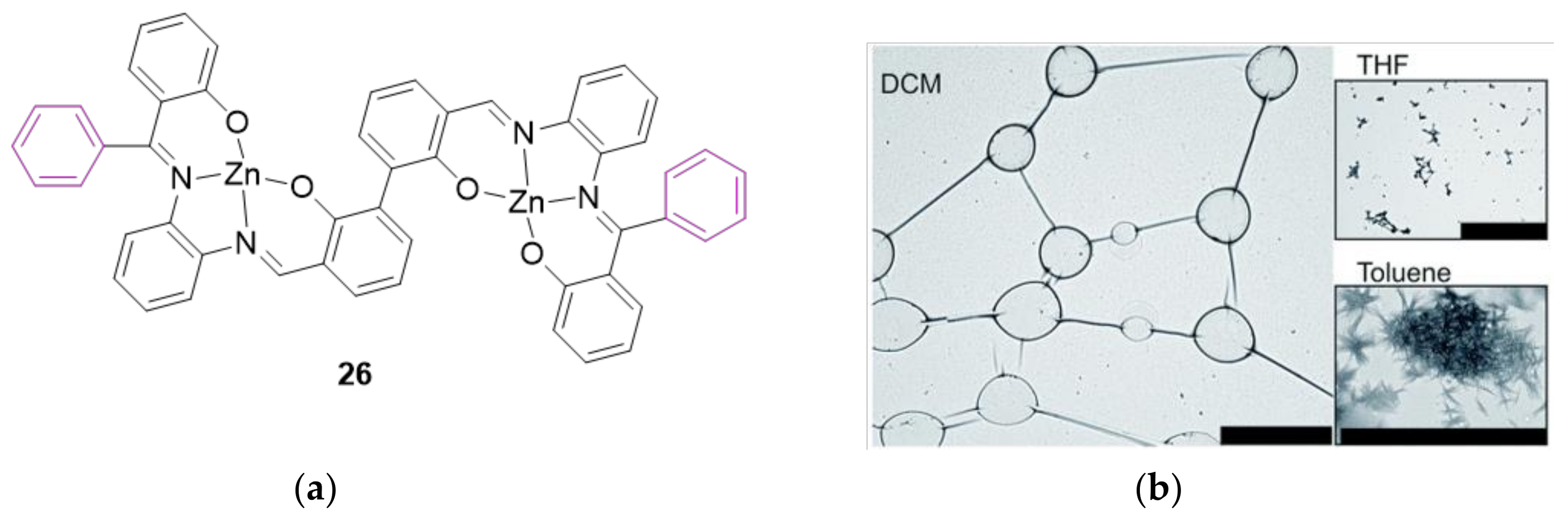
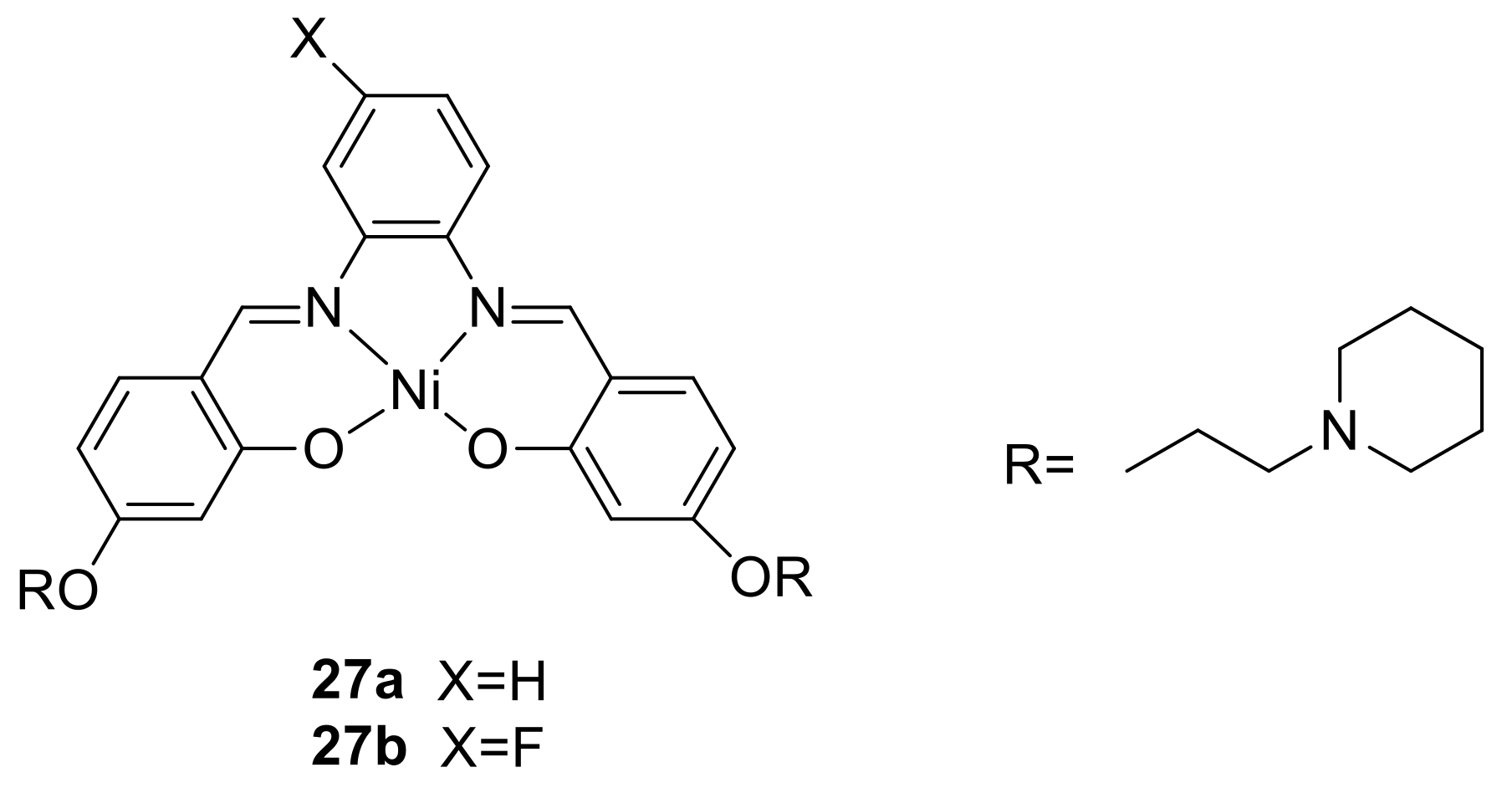
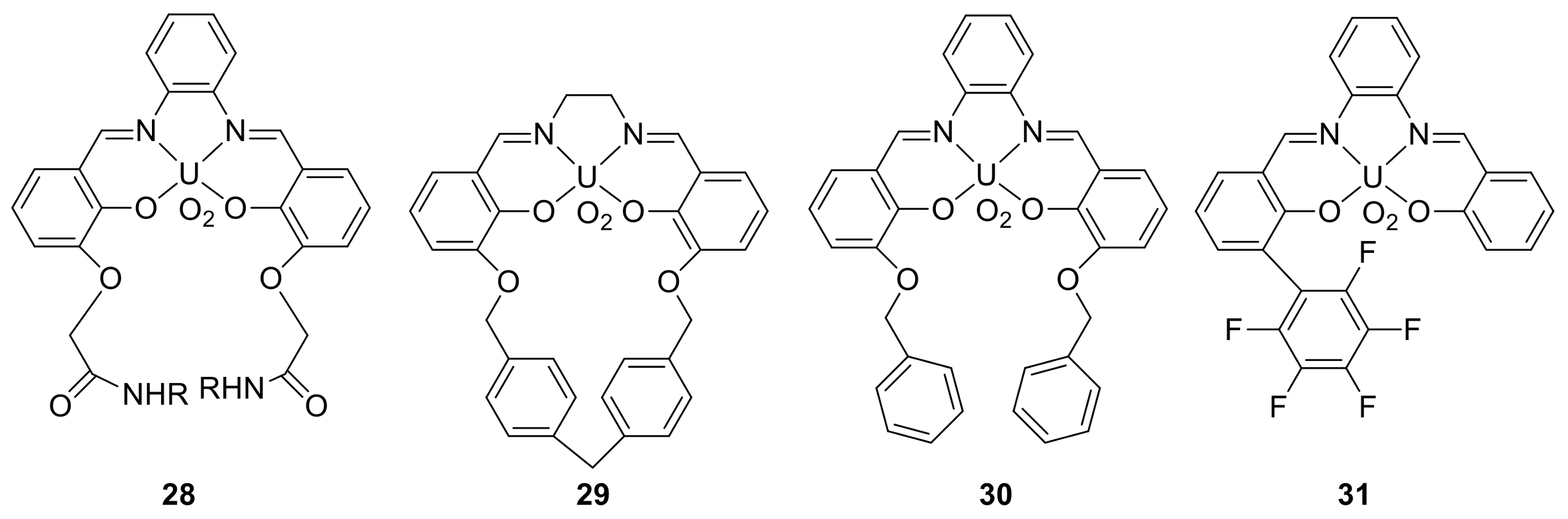
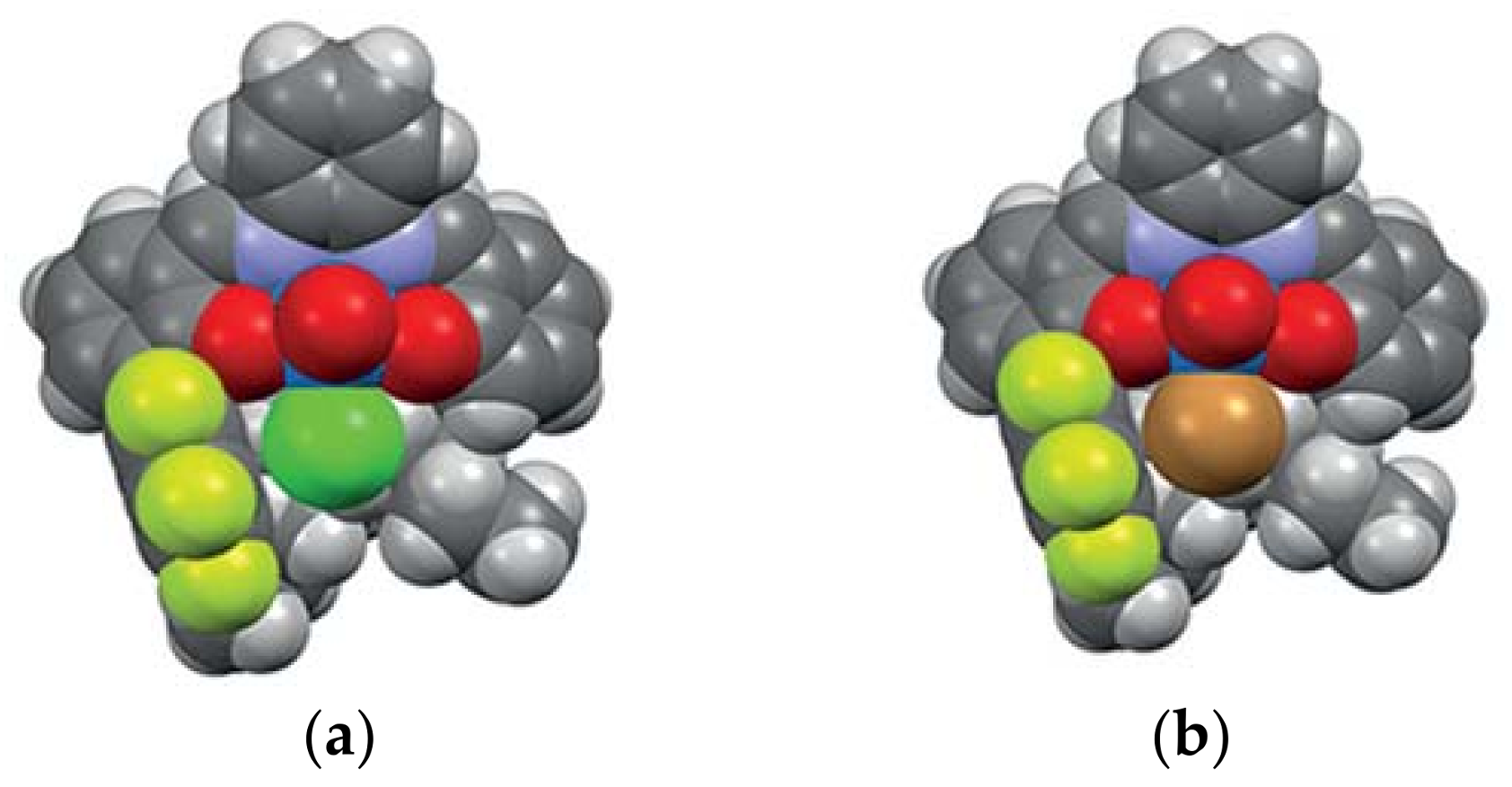
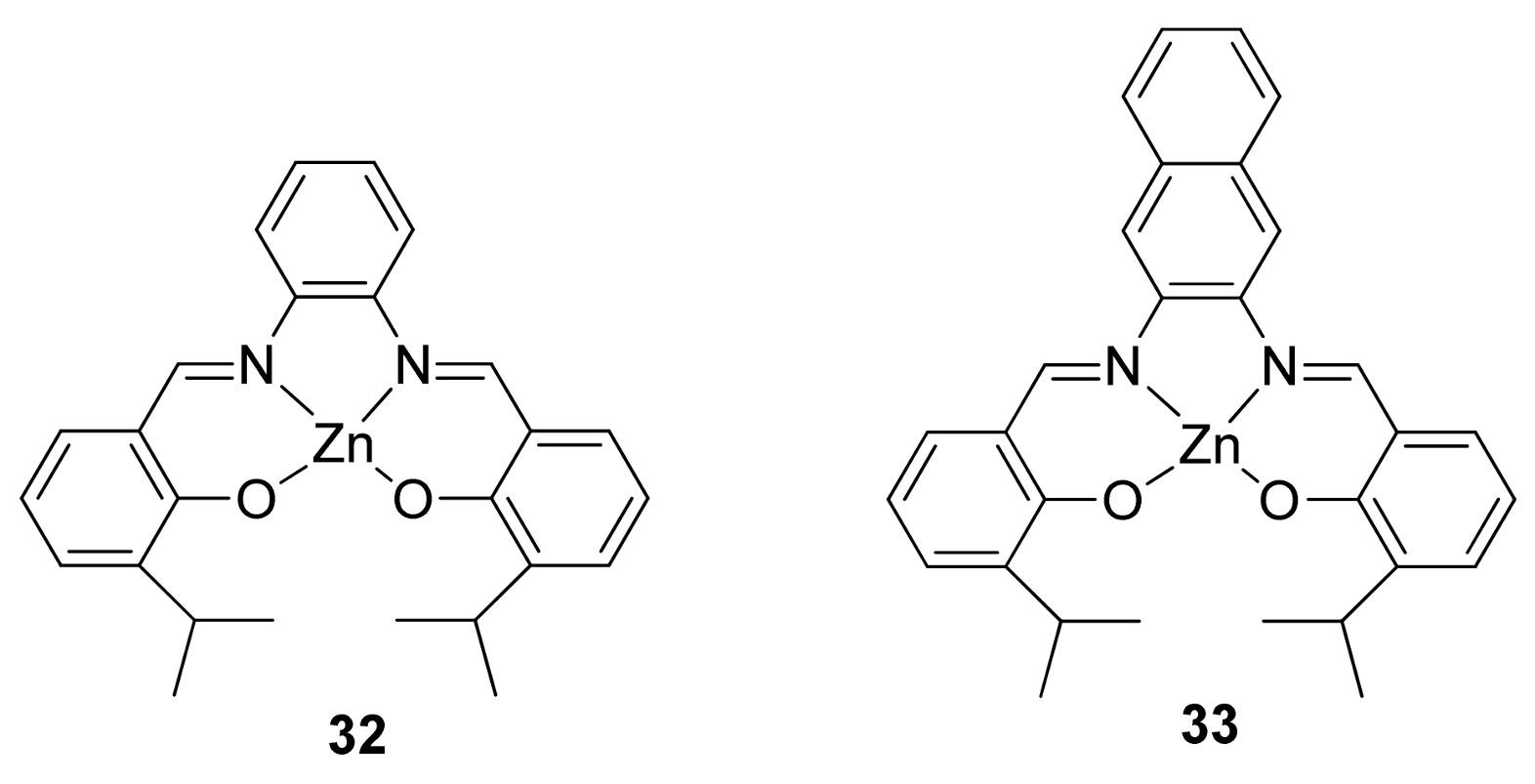
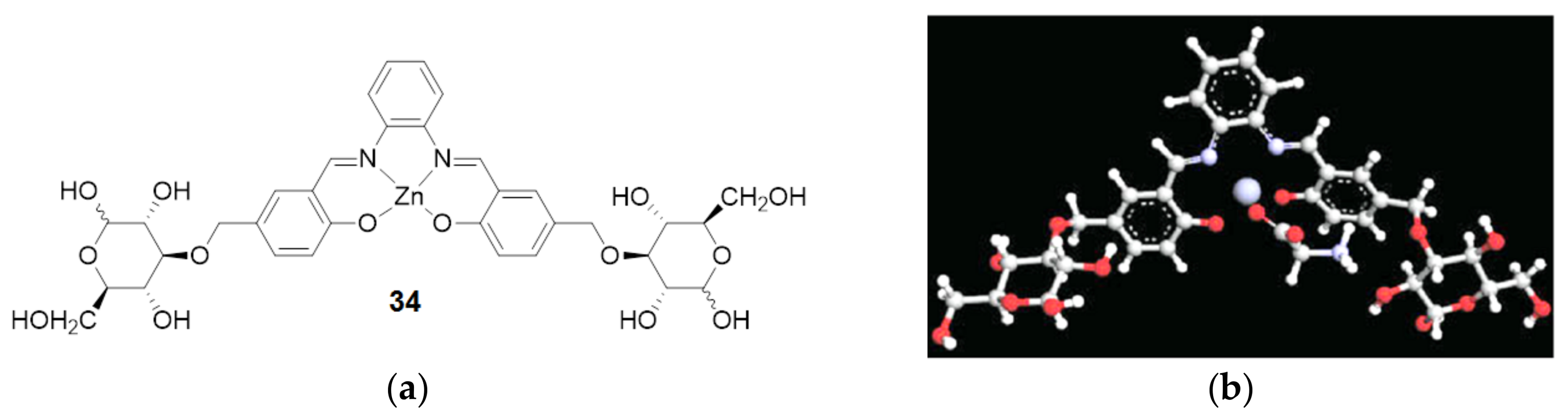
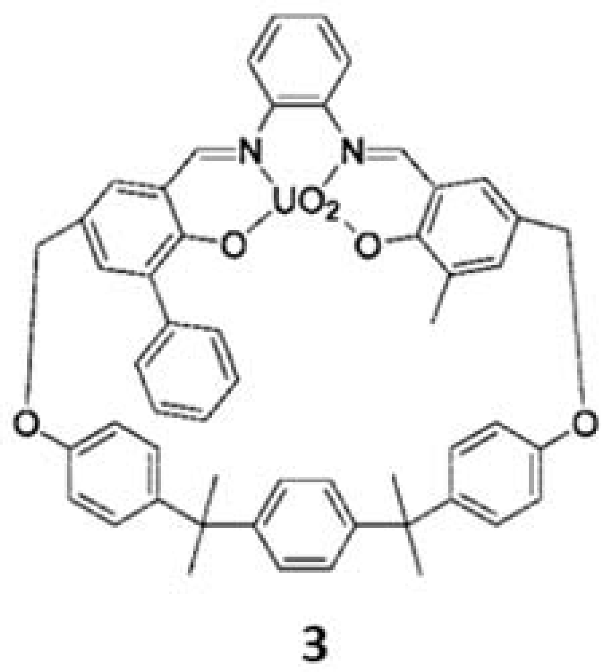
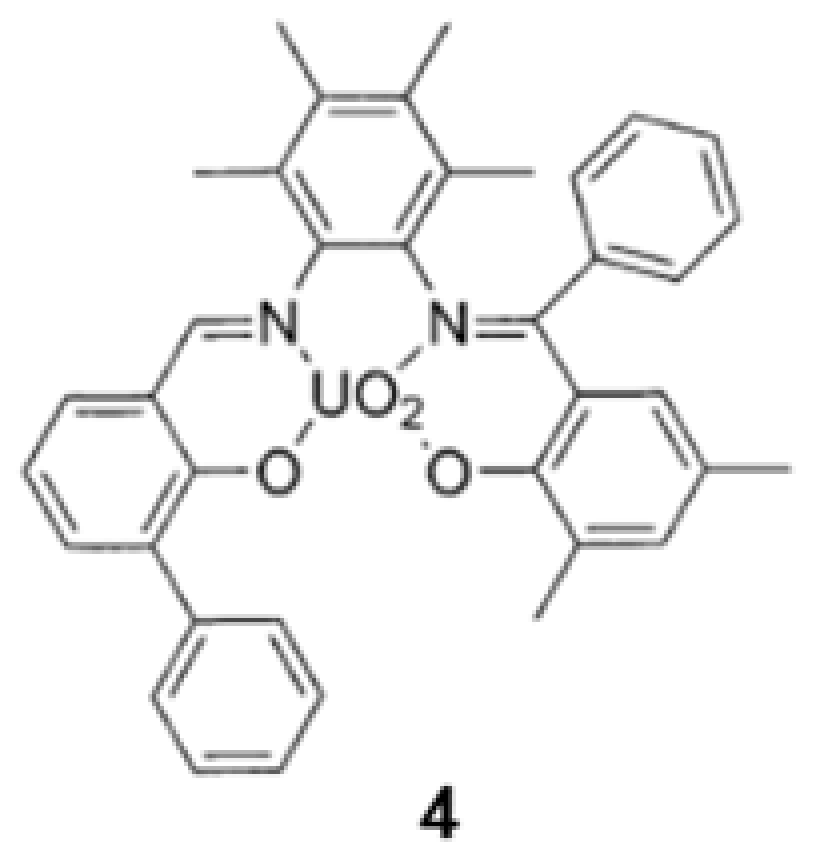
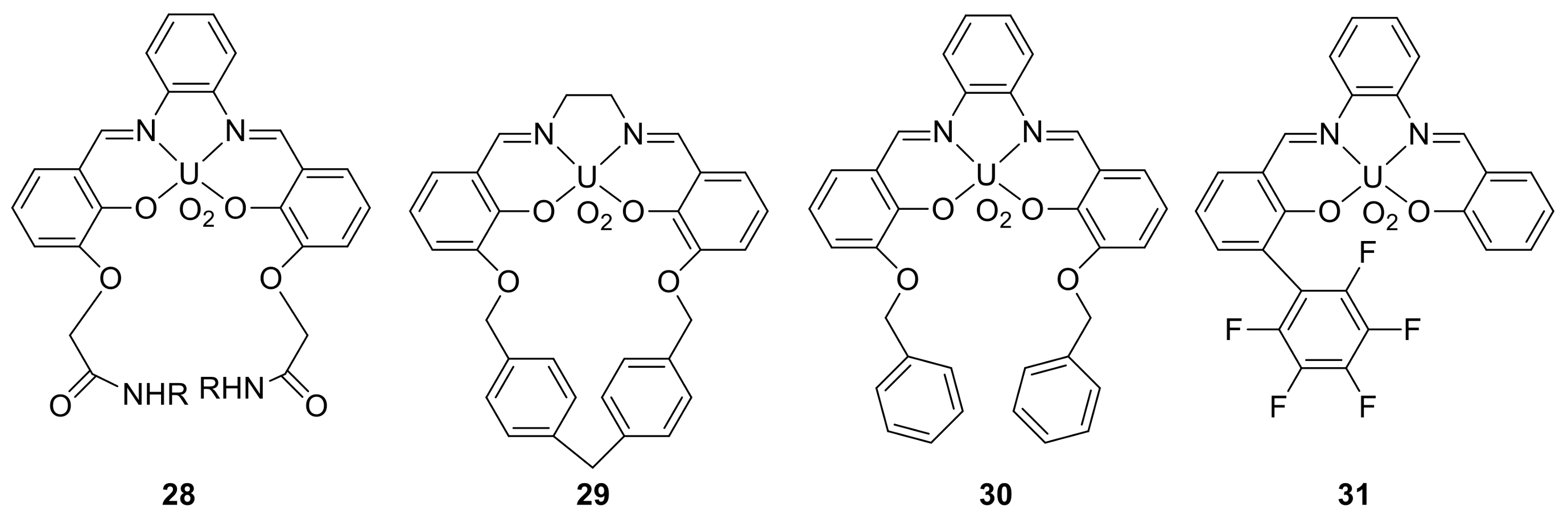
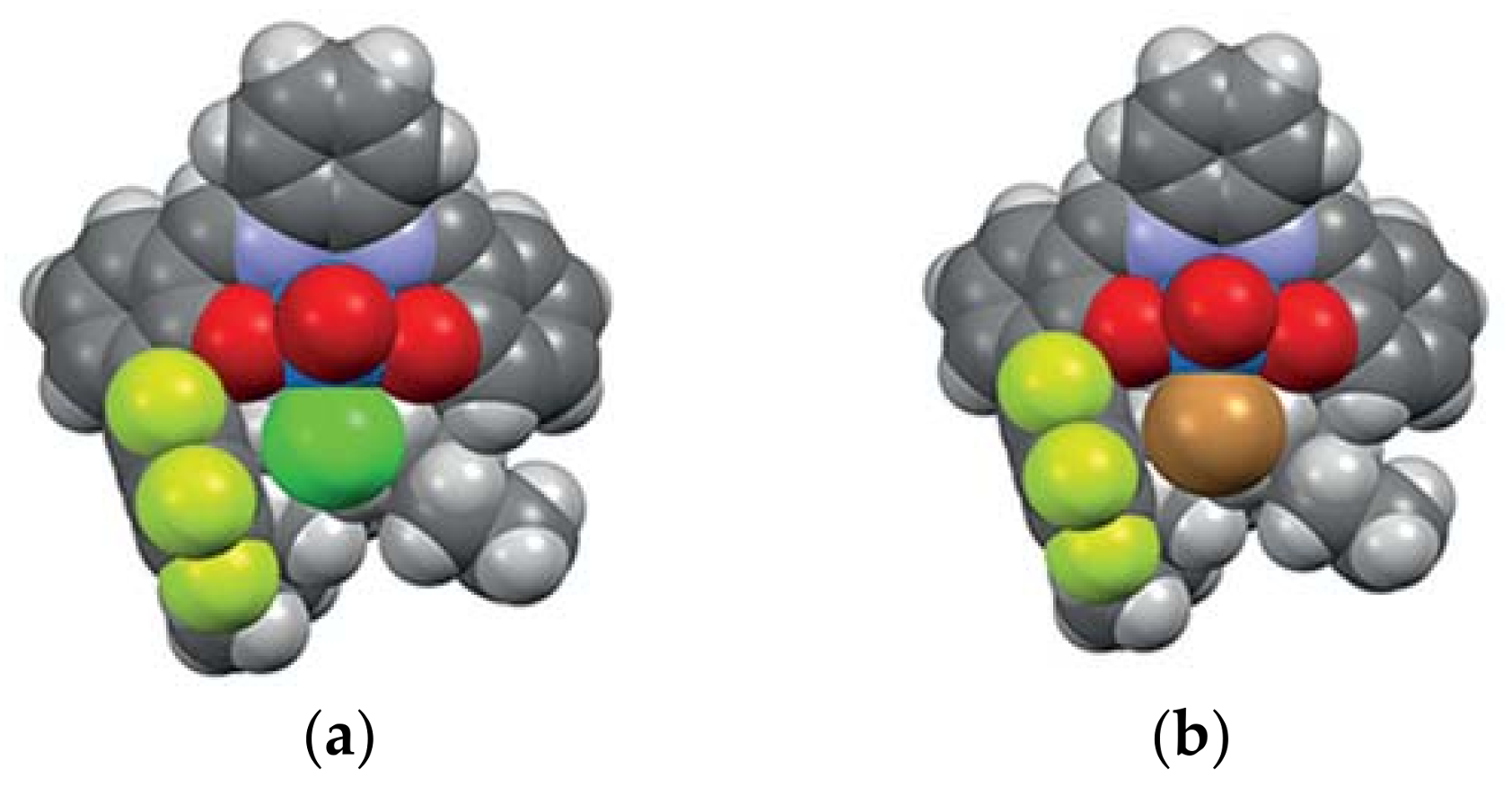
3. The Ability to Behave as Supramolecular Receptors
4. Towards Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Vigato, P.A.; Tamburini, S. The challenge of cyclic and acyclic Schiff bases and related derivatives. Coord. Chem. Rev. 2004, 248, 1717–2128. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Breith, E.; Lűbbe, E.; Tsumaki, T. Tricyclische orthokondensierte Nebenvalenzringe. Eur. J. Org. Chem. 1933, 503, 84–130. [Google Scholar] [CrossRef]
- Campbell, E.J.; Nguyen, S.T. Unsymmetrical salen-type ligands: High yield synthesis of salen-type Schiff bases containing two different benzaldehyde moieties. Tetrahedron Lett. 2001, 42, 1221–1225. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Pasquini, C.; Schiaffino, L. Nonsymmetrically Substituted Uranyl–Salophen Receptors: New Opportunities for Molecular Recognition and Catalysis. Supramol. Chem. 2007, 19, 79–87. [Google Scholar] [CrossRef]
- Omura, K.; Uchida, T.; Irie, R.; Katsuki, T. Design of a robust Ru(salen) complex: Aziridination with improved turnover number using N-arylsulfonyl azides as precursors. Chem. Commun. 2004, 2060–2061. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, A.; Tabbi, G.; Vecchio, G. Bioconjugates of cyclodextrins of manganese salen-type ligand with superoxide dismutase activity. J. Inorg. Biochem. 2004, 98, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Doctrow, S.R.; Huffman, K.; Marcus, C.B.; Tocco, G.; Malfroy, E.; Adinolfi, C.A.; Kruk, H.; Baker, K.; Lazarowych, N.; Mascarenhas, J.; et al. Salen–Manganese Complexes as Catalytic Scavengers of Hydrogen Peroxide and Cytoprotective Agents: Structure-Activity Relationship Studies. J. Med. Chem. 2002, 45, 4549–4558. [Google Scholar] [CrossRef] [PubMed]
