Metal (Hg, Pt, Ru) Bisalkynyl Bridge between Tetrathiafulvalene Electrophores and Electronic Interplay


Abstract
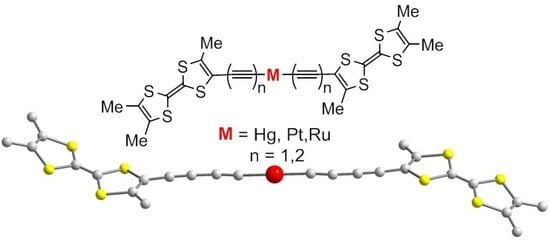
1. Introduction
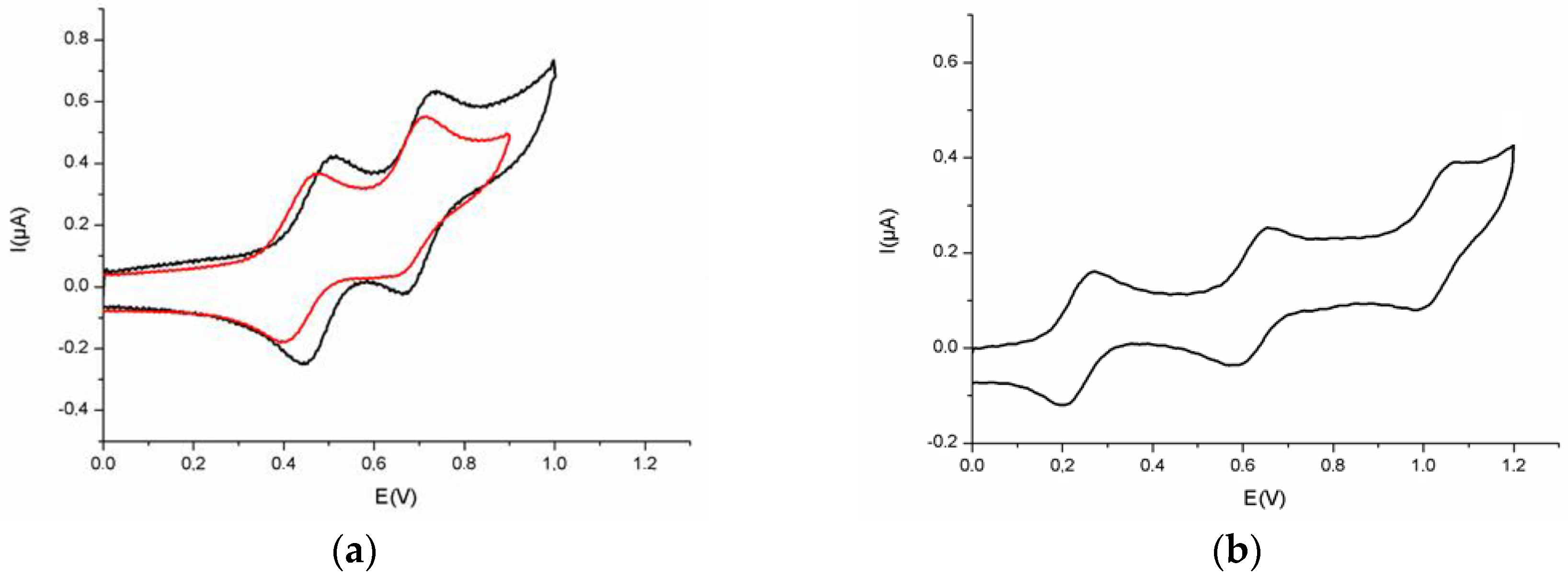
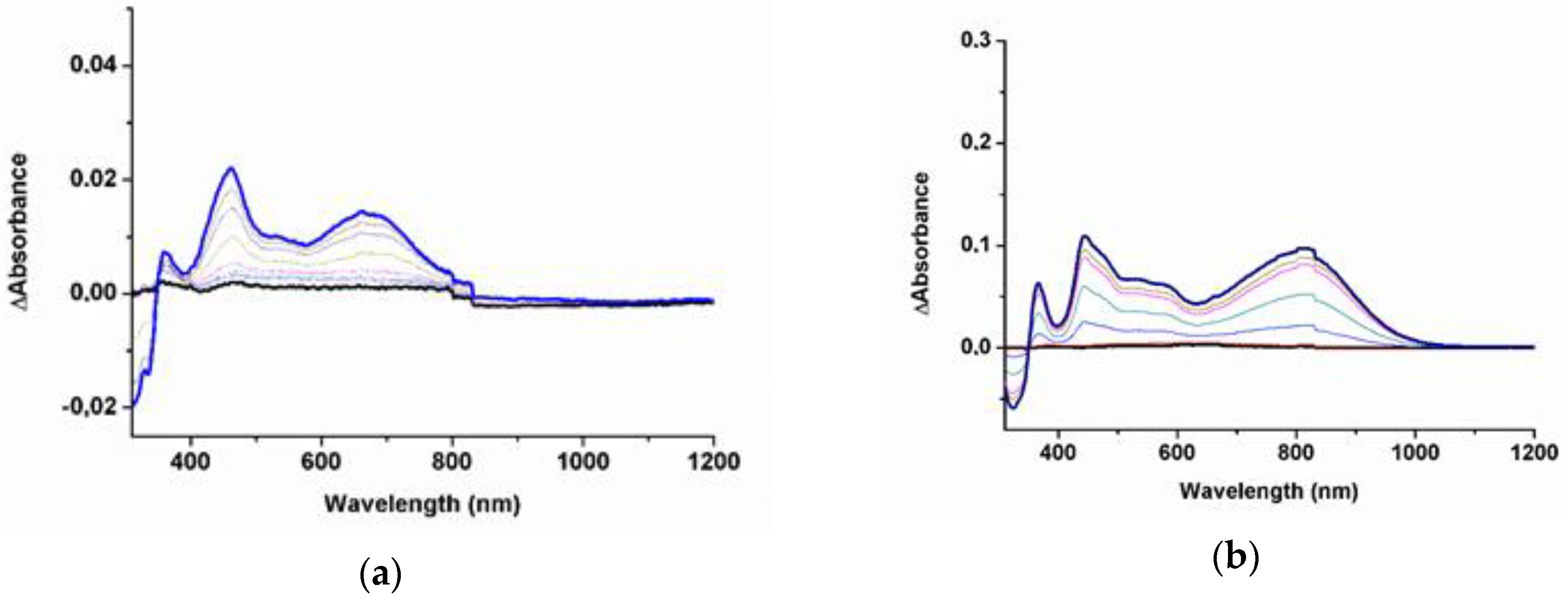
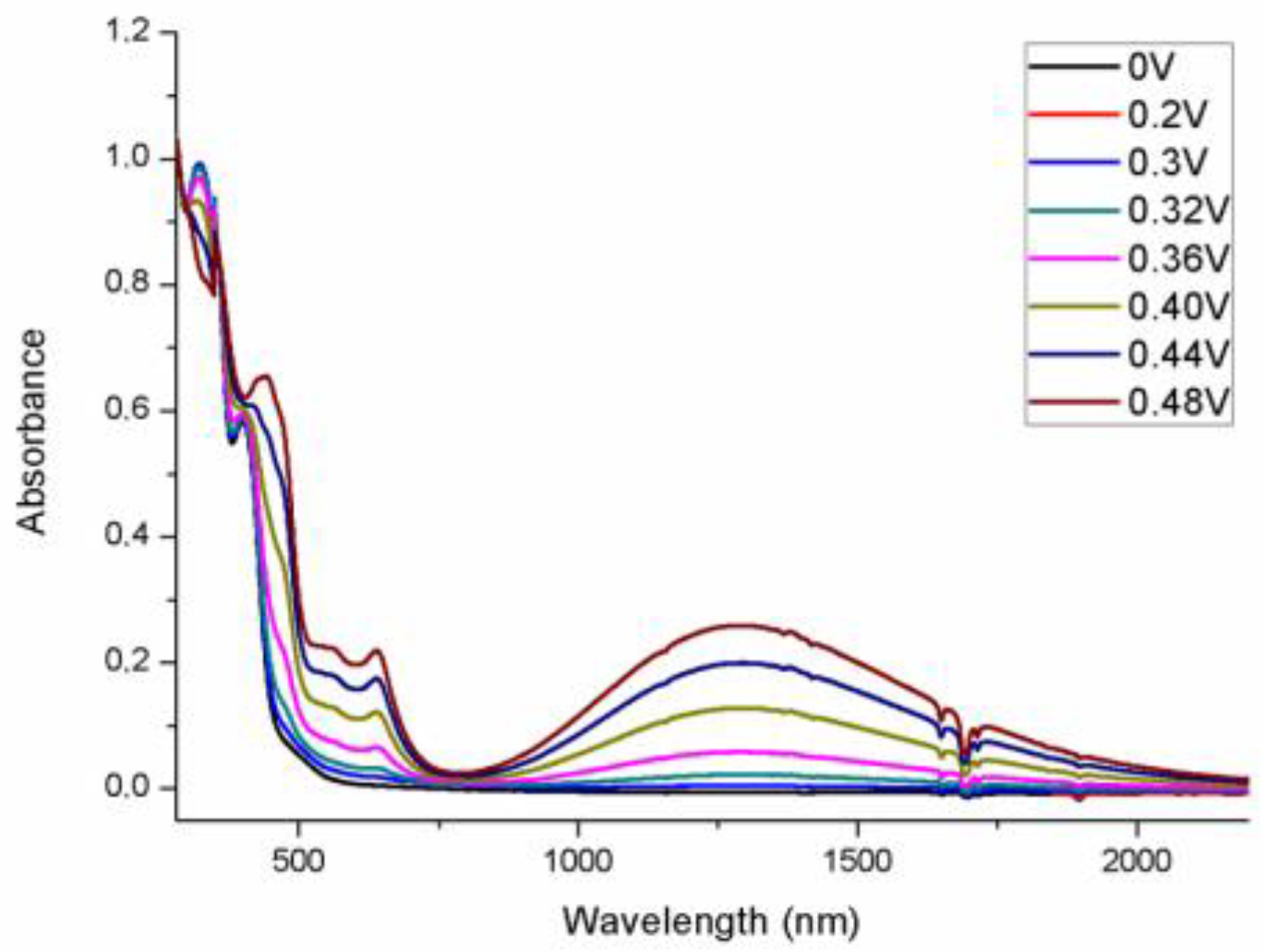
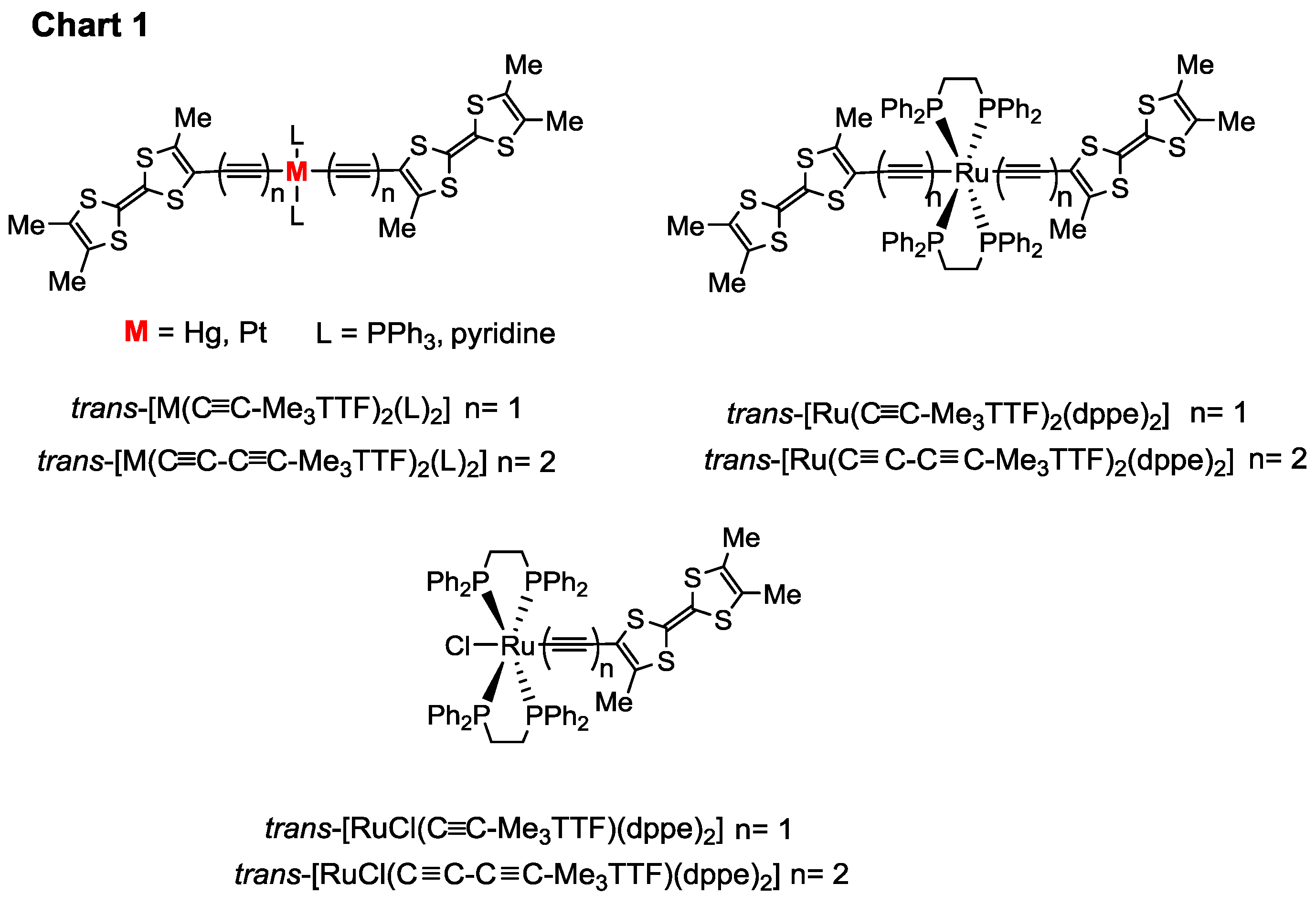
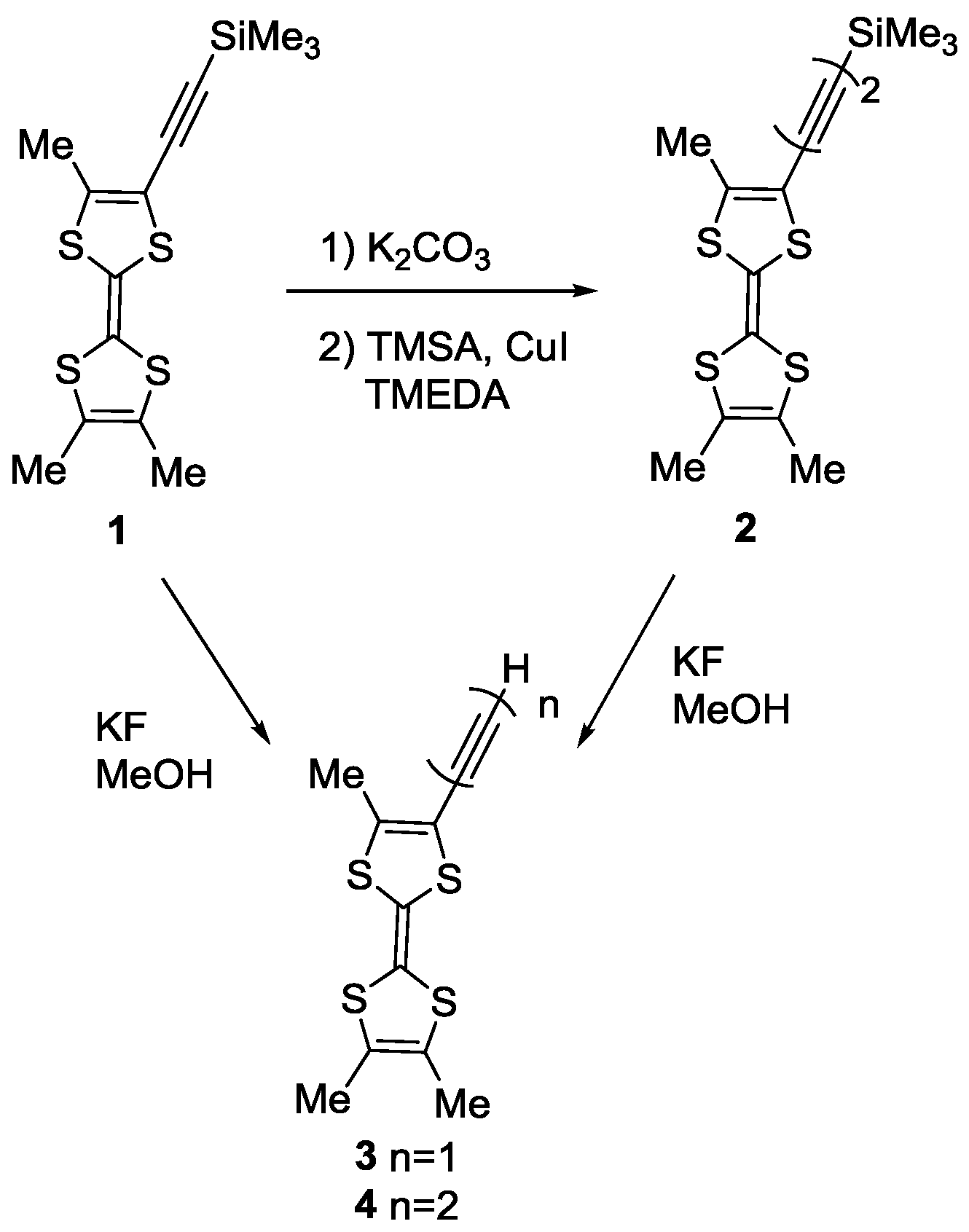
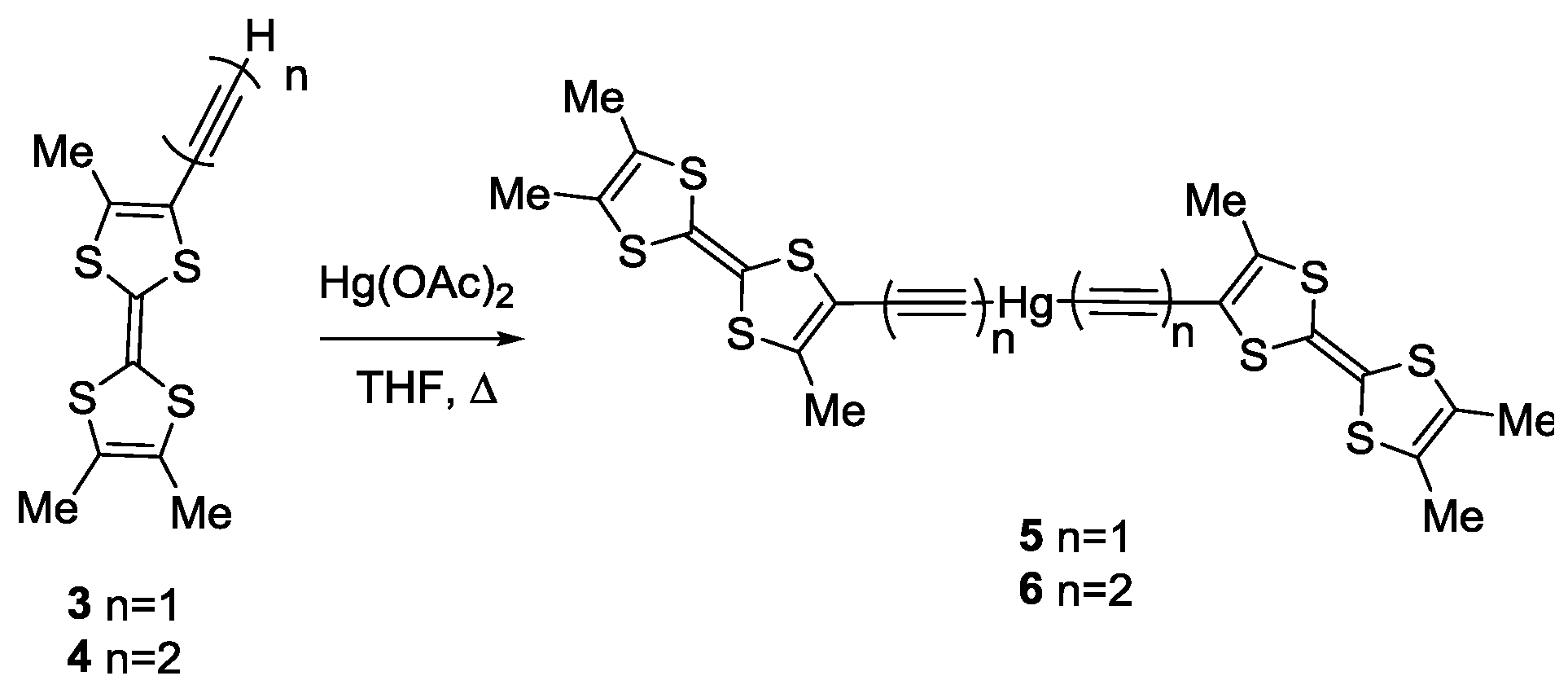
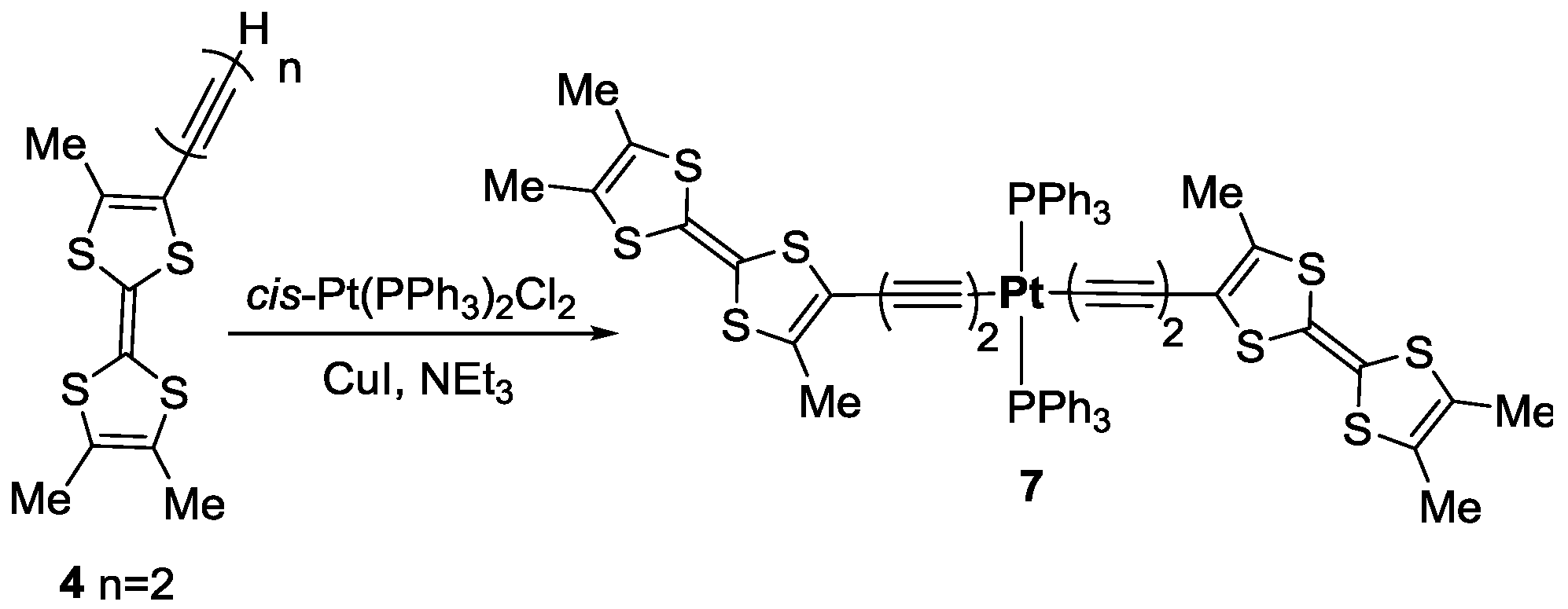
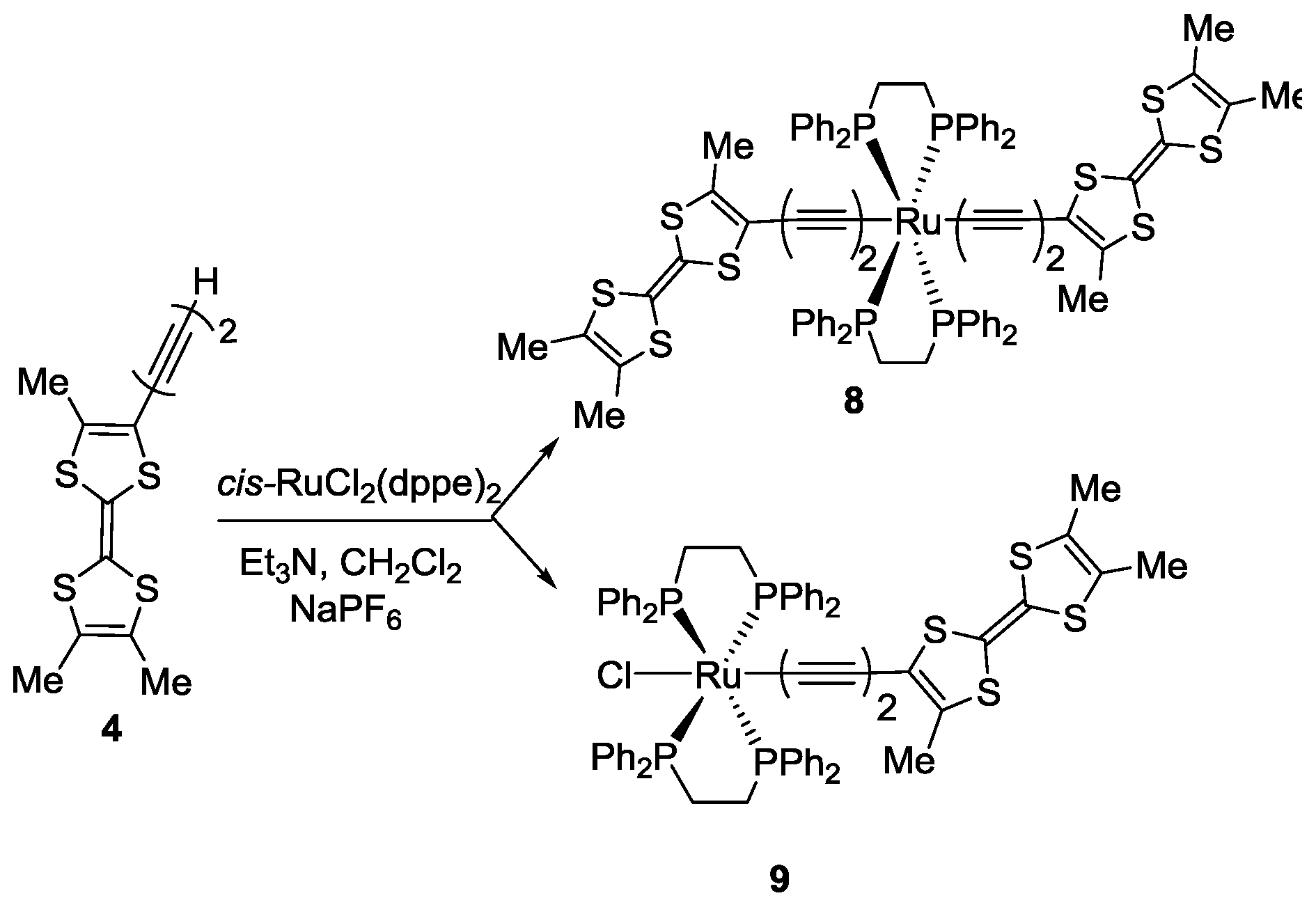
2. Results
3. Materials and Methods
3.1. Synthesis of TTF 2
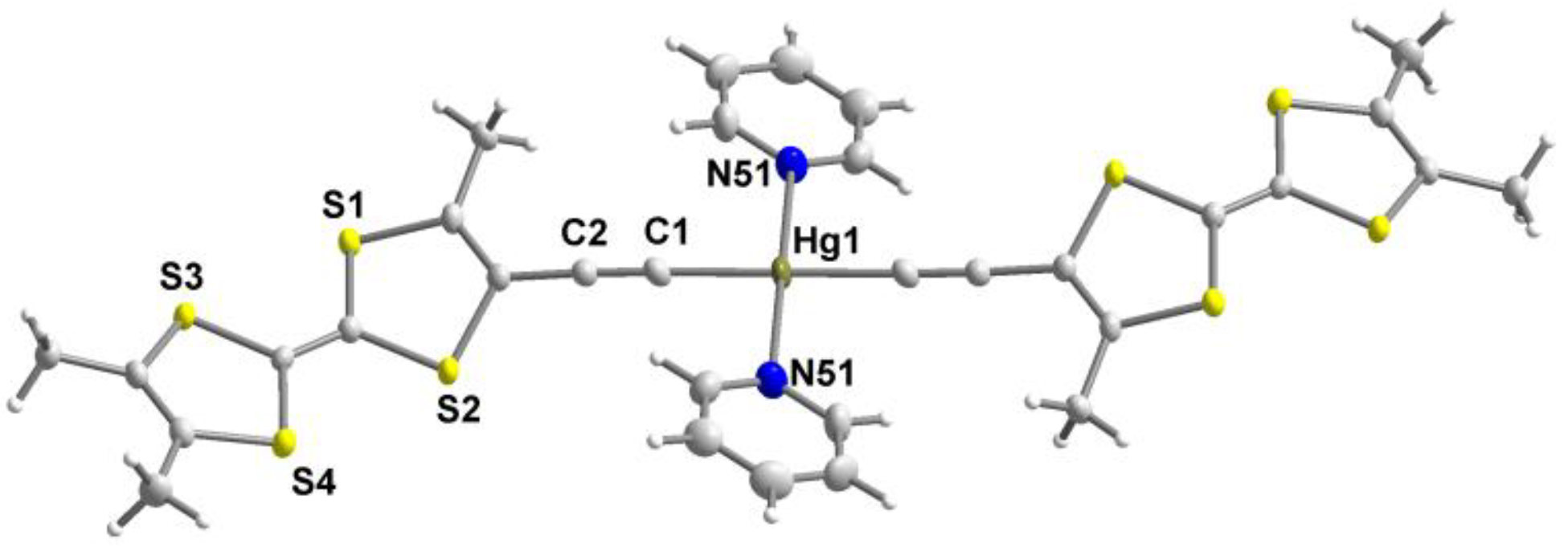
3.2. Synthesis of trans-[Hg(C≡CMe3TTF)2] 5
3.3. Synthesis of trans-[Hg(C≡C–C≡CMe3TTF)2] 6
3.4. Synthesis of trans-[Pt(C≡C–C≡CMe3TTF)2(PPh3)2] 7
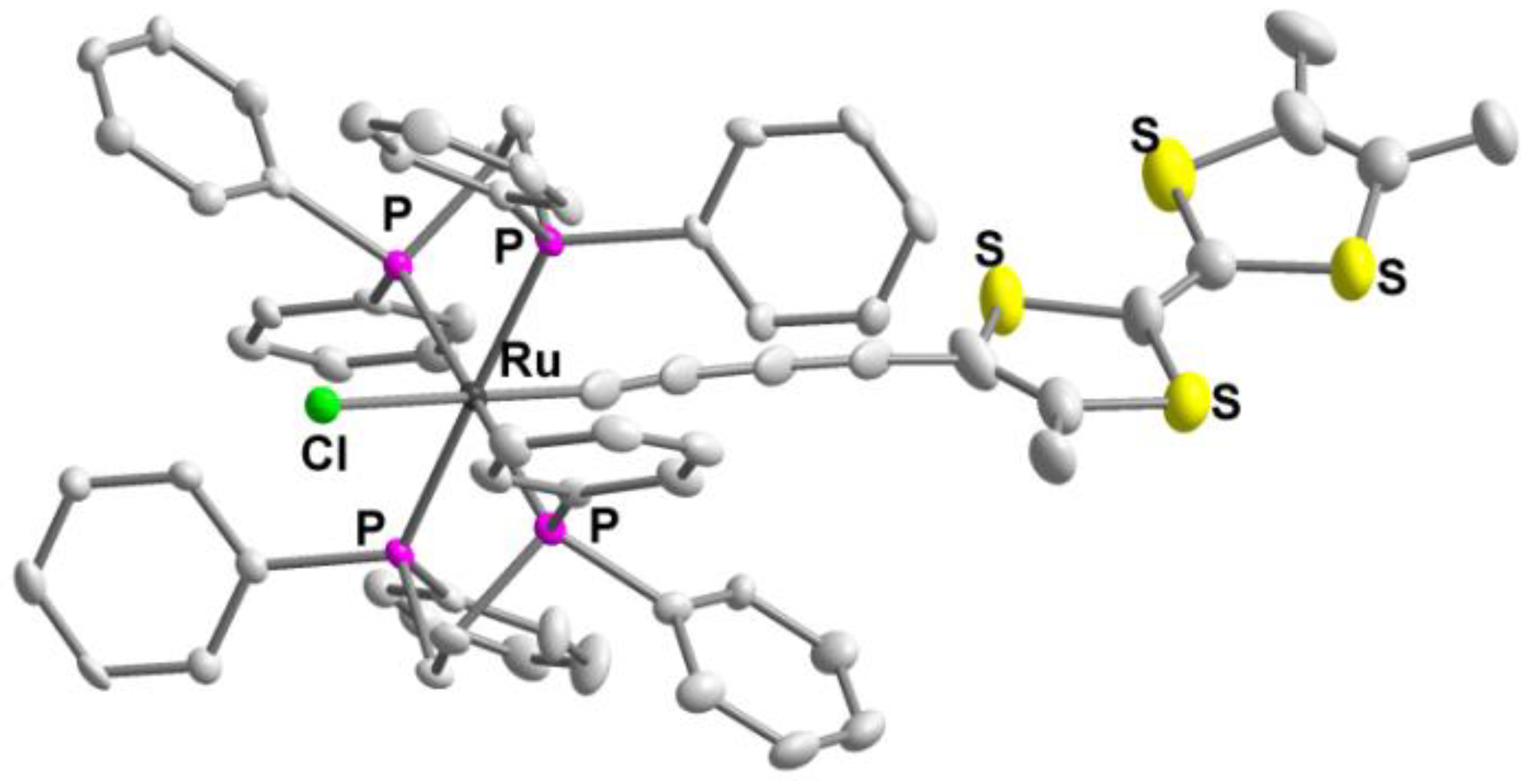
3.5. Synthesis of trans-[RuCl(C≡C–C≡C–Me3TTF)(dppe)2] 9
3.6. Crystallography
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Iyoda, M.; Hasegawa, M.; Miyake, Y. Bi-TTF, bis-TTF and related oligomers. Chem. Rev. 2004, 104, 5085–5114. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, J.O.; Nielsen, M.B.; Becher, J. Tetrathiafulvalene cyclophanes and cage molecules. Chem. Rev. 2004, 104, 5115–5131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Mazzanti, V.; Parker, C.R.; Broman, S.L.; Wallberg, J.H.; Luspai, K.; Brincko, A.; Kjaergaard, H.G.; Kadziola, A.; et al. Interactions between tetrathiafulvalene units in dimeric structures—The influence of cyclic cores. Beilstein J. Org. Chem. 2015, 11, 930–948. [Google Scholar] [CrossRef] [PubMed]
