Biodegradation of Cosmetics Products: A Computational Study of Cytochrome P450 Metabolism of Phthalates



Abstract
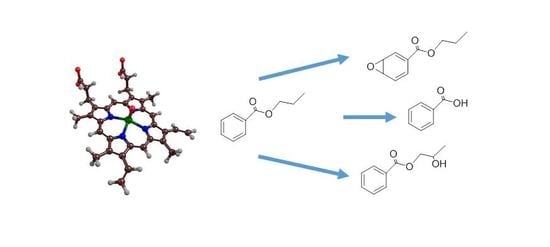
:
1. Introduction
2. Results
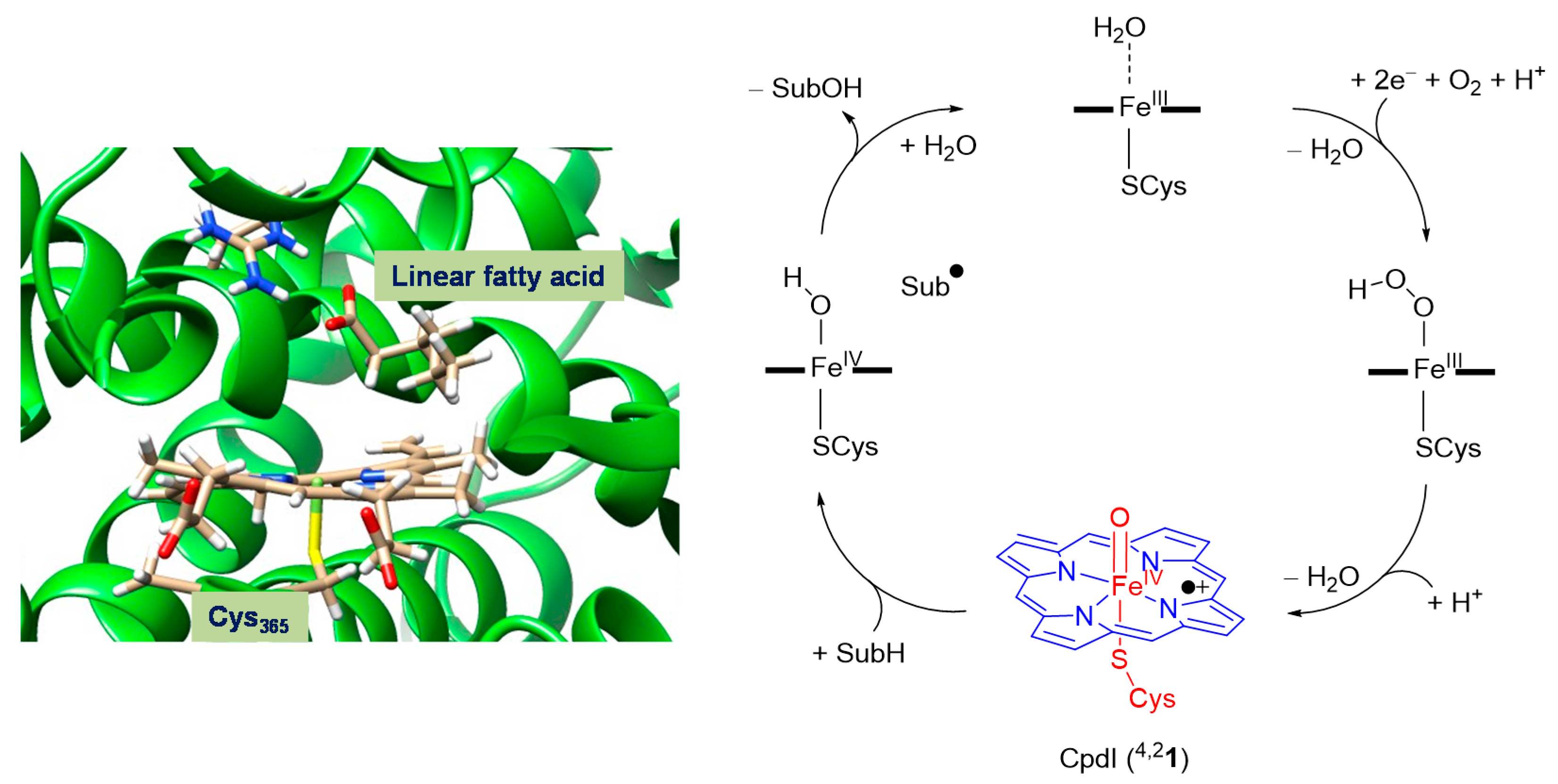
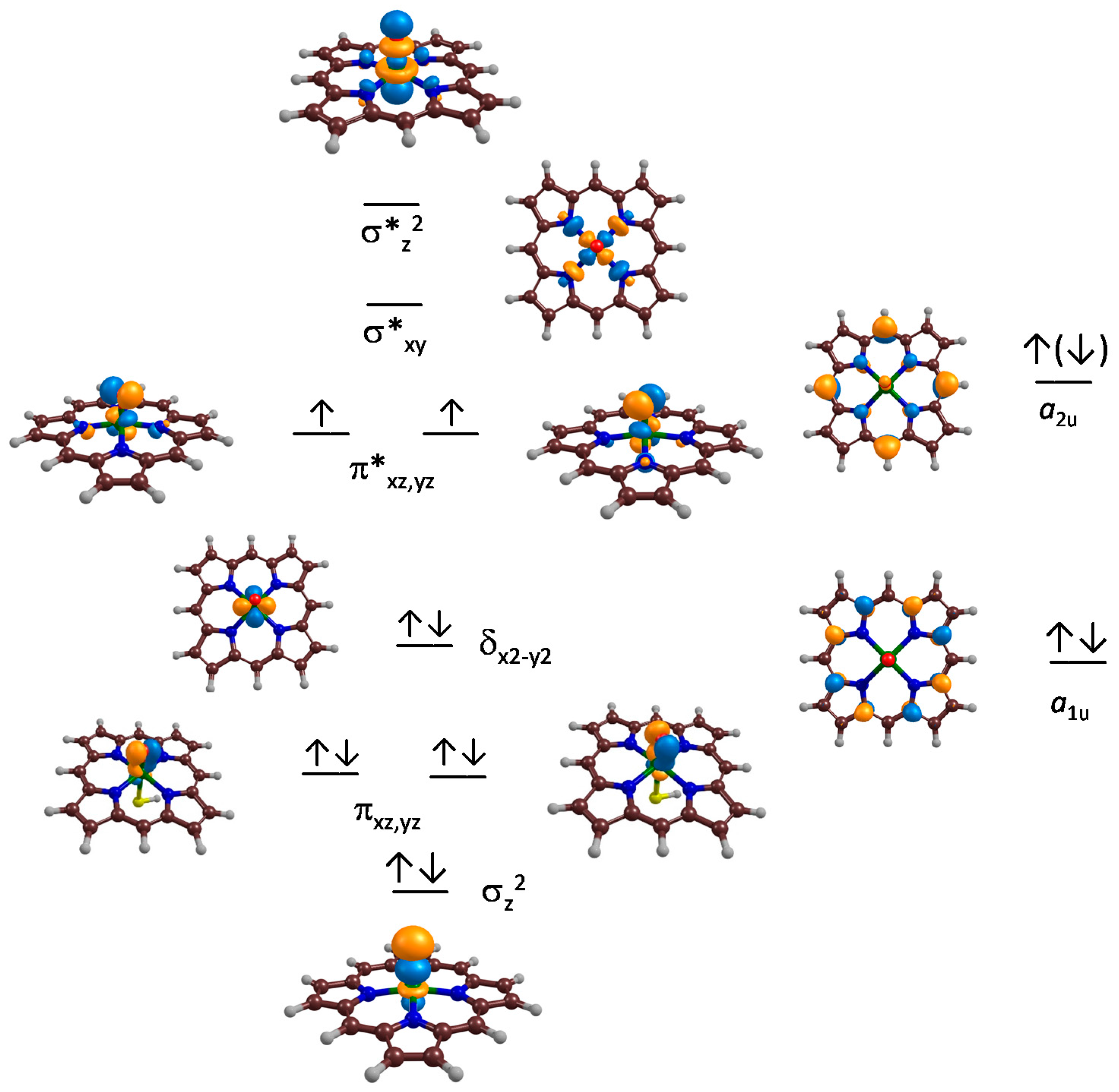