- Erxleben, A. Transition metal salen complexes in bioinorganic and medicinal chemistry. Inorg. Chim. Acta 2017, 40–57. [Google Scholar] [CrossRef]
- Sukhikh, T.S.; Vostrikova, K.E. Assembly of Mn(III) Schiff Base Complexes with Heptacyanorhenate (IV). Inorganics 2017, 5, 59. [Google Scholar] [CrossRef]
- Pinkowicz, D.; Southerland, H.I.; Avendaño, C.; Prosvirin, A.; Sanders, C.; Wernsdorfer, W.; Pedersen, K.S.; Dreiser, J.; Clérac, R.; Nehrkorn, J.; et al. Cyanide single-molecule magnets exhibiting solvent dependent reversible “on” and “off” exchange bias behavior. J. Am. Chem. Soc. 2015, 137, 14406–14422. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, T. Chiral Metallosalen Complexes: Structures and Catalyst Tuning for Asymmetric Epoxidation and Cyclopropanation. Adv. Synth. Catal. 2002, 344, 131–147. [Google Scholar] [CrossRef]
- Katsuki, T. Mn–salen catalyst, competitor of enzymes, for asymmetric epoxidation. J. Mol. Catal. 1996, 113, 87–107. [Google Scholar] [CrossRef]
- Katsuki, T. Unique asymmetric catalysis of cis-β metal complexes of salen and its related Schiff-base ligands. Chem. Soc. Rev. 2004, 33, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, M.; Nardin, G.; Randaccio, L. Structural aspects of metal complexes with some tetradentate schiff bases. Coord. Chem. Rev. 1972, 7, 385–403. [Google Scholar] [CrossRef]
- Cozzi, P.G. Metal–Salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Irie, R.; Hamada, T.; Suzuki, K.; Katsuki, T. Rational design of Mn–salen catalyst (2): Highly enantioselective epoxidation of conjugated cis olefins. Tetrahedron 1994, 50, 11827–11838. [Google Scholar] [CrossRef]