- Frasconi, M.; Kikuchi, T.; Cao, D.; Wu, Y.; Liu, W.-G.; Dyar, S.M.; Barin, G.; Sarjeant, A.A.; Stern, C.L.; Carmieli, R.; et al. Mechanical Bonds and Topological Effects in Radical Dimer Stabilization. J. Am. Chem. Soc. 2014, 136, 11011–11026. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.; Müllen, K. Oligomeric tetrathiafulvalenes: Extended donors for increasing the dimensionality of electrical conduction. Adv. Mater. 1994, 6, 439–459. [Google Scholar] [CrossRef]
- Aqad, E.; Becker, J.Y.; Bernstein, J.; Ellern, A.; Khodorkovsky, V.; Shapiro, L. New η-electron donors containing two tetrathiafulvalene units fused to 1,4-dithiine and a conducting charge transfer complex with tetracyanoquinodimethane. J. Chem. Soc. Chem. Commun. 1994, 2775–2776. [Google Scholar] [CrossRef]
- Danila, I.; Biaso, F.; Sidorenkova, H.; Geoffroy, M.; Fourmigué, M.; Levillain, E.; Avarvari, N. Rigid bis(tetrathiafulvalenes) doubly bridged by phosphino groups and derivatives: Synthesis and intramolecular mixed valence state. Organometallics 2009, 28, 3691–3699. [Google Scholar] [CrossRef][Green Version]
- Avarvari, N.; Fourmigué, M. 1,4-Dihydro-1,4-diphosphinine fused with two tétrathiafulvalènes. Chem. Commun. 2004, 2794–2795. [Google Scholar] [CrossRef] [PubMed]
- Biaso, F.; Geoffroy, M.; Canadell, E.; Auban-Senzier, P.; Levillain, E.; Fourmigué, M.; Avarvari, N. Intramolecular mixed-valence state through silicon or germanium double bridges in rigid bis(Tetrathiafulvalenes). Chem. Eur. J. 2007, 13, 5394–5400. [Google Scholar] [CrossRef] [PubMed]
- Lorcy, D.; Bellec, N.; Fourmigué, M.; Avarvari, N. Tetrathiafulvalene-based group XV ligands: Synthesis, coordination chemistry and radical cation salts. Coord. Chem. Rev. 2009, 253, 1398–1438. [Google Scholar] [CrossRef]