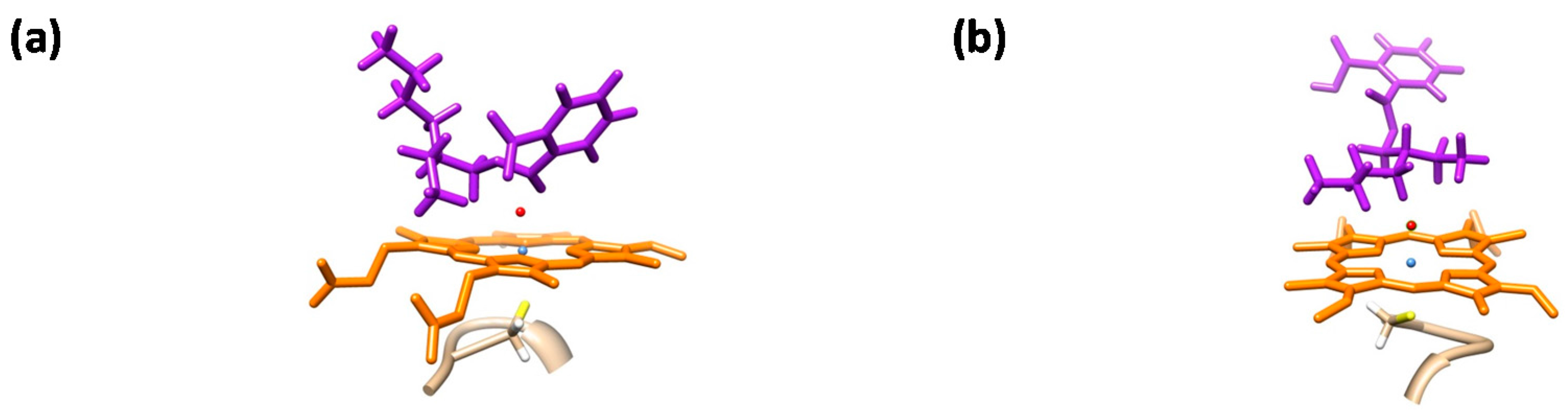
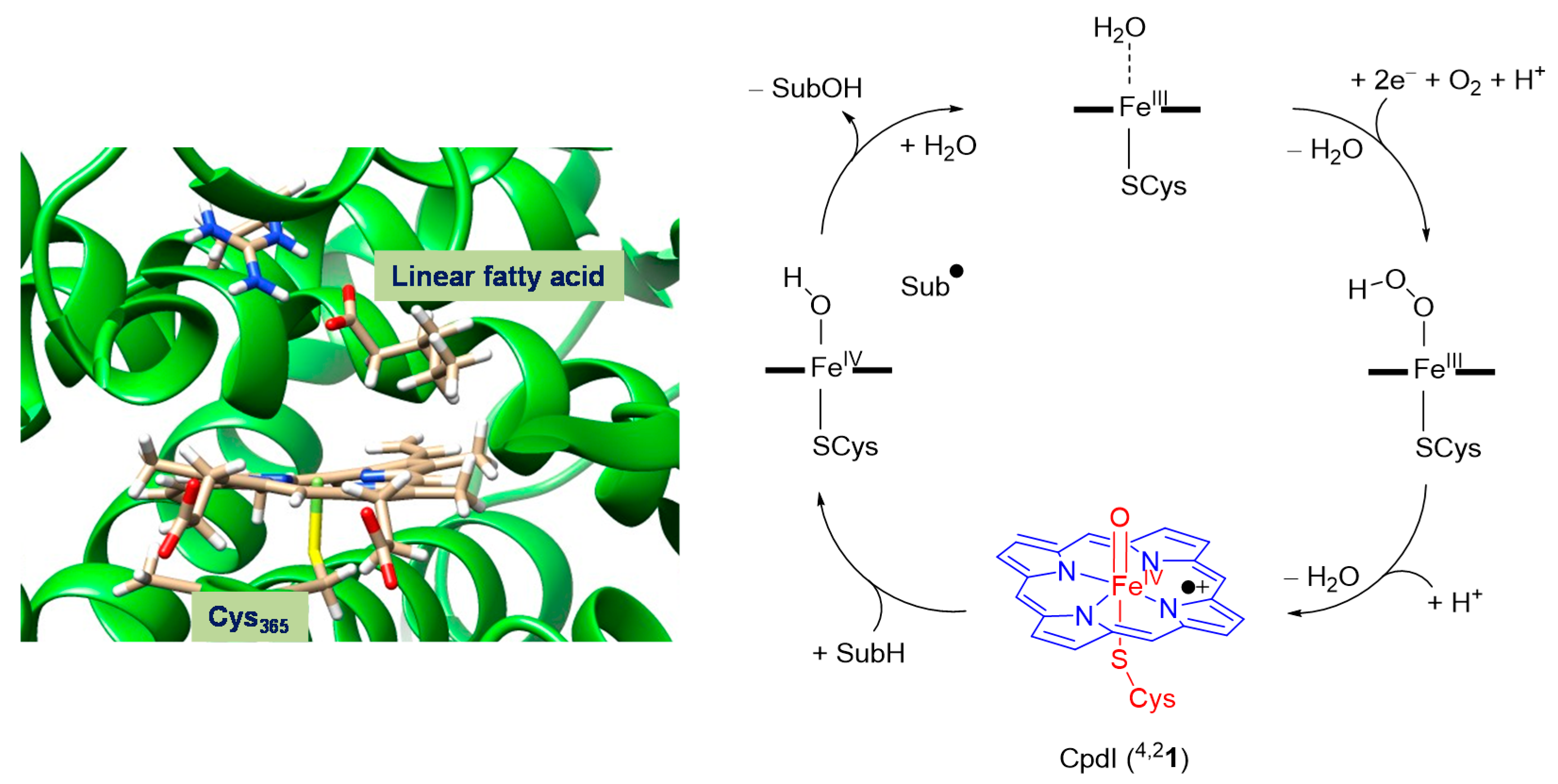