- Bandoli, G.; Clemente, D.A. Preparation and crystal structure of aqua[bis(2-hydroxyphenylimino)-ethanato-OO′NN′-]-dioxouranium. J. Chem. Soc. Dalton Trans. 1975, 612–615. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Mandolini, L.; Pasquini, C.; Schiaffino, L. “Inherent chirality” and curvature. New J. Chem. 2004, 28, 1198–1199. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Mandolini, L.; Palmieri, G.; Pasquini, C.; Schiaffino, L. Unprecedented detection of inherent chirality in uranyl–salophen complexes. Chem. Commun. 2003, 2178–2179. [Google Scholar] [CrossRef]
- Van Doorn, A.R.; Schaafstra, R.; Bos, M.; Harkema, S.; Van Eerden, J.; Verboom, W.; Reinhoudt, D.N. Molecular recognition of polar neutral molecules by metallomacrocycles: Synthesis, proton NMR spectroscopy, X-ray structure, electrochemistry, and ab initio calculations. J. Org. Chem. 1991, 56, 6083–6094. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Mandolini, L.; Pasquini, C.; Schiaffino, L. Inherently Chiral Uranyl–Salophen Macrocycles: Computer-Aided Design and Resolution. J. Org. Chem. 2005, 70, 9814–9821. [Google Scholar] [CrossRef] [PubMed]
- Ciogli, A.; Dalla Cort, A.; Gasparrini, F.; Lunazzi, L.; Mandolini, L.; Mazzanti, A.; Pasquini, C.; Pierini, M.; Schiaffino, L.; Yafteh Mihan, F. Enantiomerization of Chiral Uranyl–Salophen Complexes via Unprecedented Ligand Hemilability: Toward Configurationally Stable Derivatives. J. Org. Chem. 2008, 73, 6108–6118. [Google Scholar] [CrossRef] [PubMed]
- Dalla Cort, A.; Miranda Murua, J.I.; Pasquini, C.; Pons, M.; Schiaffino, L. Evaluation of Chiral Recognition Ability of a Novel Uranyl-Salophen-Based Receptor: An Easy and Rapid Testing Protocol. Chem. Eur. J. 2004, 10, 3301–3307. [Google Scholar] [CrossRef] [PubMed]
- Bera, M.K.; Chakraborty, C.; Malik, S. How the stereochemistry decides the selectivity: An approach towards metal ion detection. New J. Chem. 2015, 39, 9207–9214. [Google Scholar] [CrossRef]