- Bergkamp, J.J.; Decurtins, S.; Liu, S.-X. Current advances in fused tetrathiafulvalene donor–acceptor systems. Chem. Soc. Rev. 2015, 44, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Vajpayee, V.; Bivaud, S.; Goeb, S.; Croué, V.; Allain, M.; Popp, B.V.; Garci, A.; Therrien, B.; Sallé, M. Electron-rich arene–ruthenium metalla-architectures incorporating tetrapyridyl–tetrathiafulvene donor moieties. Organometallics 2014, 33, 1651–1658. [Google Scholar] [CrossRef]
- Massue, J.; Bellec, N.; Chopin, S.; Levillain, E.; Roisnel, T.; Clérac, R.; Lorcy, D. Electroactive ligands: The first metal complexes of tetrathiafulvenyl−acetylacetonate. Inorg. Chem. 2005, 44, 8740–8748. [Google Scholar] [CrossRef] [PubMed]
- Bellec, N.; Massue, J.; Roisnel, T.; Lorcy, D. Chelating ability of a conjugated redox active tetrathiafulvalenyl-acetylacetonate ligand. Inorg. Chem. Comm. 2007, 10, 1172–1176. [Google Scholar] [CrossRef]
- Liu, W.; Xiong, J.; Wang, Y.; Zhou, X.-H.; Wang, R.; Zuo, J.-L.; You, X.-Z. Syntheses, structures, and properties of tricarbonyl (chloro) rhenium(I) complexes with redox-active tetrathiafulvalene−pyrazole ligands. Organometallics 2009, 28, 755–762. [Google Scholar] [CrossRef]
- Vacher, A.; Barriere, F.; Roisnel, T.; Piekara-Sady, L.; Lorcy, D. Electronically coupled tetrathiafulvalene electrophores across a non-innocent acetylide–ruthenium bridge. Organometallics 2011, 30, 3570–3578. [Google Scholar] [CrossRef]
- Vacher, A.; Barrière, F.; Roisnel, T.; Lorcy, D. Electronic communication between metal–organic electrophores in an organometallic ruthenium–acetylide–tetrathiafulvalene complex. Chem. Commun. 2009, 7200–7202. [Google Scholar] [CrossRef] [PubMed]