2.1. CpdI Structure and Electronic Configuration
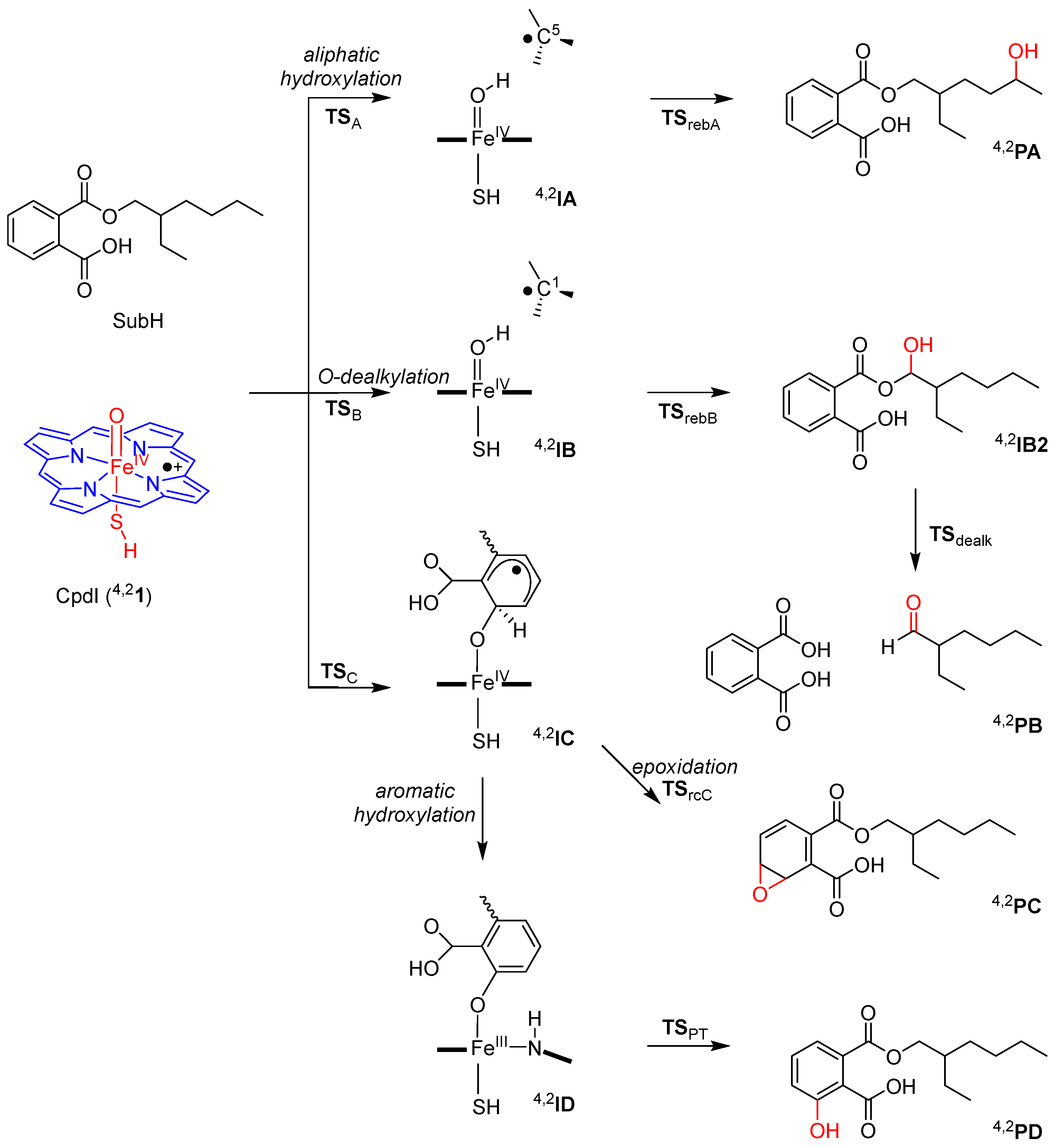