- Salassa, G.; Coenen, M.J.J.; Wezenberg, S.J.; Hendriksen, B.L.M.; Speller, S.; Elemans, J.A.A.W.; Kleij, A.W. Extremely Strong Self-Assembly of a Bimetallic Salen Complex Visualized at the Single-Molecule Level. J. Am. Chem. Soc. 2012, 134, 7186–7192. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Ikeda, Y. Structural Characterization and Reactivity of UO2(salophen)L and [UO2(salophen)]2: Dimerization of UO2(salophen) Fragments in Noncoordinating Solvents (salophen = N,N′-Disalicylidene-o-phenylenediaminate, L = N,N-Dimethylformamide, Dimethyl Sulfoxide). Inorg. Chem. 2007, 46, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
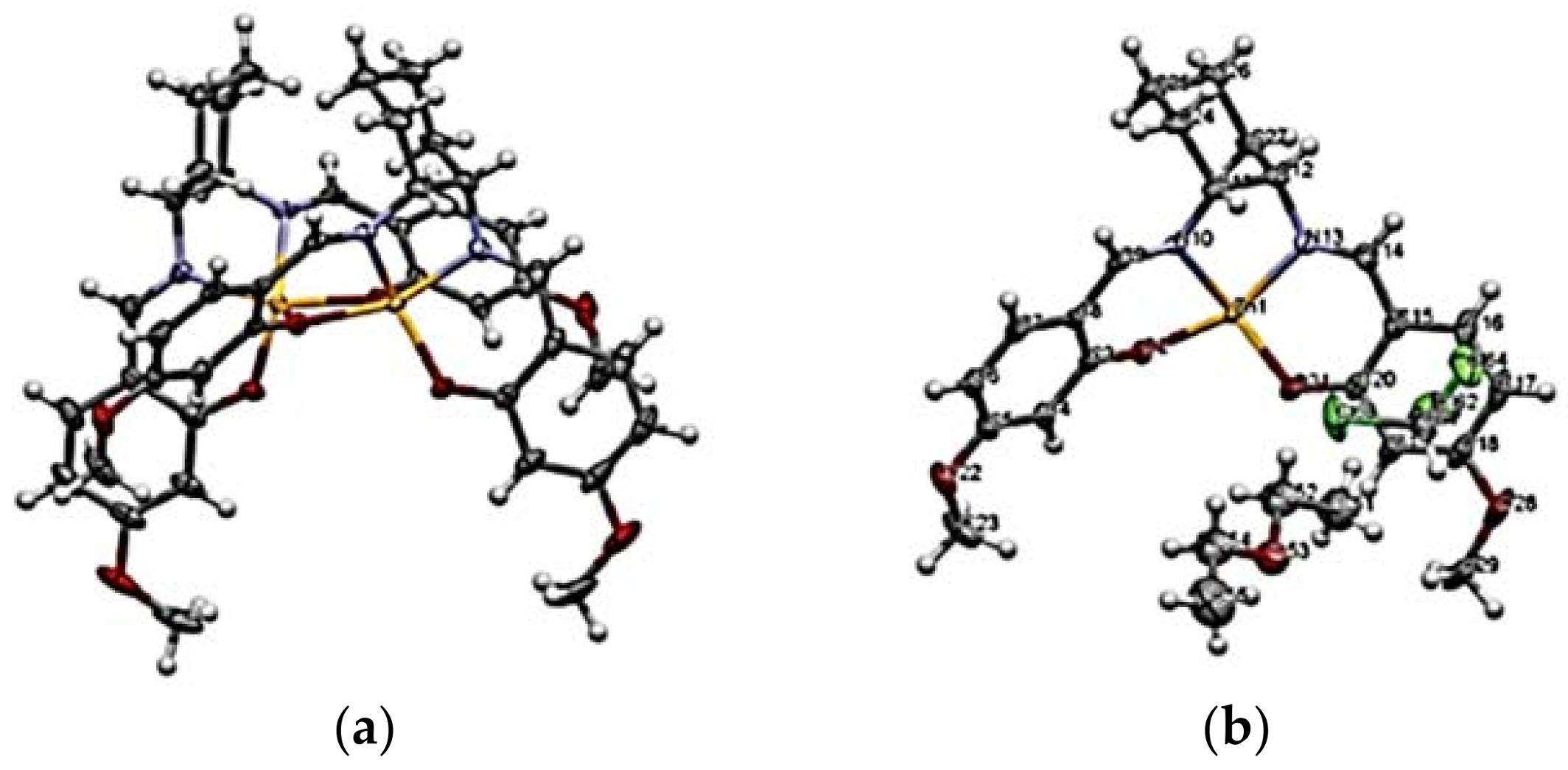
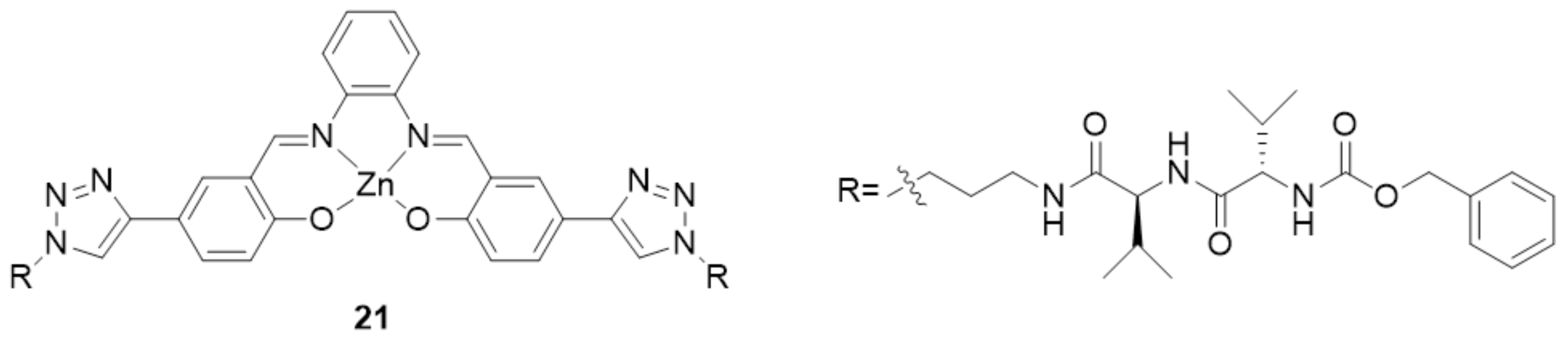
- Piccinno, M.; Angulo-Pachón, C.A.; Ballester, P.; Escuder, B.; Dalla Cort, A. Rational Design of a Supramolecular Gel Based on a Zn(II)–salophen Bis-dipeptide Derivative. RSC Adv. 2016, 6, 57306–57309. [Google Scholar] [CrossRef]
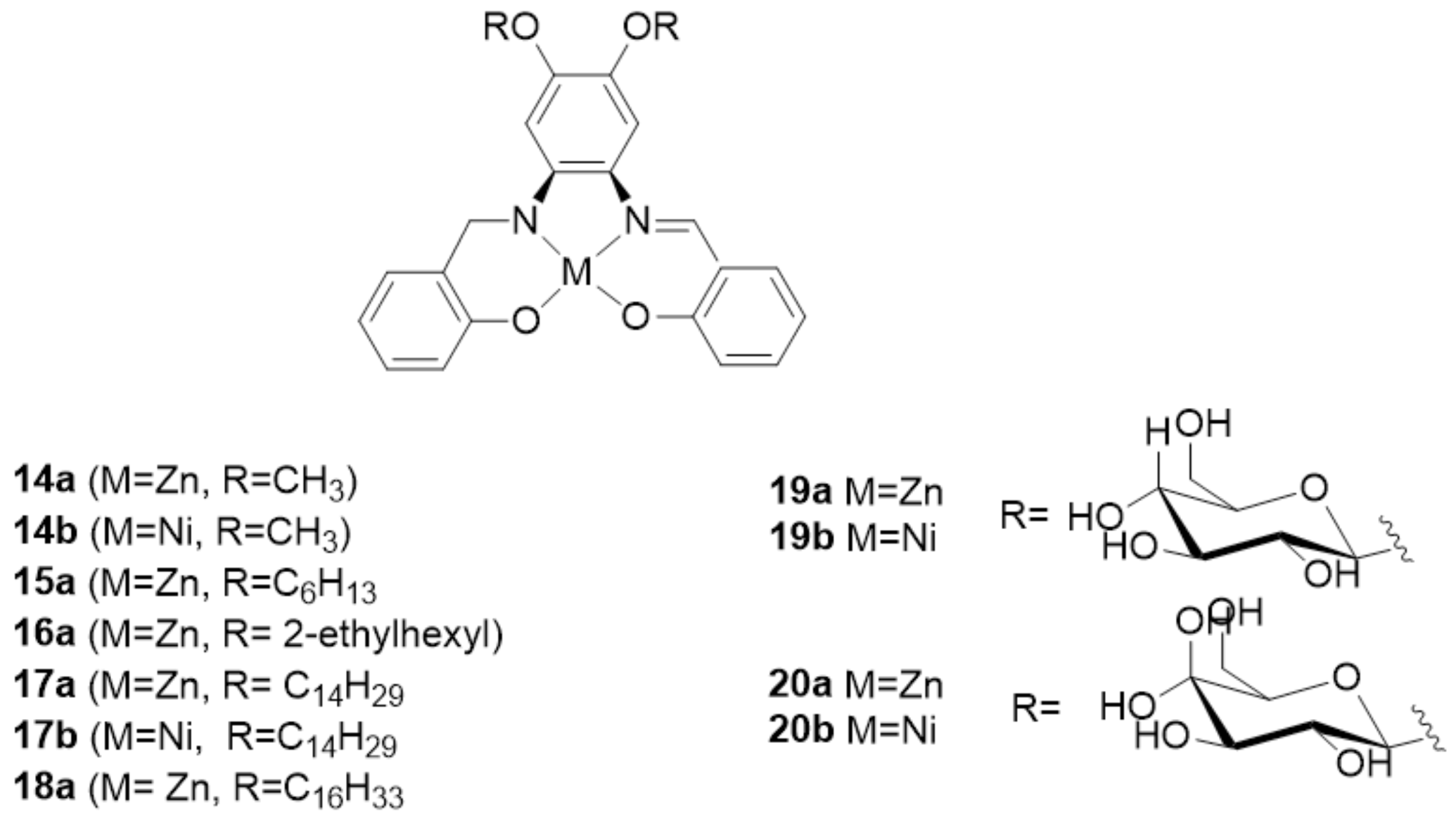
- Consiglio, G.; Failla, S.; Oliveri, I.P.; Purrello, R.; Di Bella, S. Controlling the molecular aggregation. An amphiphilic Schiff-base zinc(II)complex as supramolecular fluorescent probe. Dalton Trans. 2009, 10426–10428,. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, G.; Oliveri, I.P.; Punzo, F.; Thompson, A.L.; Di Bella, S.; Failla, S. Structure and Aggregation Properties of a Schiff-Base Zinc(II) Complex Derived from cis-1,2-Diaminocyclohexane. Dalton Trans. 2015, 44, 13040–13048. [Google Scholar] [CrossRef] [PubMed]
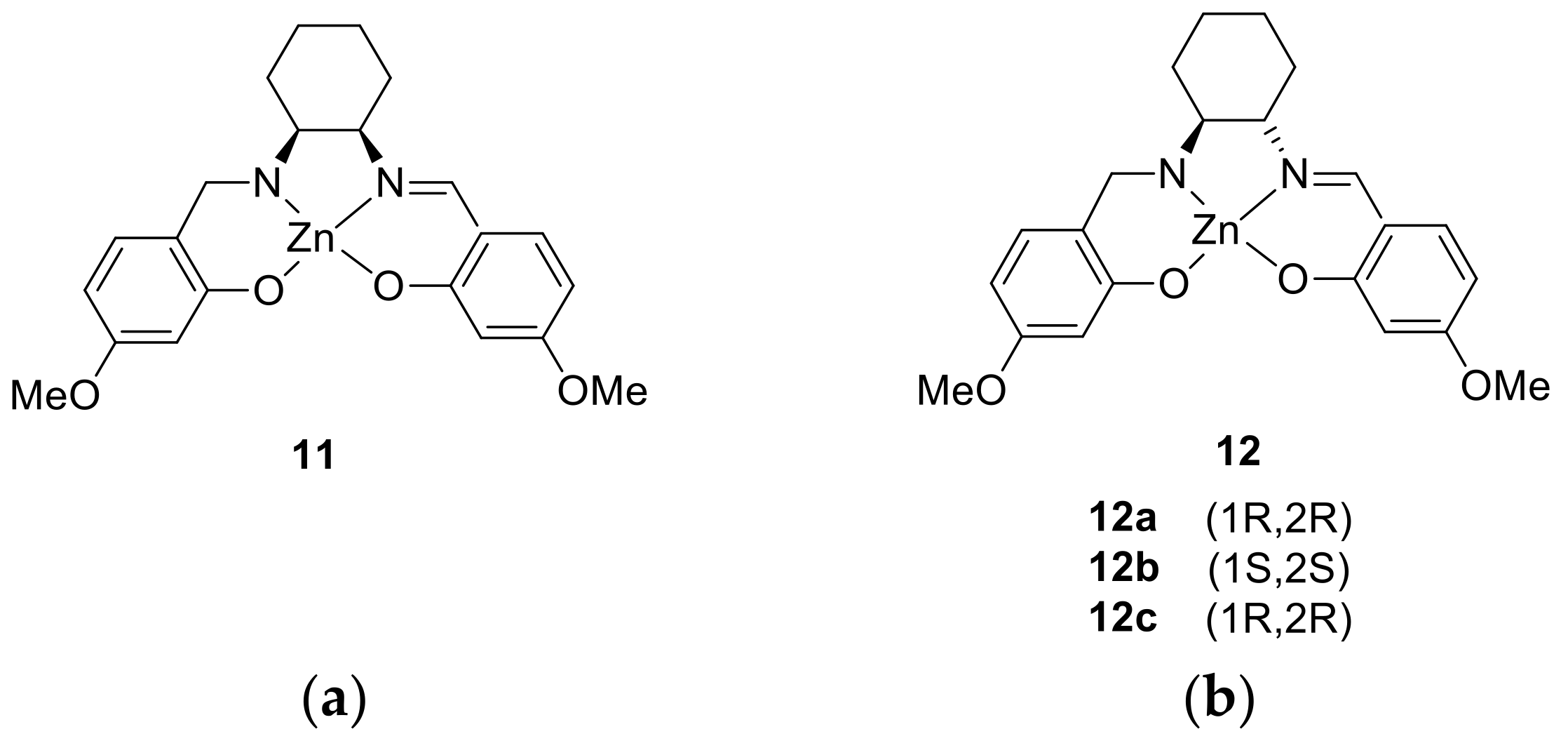
- Consiglio, G.; Oliveri, I.P.; Failla, S.; Di Bella, S. Supramolecular Aggregation of a New Substituted Bis(salicylaldiminato)zinc(II) Schiff-Base Complex Derived from trans-1,2-Diaminocyclohexane. Inorganics 2018, 6, 8. [Google Scholar] [CrossRef]
- Consiglio, G.; Oliveri, I.P.; Failla, S.; Di Bella, S. Supramolecular Aggregates of Defined Stereochemical Scaffolds: Aggregation/Deaggregation in Schiff-Base Zinc(II) Complexes Derived from Enantiopure trans-1,2-Diaminocyclohexane. Inorg. Chem. 2016, 55, 10320–10328. [Google Scholar] [CrossRef] [PubMed]
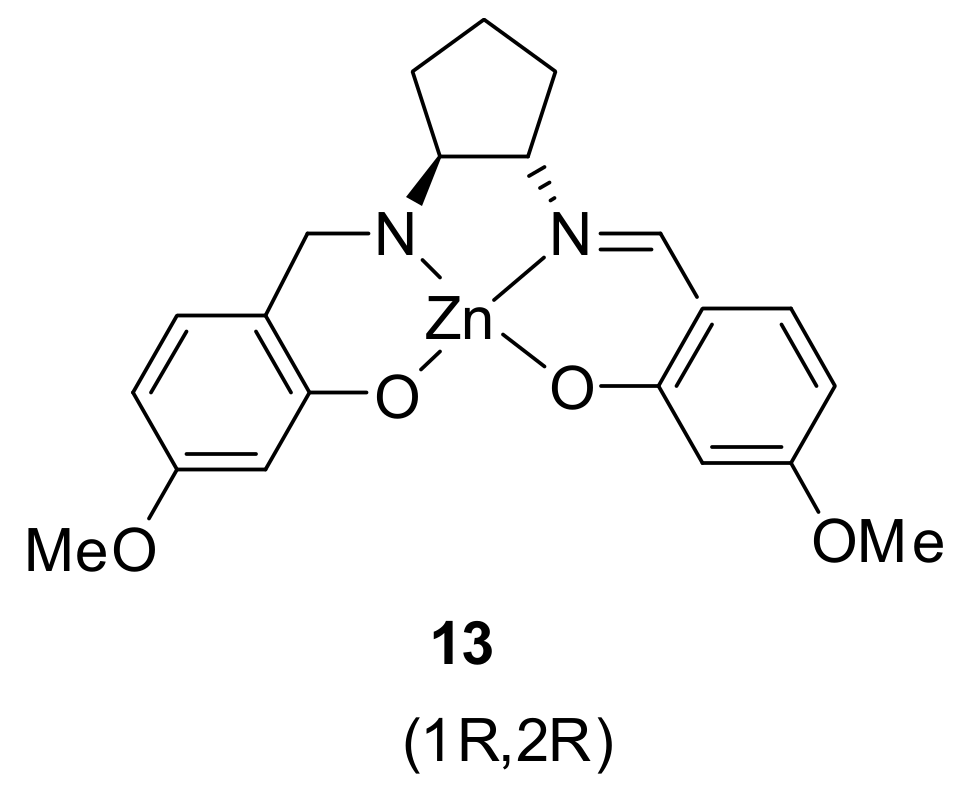