- Vacher, A.; Barrière, F.; Lorcy, D. Ferrocene and tetrathiafulvalene redox interplay across a bis-acetylide–ruthenium bridge. Organometallics 2013, 32, 6130–6135. [Google Scholar] [CrossRef]
- Nishijo, J.; Judai, K.; Nishi, N. Weak ferromagnetism and strong spin−spin interaction mediated by the mixed-valence ethynyltetrathiafulvalene-type ligand. Inorg. Chem. 2011, 50, 3464–3470. [Google Scholar] [CrossRef] [PubMed]
- Nishijo, J.; Enomoto, M. A Series of weak ferromagnets based on a chromium–acetylide–TTF type complex: Correlation of the structures and magnetic properties and origin of the weak ferromagnetism. Inorg. Chem. 2013, 52, 13263–13268. [Google Scholar] [CrossRef] [PubMed]
- Nishijo, J. Chromium–ethynyltetrathiafulvalene complex based magnetic materials. Polyhedron 2013, 66, 43–47. [Google Scholar] [CrossRef]
- Nishijo, J.; Shima, Y.; Enomoto, M. Synthesis, crystal structures and magnetic properties of new chromium(III)–acetylide–TTF type complexes. Polyhedron 2017, 136, 35–41. [Google Scholar] [CrossRef]
- Vacher, A.; Barrière, F.; Camerel, F.; Bergamini, J.F.; Roisnel, T.; Lorcy, D. Cis and trans-bis(tetrathiafulvalene-acetylide) platinum(II) complexes: Syntheses, crystal structures, and influence of the ancillary ligands on their electronic properties. Dalton Trans. 2013, 42, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Fourmigué, M.; Huang, Y.-S. Evaluation of the extent of interaction within dimeric tetrathiafulvalenes (TTF) incorporating organometallic -Hg-, -SiMe2-, and -PPh- links. Organometallics 1993, 12, 797–802. [Google Scholar] [CrossRef]
- Buschbeck, R.; Low, P.J.; Lang, H. Homoleptic transition metal acetylides. Coord. Chem. Rev. 2011, 255, 241–272. [Google Scholar] [CrossRef]