- Oliveri, I.P.; Forte, G.; Consiglio, G.; Failla, S.; Di Bella, S. Aggregates of Defined Stereochemical Scaffolds: A Study in Solution of a Zinc(II) Schiff Base Complex Derived from the Enantiopure trans-1,2-Cyclopentanediamine. Inorg. Chem. 2017, 56, 14206–14213. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.S.; Ren, G.Q.; Jenekhe, S.A. One-Dimensional Nanostructures of π-Conjugated Molecular Systems: Assembly, Properties, and Applications from Photovoltaics, Sensors, and Nanophotonics to Nanoelectronics. Chem. Mater. 2011, 23, 682–732. [Google Scholar] [CrossRef]
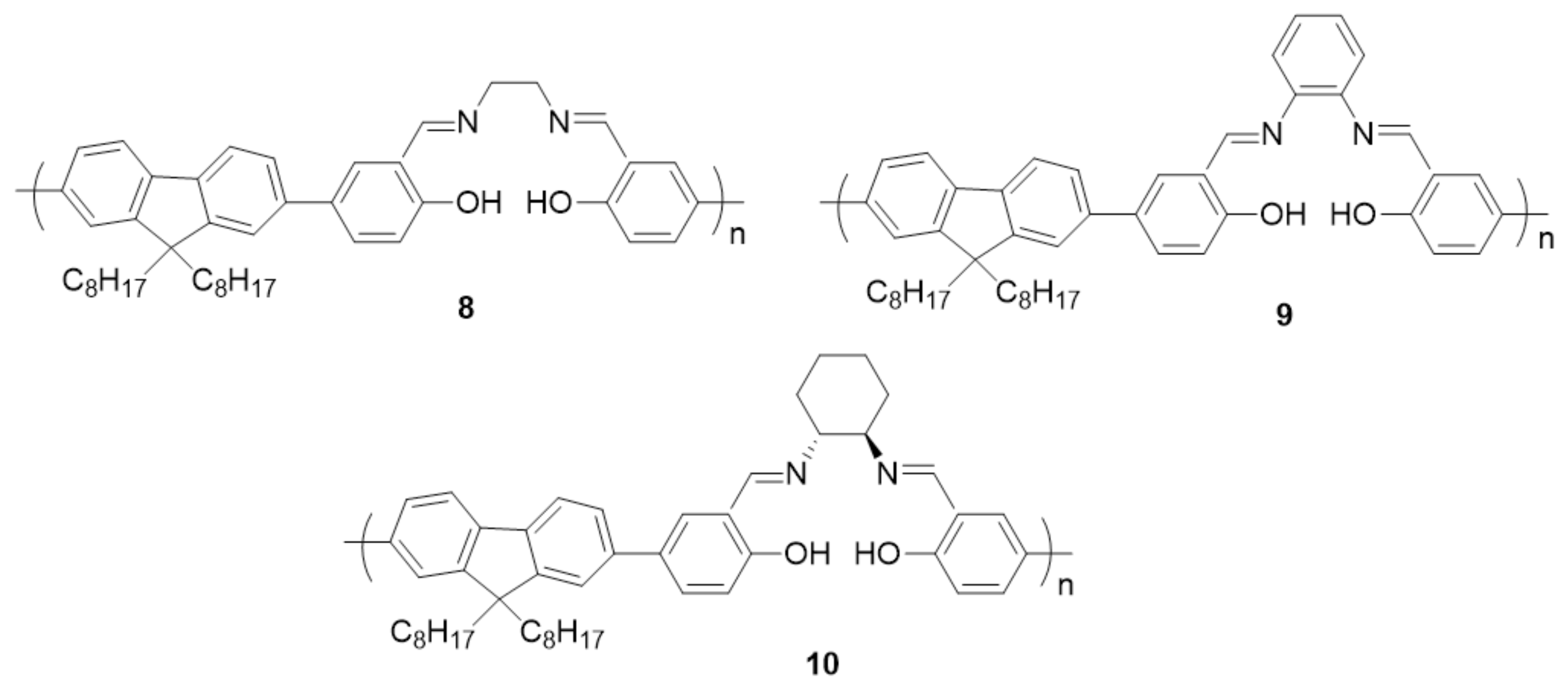
- Hui, J.K.-H.; Yu, Z.; MacLachlan, M.J. Supramolecular Assembly of Zinc Salphen Complexes: Access to Metal-Containing Gels and Nanofibers. Angew. Chem. Int. Ed. 2007, 46, 7980–7983. [Google Scholar] [CrossRef] [PubMed]
- Dalla Cort, A.; Mandolini, L.; Pasquini, C.; Rissanen, K.; Russo, L.; Schiaffino, L. Zinc–salophen complexes as selective receptors for tertiary amines. New J. Chem. 2007, 31, 1633–1638. [Google Scholar] [CrossRef]
- Pyrlin, S.V.; Hine, N.D.M.; Kleij, A.W.; Ramos, M.M.D. Self-assembly of bis-salphen compounds: From semiflexible chains to webs of nanorings. Soft Matter 2018, 14, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.S.; Varshney, U.; Bhattacharya, S. Role of the Central Metal Ion and Ligand Charge in the DNA Binding and Modification by Metallosalen Complexes. Bioconjug. Chem. 1997, 8, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.-M.; Lu, Y.-J.; Tan, J.-H.; Huang, Z.-S.; Wong, K.-Y.; Gu, L.-Q. G-Quadruplexes: Targets in Anticancer Drug Design. ChemMedChem 2008, 3, 690–713. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.E.; Arnal, A.A.; Neidle, S.; Vilar, R. Stabilization of G-Quadruplex DNA and Inhibition of Telomerase Activity by Square-Planar Nickel(II) Complexes. J. Am. Chem. Soc. 2006, 128, 5992–5993. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-Q.; Liao, T.-C.; Li, Z.-Q.; Gonzalez-Garcia, J.; Reynolds, M.; Zou, M.; Vilar, R. Dinickel–Salphen Complexes as Binders of Human Telomeric Dimeric G-Quadruplexes. Chem. Eur. J. 2017, 23, 4713–4722. [Google Scholar] [CrossRef] [PubMed]
- Dalla Cort, A.; De Bernardin, P.; Forte, G.; Yafteh Mihan, F. Metal–salophen-based receptors for anions. Chem. Soc. Rev. 2010, 39, 3863–3874. [Google Scholar] [CrossRef] [PubMed]
- Rudkevich, D.M.; Verboom, W.; Brzozka, Z.; Palys, M.J.; Stauthamer, W.P.R.V.; van Humme, G.J.; Franken, S.M.; Harkema, S.; Engbersen, J.F.J.; Reinhoudt, D.N. Functionalized UO2 Salenes: Neutral Receptors for Anions. J. Am. Chem. Soc. 1994, 116, 4341–4351. [Google Scholar] [CrossRef]
- Cametti, M.; Dalla Cort, A.; Mandolini, L.; Nissinen, M.; Rissanen, K. Specific recognition of fluoride anion using a metallamacrocycle incorporating a uranyl–salen unit. New J. Chem. 2008, 32, 1113–1116. [Google Scholar] [CrossRef]
- Cametti, M.; Nissinen, M.; Dalla Cort, A.; Mandolini, L.; Rissanen, K. Recognition of Alkali Metal Halide Contact Ion Pairs by Uranyl−Salophen Receptors Bearing Aromatic Sidearms. The Role of Cation–π Interactions. J. Am. Chem. Soc. 2005, 127, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Cametti, M.; Nissinen, M.; Dalla Cort, A.; Mandolini, L.; Rissanen, K. Ion Pair Recognition of Quaternary Ammonium and Iminium Salts by Uranyl−Salophen Compounds in Solution and in the Solid State. J. Am. Chem. Soc. 2007, 129, 3641–3648. [Google Scholar] [CrossRef] [PubMed]
- Leoni, L.; Puttreddy, R.; Jurcêk, O.; Mele, A.; Giannicchi, I.; Yafteh Mihan, F.; Rissanen, K.; Dalla Cort, A. Solution and Solid-State Studies on the Halide Binding Affinity of Perfluorophenyl-Armed Uranyl–Salophen Receptors Enhanced by Anion–π Interactions. Chem. Eur. J. 2016, 22, 18714–18717. [Google Scholar] [CrossRef] [PubMed]
- Forte, F.; Oliveri, I.P.; Consiglio, G.; Failla, S.; Di Bella, S. On the Lewis acidic character of bis(salicylaldiminato)zinc(II) Schiff-base complexes: A computational and experimental investigation on a series of compounds varying the bridging diimine. Dalton Trans. 2017, 46, 4571–4581. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.; Rodriguez, L.; Lima, J.C.; Pina, F.; Dalla Cort, A.; Pasquini, C.; Schiaffino, L. Specific Supramolecular Interactions between Zn2+–Salophen Complexes and Biologically Relevant Anions. Inorg. Chem. 2009, 48, 6229–6235. [Google Scholar] [CrossRef] [PubMed]