- Wong, W.-Y. Mercury alkynyls as versatile templates for new organometallic materials and polymers. Coord. Chem. Rev. 2007, 251, 2400–2427. [Google Scholar] [CrossRef]
- Bruce, M.I.; Halet, J.-F.; Le Guennic, B.; Skelton, B.W.; Smith, M.E.; White, A.H. Preparation and molecular structure of Hg{C≡CC≡C[Ru(dppe)Cp*]}2-non linearity in a molecular rod. Inorg. Chim. Acta 2003, 350, 175–181. [Google Scholar] [CrossRef]
- Vacher, A.; Auffray, M.; Barrière, F.; Roisnel, T.; Lorcy, D. Electronic interplay between TTF and extended-TCNQ electrophores along a ruthenium bis(acetylide) linker. Org. Lett. 2017, 19, 6060–6063. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, C.; Touchard, D.; Le Pichon, L.; Daridor, A.; Toupet, L.; Dixneuf, P.H. Mono- and bis-alkynyl ruthenium (II) complexes containing the ferrocenyl moiety; crystal structure of trans-[Ru(C≡CC5H4FeC5H5)2(Ph2PCH2CH2PPh2)] and electrochemical studies. Inorg. Chim. Acta 1998, 272, 18–196. [Google Scholar] [CrossRef]
- Yzambart, G.; Fabre, B.; Camerel, F.; Roisnel, T.; Lorcy, D. Controlled grafting of tetrathiafulvalene (TTF) containing diacetylenic units on hydrogen-terminated silicon surfaces: From redox-active TTF monolayer to polymer films. J. Phys. Chem. C 2012, 116, 12093–12102. [Google Scholar] [CrossRef]
- Jones, G.E.; Kendrick, D.A.; Holmes, A.B. 1,4-Bis(trimethylsilyl)buta-1,3-diyne. Org. Synth. 1987, 65, 52. [Google Scholar] [CrossRef]
- Basseti, M.; Floris, B.; Illuminati, G. The reaction of ethynylferrocene with mercuric acetate. Organometallics 1985, 4, 617–623. [Google Scholar] [CrossRef]