- Dalla Cort, A.; De Bernardin, P.; Schiaffino, L. A new water soluble Zn–salophen derivative as a receptor for α-aminoacids: Unexpected chiral discrimination. Chirality 2009, 21, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Dalla Cort, A.; Mandolini, L.; Schiaffino, L. Exclusive transition state stabilization in the supramolecular catalysis of Diels–Alder reaction by a uranyl salophen complex. Chem. Commun. 2005, 3867–3869. [Google Scholar] [CrossRef] [PubMed]
- Schowen, R.L.; Gandour, R.D. Catalytic Power and Transition State Stabilization, in Transition States of Biochemical Processes; Gandour, R.D., Schowen, R.L., Eds.; Plenum Press: New York, NY, USA, 1978; Chapter 2; pp. 77–113. [Google Scholar]
- Mendes, R.A.; Germino, J.C.; Fazolo, B.R.; Thaines, E.H.N.S.; Ferraro, F.; Santana, A.M.; Ramos, R.J.; de Souza, G.L.C.; Freitas, R.G.; Vazquez, P.A.M.; et al. Electronic and magnetic properties of the [Ni(salophen)]: An experimental and DFT study. J. Adv. Res. 2018, 9, 27–33. [Google Scholar] [CrossRef]
- Miyasaka, H.; Saitoh, A.; Abe, S. Magnetic assemblies based on Mn(III) salen analogues. Coord. Chem. Rev. 2007, 251, 2622–2664. [Google Scholar] [CrossRef]
- Yin, H.-Y.; Tang, J.; Zhang, J.-L. Introducing Metallosalens into Biological Studies: The Renaissance of Traditional Coordination Complexes. Eur. J. Inorg. Chem. 2017, 5085–5093. [Google Scholar] [CrossRef]
- Yin, H.-Y.; Lai, J.; Tang, J.; Shang, Y.; Zhang, J.-L. A Cryptand-Type Aluminum Tris(salophen) Complex: Synthesis, Characterization, and Cell Imaging Application. Inorganics 2018, 6, 20. [Google Scholar] [CrossRef]
- Tzubery, A.; Melamed-Bookb, N.; Tshuva, E.Y. Fluorescent antitumor titanium(IV) salen complexes for cell imaging. Dalton Trans. 2018, 47, 3669–3673. [Google Scholar] [CrossRef] [PubMed]
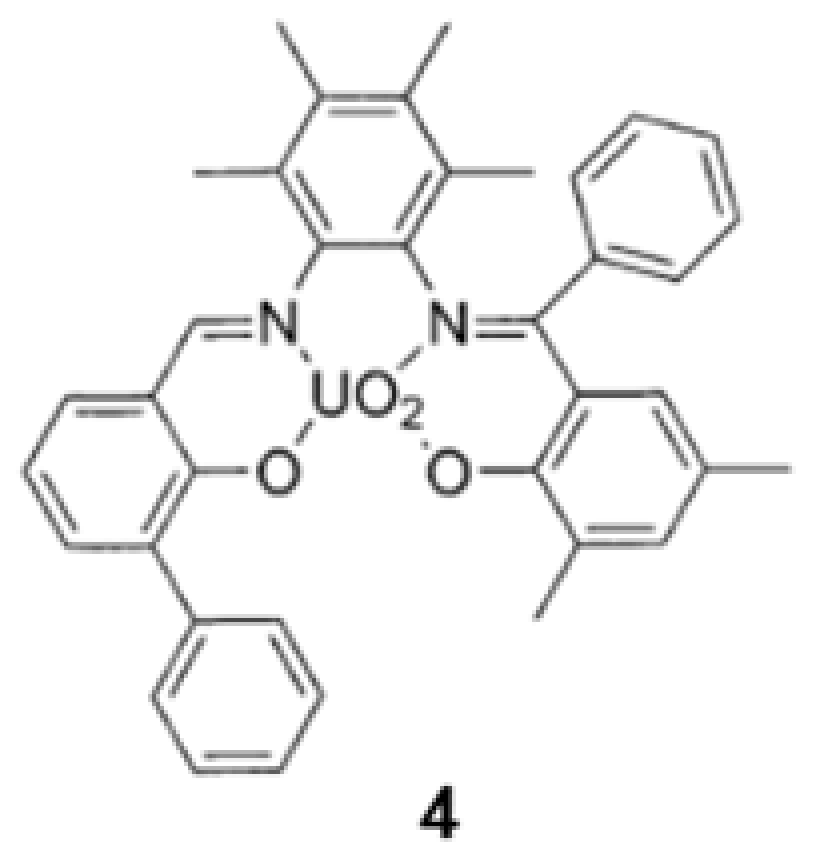
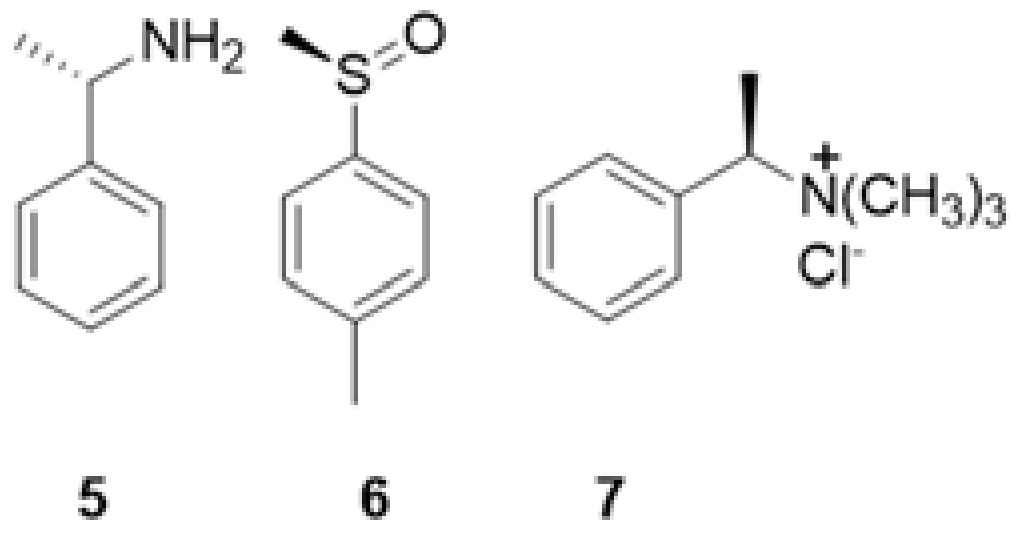
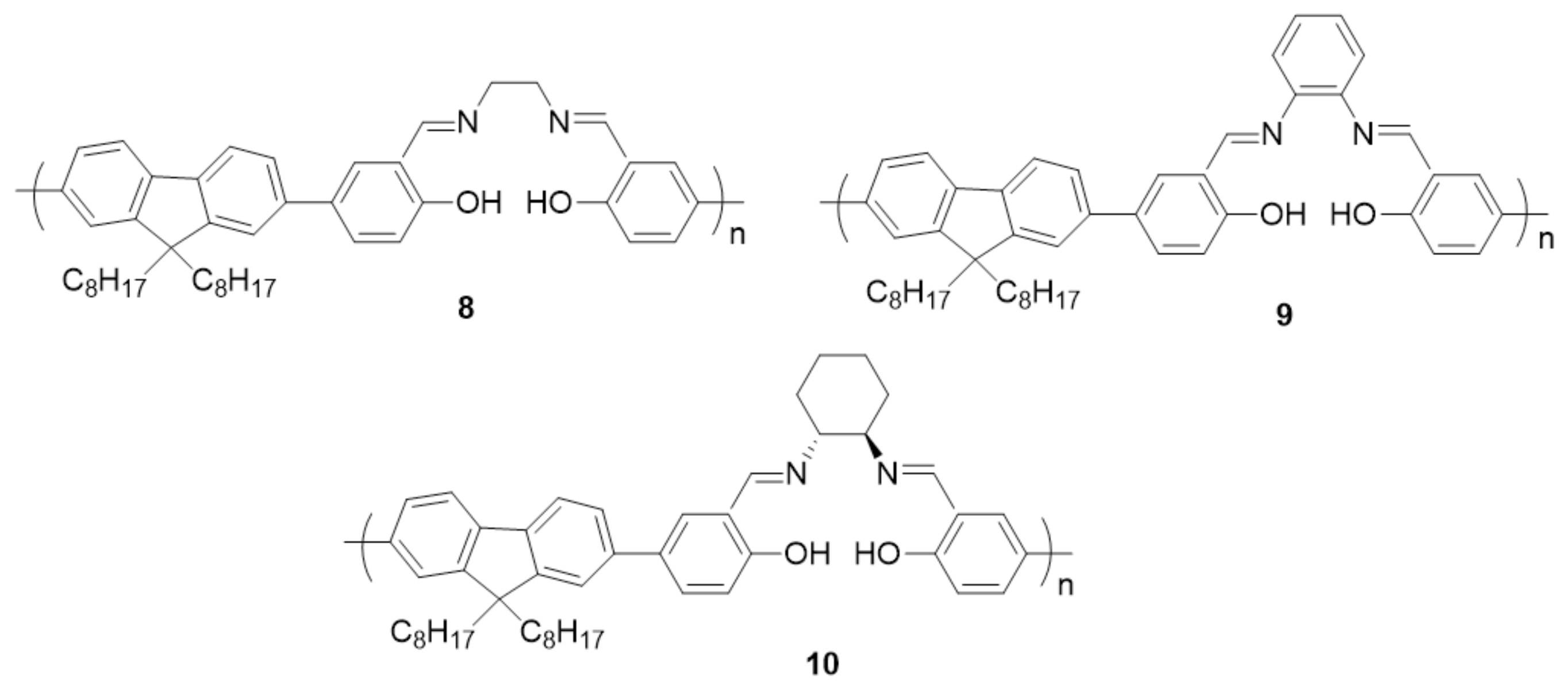
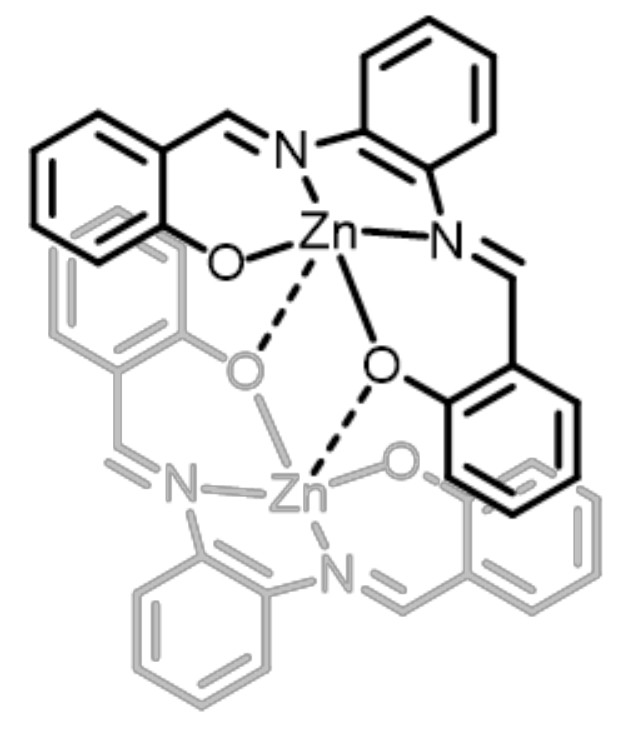
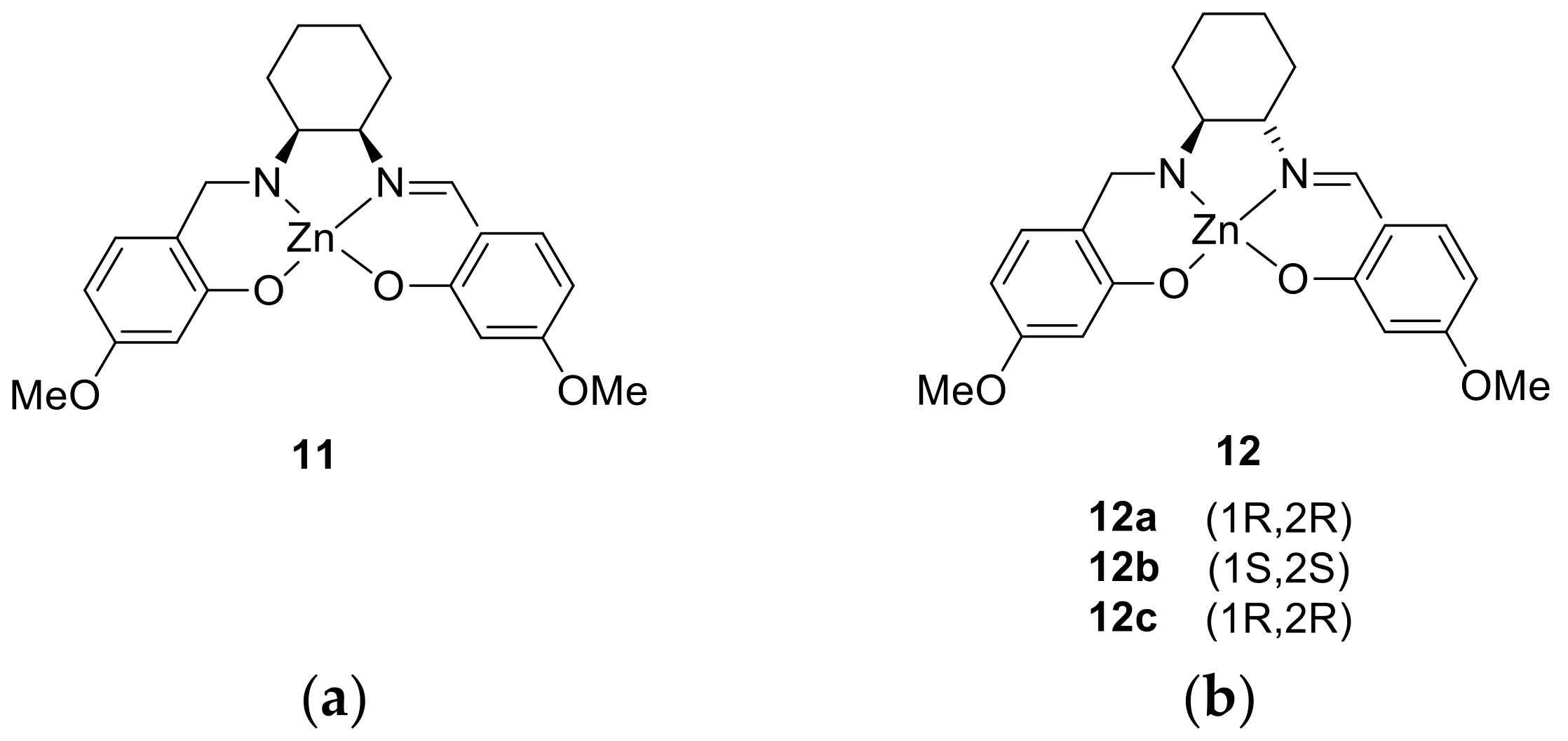
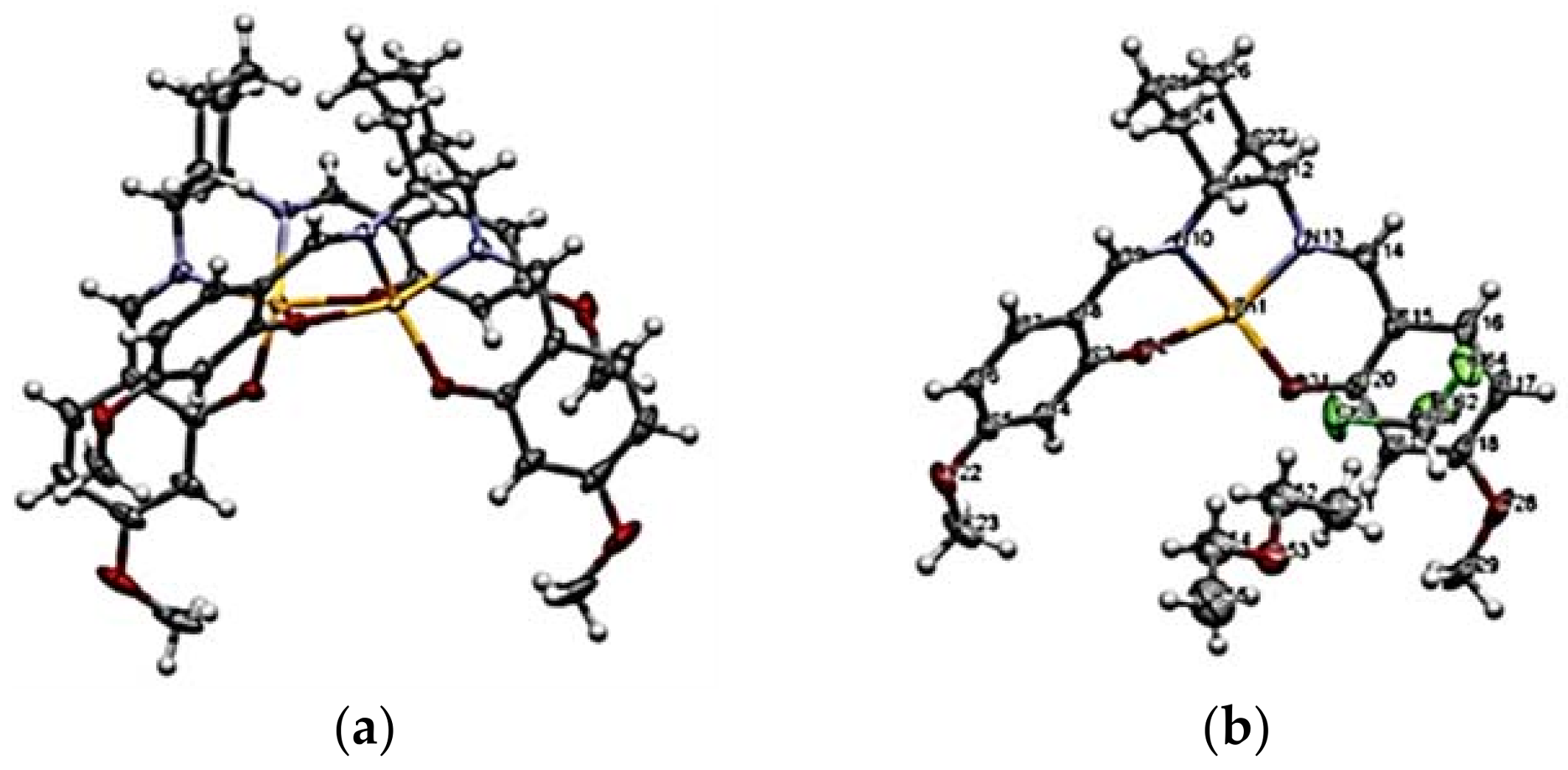
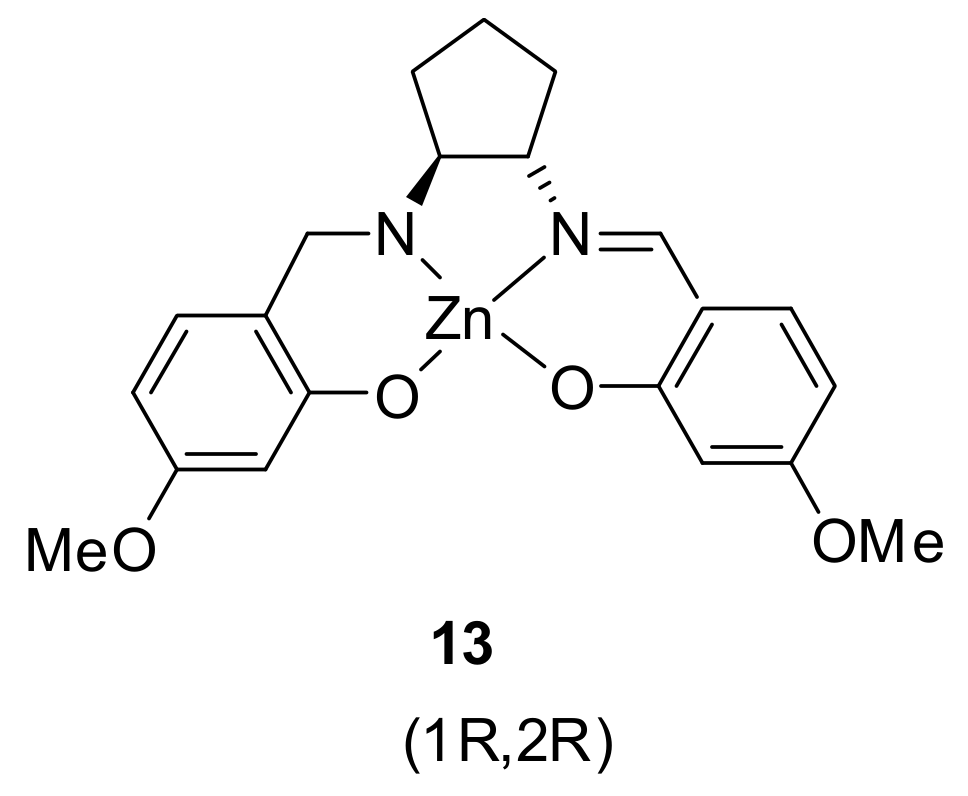
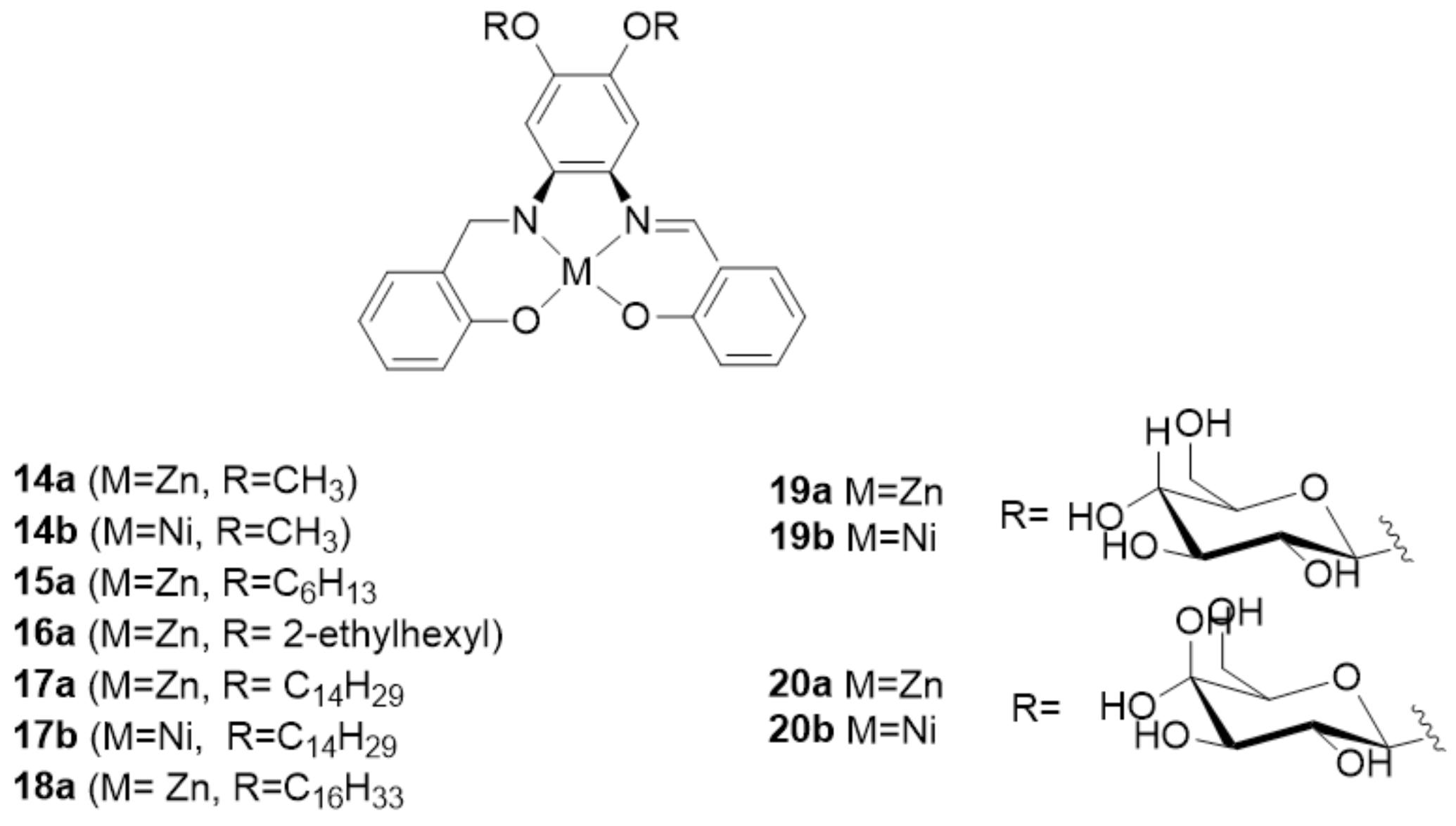



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
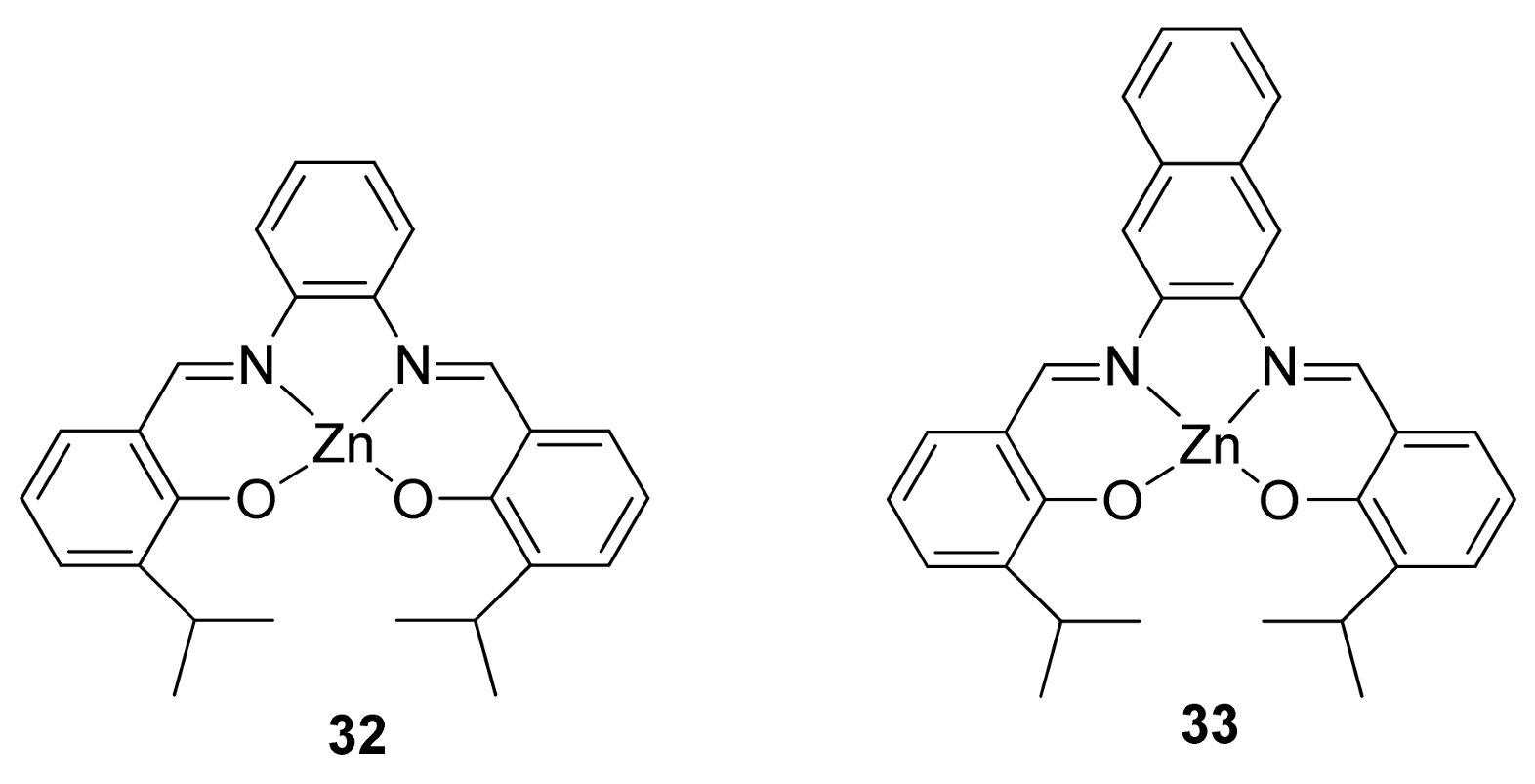{kind=link}
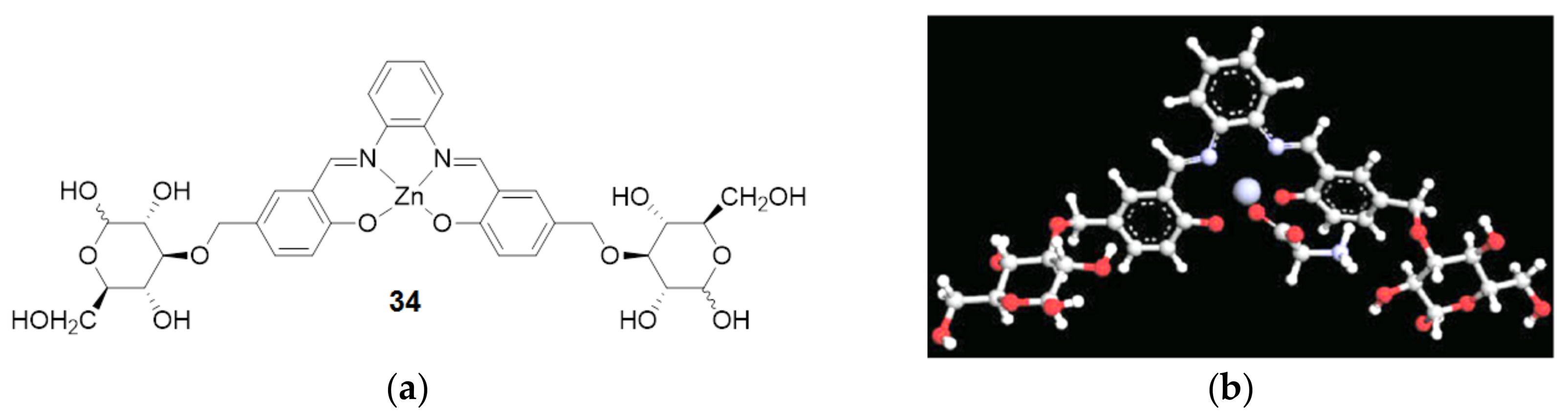{kind=link}
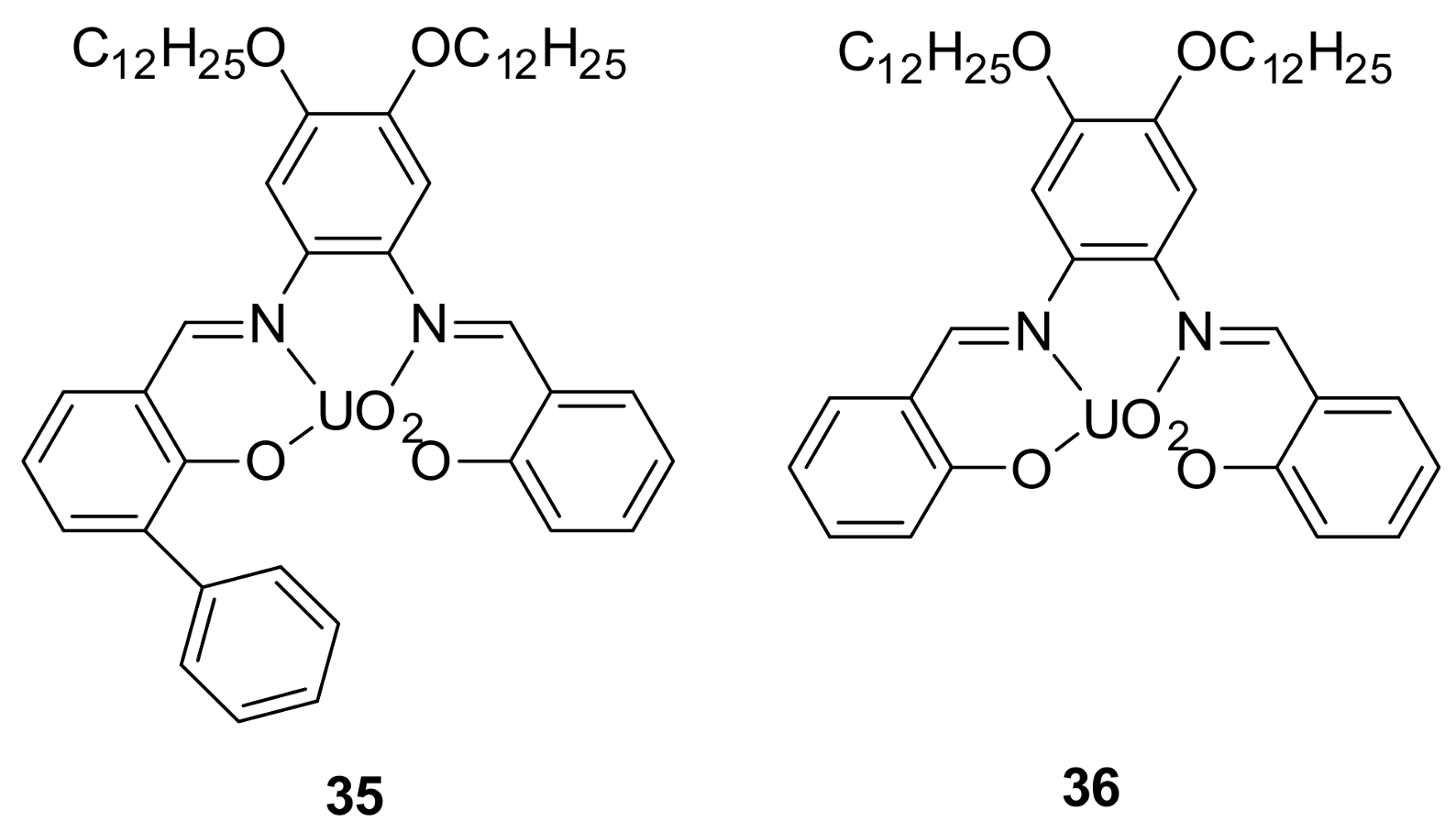{kind=link}
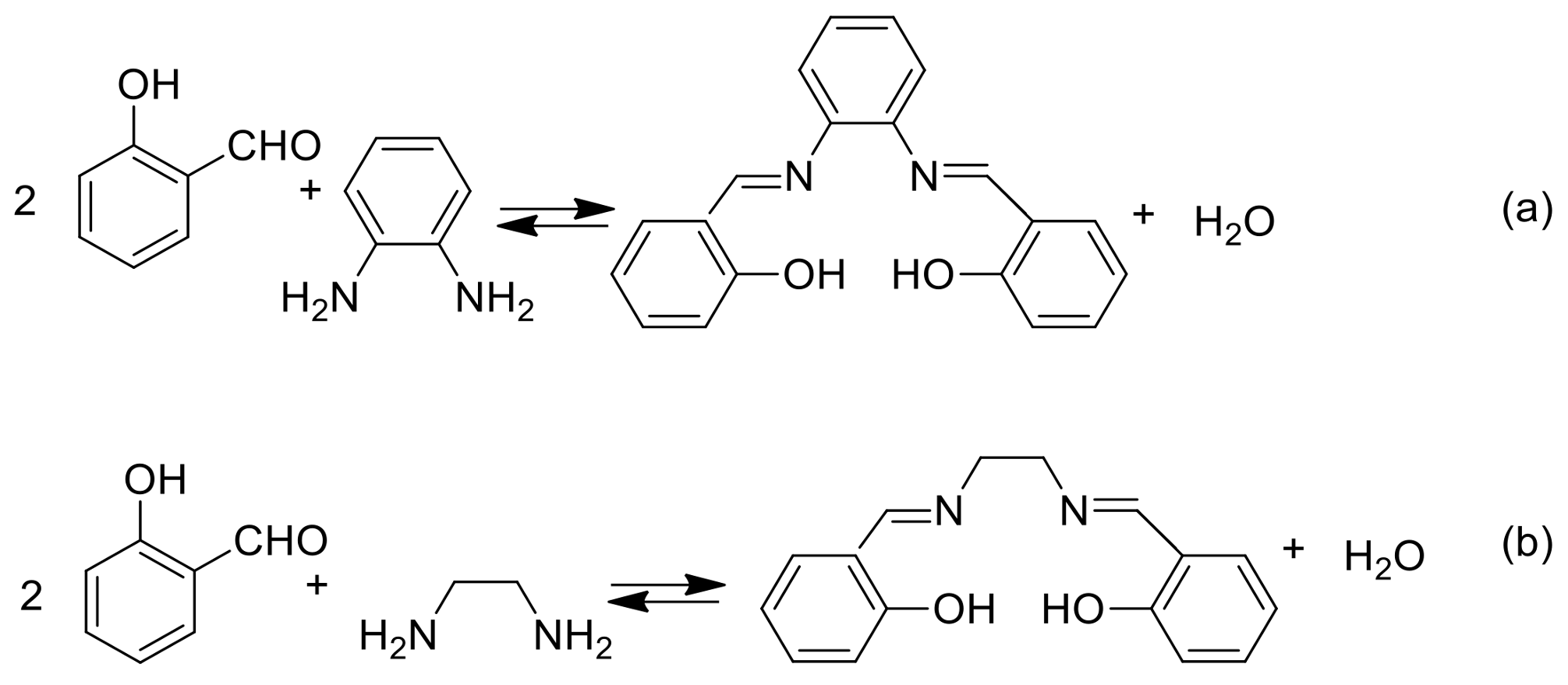{kind=link}
{kind=link}
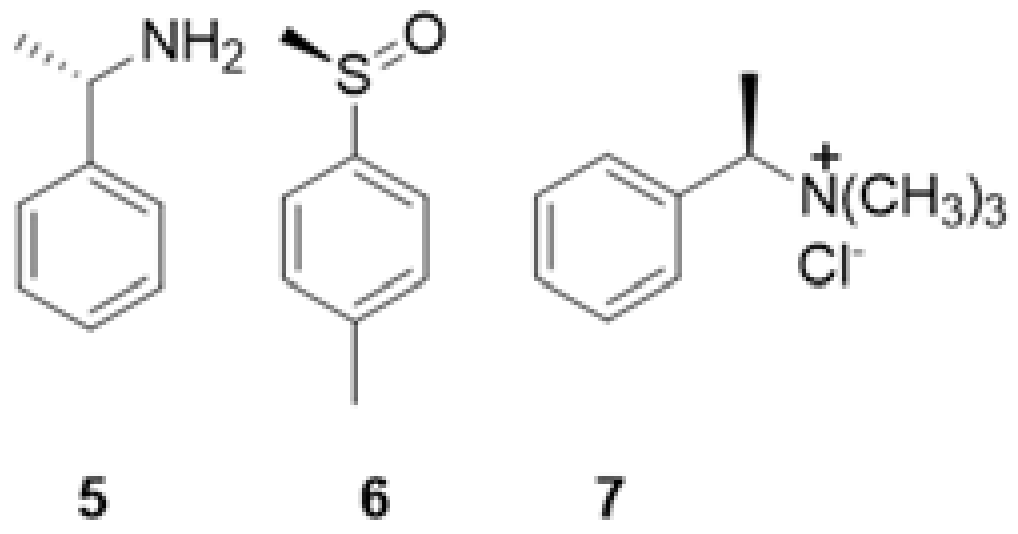
| 5 | 6 | 7 | |
|---|---|---|---|
| CDCl3/CD3OD 97:3 (v/v) | Kmaj = 46 ± 3 a Kmin = 21 ± 1 | - | - |
| CDCl3/CD3OD 99:1 (v/v) | Kmaj = 970 ± 40 Kmin = 450 ± 20 | Kmaj = 710 ± 20 Kmin = 420 ± 10 | Kmaj = 68 ± 6 Kmin = 52 ± 5 |
| L | D | ||
|---|---|---|---|
| Formiate | >106 | ||
| Acetate | >106 | ||
| Glycine | 3800 ± 500 | ||
| Alanine | 2010 ± 60 | 2900 ± 500 | |
| Valine | 1500 ± 200 | 1070 ± 40 | |
| Phenylalanine | 2500 ± 600 | 260 ± 10 | |
| Leucine | 58 ± 7 | 26 ± 7 | |
| Proline | <10 | 62 ± 6 | |
| Tryptophan | <5 | <5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leoni, L.; Dalla Cort, A. The Supramolecular Attitude of Metal–Salophen and Metal–Salen Complexes. Inorganics 2018, 6, 42. https://doi.org/10.3390/inorganics6020042
Leoni L, Dalla Cort A. The Supramolecular Attitude of Metal–Salophen and Metal–Salen Complexes. Inorganics. 2018; 6(2):42. https://doi.org/10.3390/inorganics6020042
Chicago/Turabian StyleLeoni, Luca, and Antonella Dalla Cort. 2018. "The Supramolecular Attitude of Metal–Salophen and Metal–Salen Complexes" Inorganics 6, no. 2: 42. https://doi.org/10.3390/inorganics6020042
APA StyleLeoni, L., & Dalla Cort, A. (2018). The Supramolecular Attitude of Metal–Salophen and Metal–Salen Complexes. Inorganics, 6(2), 42. https://doi.org/10.3390/inorganics6020042