- Dietrich, S.; Mansilla, N.; Hildebrandt, A.; Wetzold, N.; Rheinwald, G. Alkynyl Ti–M complexes with M=Cd and Hg: Synthesis, characterization, and reaction chemistry. J. Organomet. Chem. 2011, 696, 2491–2498. [Google Scholar] [CrossRef]
- Dewhurst, R.D.; Hill, A.F.; Smith, M.K. Hazards associated with bis(alkynyl)mercurials. Organometallics 2006, 25, 2388–2389. [Google Scholar] [CrossRef]
- Sadowy, A.L.; Ferguson, M.J.; McDonald, R.; Tykwinski, R.R. Chiral cis-platinum acetylide complexes via diphosphine ligand exchange: Effect of the ligand. Organometallics 2008, 27, 6321–6325. [Google Scholar] [CrossRef]
- Colbert, M.C.B.; Lewis, J.; Long, N.J.; Raithby, P.R.; White, A.J.P.; Williams, D.J. Synthetic, structural, electrochemical and electronic characterisation of heterobimetallic bis(acetylide) ferrocene complexes. J. Chem. Soc. Dalton Trans. 1997, 99–104. [Google Scholar] [CrossRef]
- Miyazaki, A.; Ogyu, Y.; Justaud, F.; Ouahab, L.; Cauchy, T.; Halet, J.-F.; Lapinte, C. Synthesis, Molecular Structure, Properties, and Electronic Structures of [Cp*(dppe)FeC≡C–TTFMe3][PF6]n (n = 0, 1): Electronic Coupling between the Inorganic and Organic Electrophores. Organonetallics 2010, 29, 4628–4638. [Google Scholar] [CrossRef]
- Justaud, F.; Gendron, F.; Ogyu, Y.; Kumamoto, Y.; Miyazaki, A.; Ouahab, L.; Costuas, K.; Halet, J.-F.; Lapinte, C. Hybrid Molecular Systems Containing Tetrathiafulvalene and Iron-Alkynyl Electrophores: Five-Component Functional Molecules Obtained from C–H Bond Activation. Chem. Eur. J. 2013, 19, 5742–5757. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R.J. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | a | b | c | νC≡C (cm−1) | Ref. |
|---|---|---|---|---|---|
| TTF 1 (R = SiMe3) | 1.422(3) | 1.209(3) | 1.841(3) | 2140 | [16] |
| TTF 3 (R = H) | 1.408(6) | 1.152(8) | 0.950(6) | 2090 | [16] |
| trans-[Pt(C≡CMe3TTF)2(PPh3)2] | 1.422(6) | 1.220(5) | 1.999(5) | 2086 | [23] |
| trans-[Ru(C≡CMe3TTF)2(dppe)2] | 1.423(3) | 1.203(3) | 2.069(3) | 2029 | [16] |
| trans-[Hg(C≡CMe3TTF)2(Pyr)2] | 1.431(9) | 1.196(9) | 2.032(6) | 2141 | this work |

| Compound | a | b | c | d | e | νC≡C (cm−1) | Ref. |
|---|---|---|---|---|---|---|---|
| TTF 2 (R = SiMe3) | 1.414(5) | 1.205(5) | 1.376(5) | 1.208(5) | 1.845(4) | 2181, 2097 | [30] |
| TTF 4 (R = H) | 1.416(4) | 1.199(4) | 1.376(4) | 1.177(4) | 0.950(3) | 2201 | [30] |
| 8 (R = Ru) | 1.414(3) | 1.215(3) | 1.370(3) | 1.218(3) | 2.051(2) | 2129, 1993 | [28] |
| 9 (R = Ru) | 1.368(10) | 1.272(9) | 1.253(9) | 1.202(9) | 2.075(6) | 2134, 2011 | this work |
| Compound | E1 (TTF) | E2 (TTF) | Eox (Ru) | Ref. |
|---|---|---|---|---|
| TTF 3 | 0.38 (0.47 a) | 0.88 (0.70 a) | - | [17] (a this work) |
| TTF 4 | 0.40 (0.50 a) | 0.90 (0.71 a) | - | [30] (a this work) |
| trans-[Hg(C≡CMe3TTF)2] 5 | 0.43 a | 0.67 a | - | this work |
| trans-[Hg(C≡CMe3TTF)2(pyr)2] | 0.47 a | 0.72 a | - | this work |
| trans-[Hg((C≡C–)2Me3TTF)2] 6 | 0.47 | 0.70 | - | this work |
| trans-[Pt(C≡CMe3TTF)2(PPh3)2] | 0.21 | 0.72 | - | [23] |
| trans-[Pt((C≡C–)2Me3TTF)2(PPh3)2] 7 | 0.33 | 0.83 | - | this work |
| trans-[Ru(C≡CMe3TTF)2(dppe)2] | 0.05/0.16 | 0.58/0.69 | 1.33 | [16] |
| trans-[Ru((C≡C–)2Me3TTF)2(dppe)2] 8 | 0.24 | 0.69/0.76 | 1.13 | [28] |
| trans-[RuCl(C≡CMe3TTF)(dppe)2] | 0.07 | 0.52 | 1.07 | [17] |
| trans-[RuCl((C≡C–)2Me3TTF)(dppe)2] 9 | 0.23 | 0.62 | 1.02 | this work |
| Compound | 5 | 9 |
|---|---|---|
| Formula | C22H18HgS8, 2C5H5N | C65H57ClP4RuS4, 2CH2Cl2 |
| FW (g·mol−1) | 897.63 | 1396.6 |
| Crystal system | triclinic | triclinic |
| space group | ||
| a (Å) | 8.8323(5) | 9.1798(4) |
| b (Å) | 10.0293(5) | 12.9853(7) |
| c (Å) | 11.0770(6) | 27.0066(14) |
| α (°) | 63.264(2) | 77.795(3) |
| β (°) | 73.055(3) | 83.777(2) |
| γ (°) | 80.624(2) | 80.909(2) |
| V (Å3) | 837.69(8) | 3097.6(3) |
| T(K) | 150(2) | 150(2) |
| Z | 1 | 2 |
| Dcalc (g·cm−3) | 1.779 | 1.497 |
| μ (mm−1) | 5.119 | 0.749 |
| Total refls | 11,468 | 40,676 |
| Uniq. refls.(Rint) | 3813(0.0346) | 14,112(0.0949) |
| Unique refls. (I > 2s(I)) | 3641 | 8399 |
| R1, wR2 | 0.0350, 0.0904 | 0.0783, 0.1977 |
| R1, wR2 (all data) | 0.0373, 0.0918 | 0.1298, 0.2281 |
| GoF | 1.065 | 1.071 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auffray, M.; Vacher, A.; Roisnel, T.; Lorcy, D. Metal (Hg, Pt, Ru) Bisalkynyl Bridge between Tetrathiafulvalene Electrophores and Electronic Interplay. Inorganics 2018, 6, 43. https://doi.org/10.3390/inorganics6020043
Auffray M, Vacher A, Roisnel T, Lorcy D. Metal (Hg, Pt, Ru) Bisalkynyl Bridge between Tetrathiafulvalene Electrophores and Electronic Interplay. Inorganics. 2018; 6(2):43. https://doi.org/10.3390/inorganics6020043
Chicago/Turabian StyleAuffray, Morgan, Antoine Vacher, Thierry Roisnel, and Dominique Lorcy. 2018. "Metal (Hg, Pt, Ru) Bisalkynyl Bridge between Tetrathiafulvalene Electrophores and Electronic Interplay" Inorganics 6, no. 2: 43. https://doi.org/10.3390/inorganics6020043
APA StyleAuffray, M., Vacher, A., Roisnel, T., & Lorcy, D. (2018). Metal (Hg, Pt, Ru) Bisalkynyl Bridge between Tetrathiafulvalene Electrophores and Electronic Interplay. Inorganics, 6(2), 43. https://doi.org/10.3390/inorganics6020043