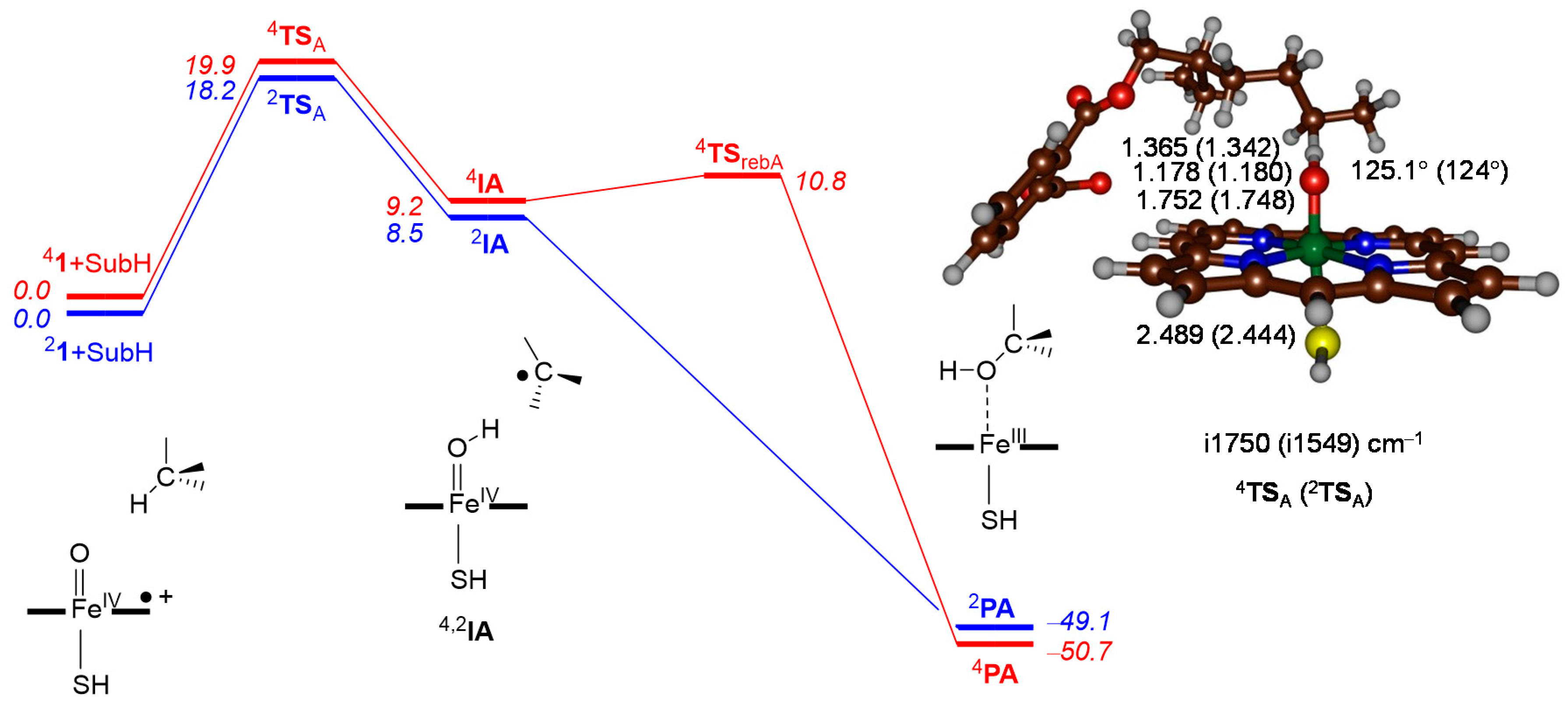
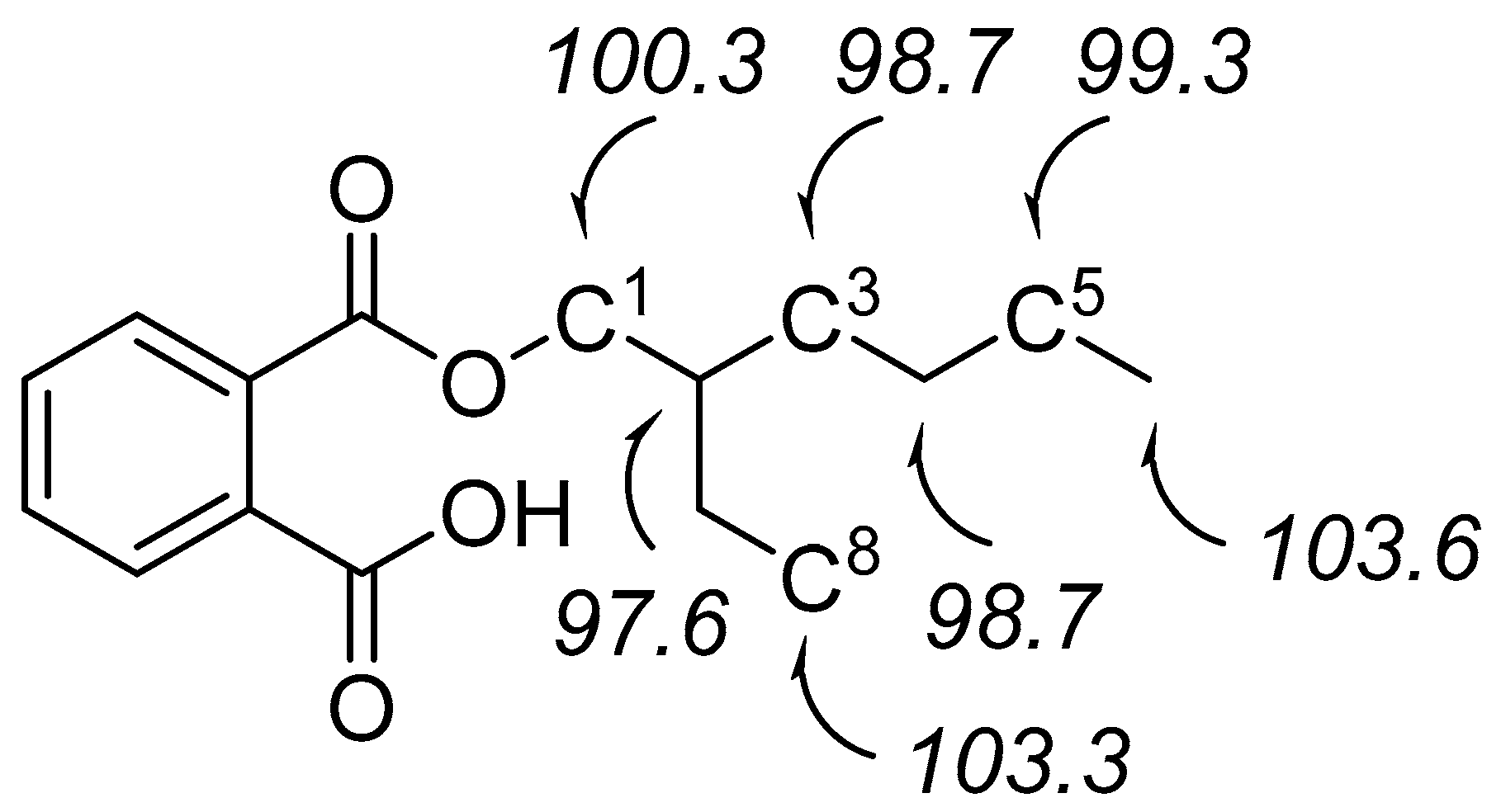
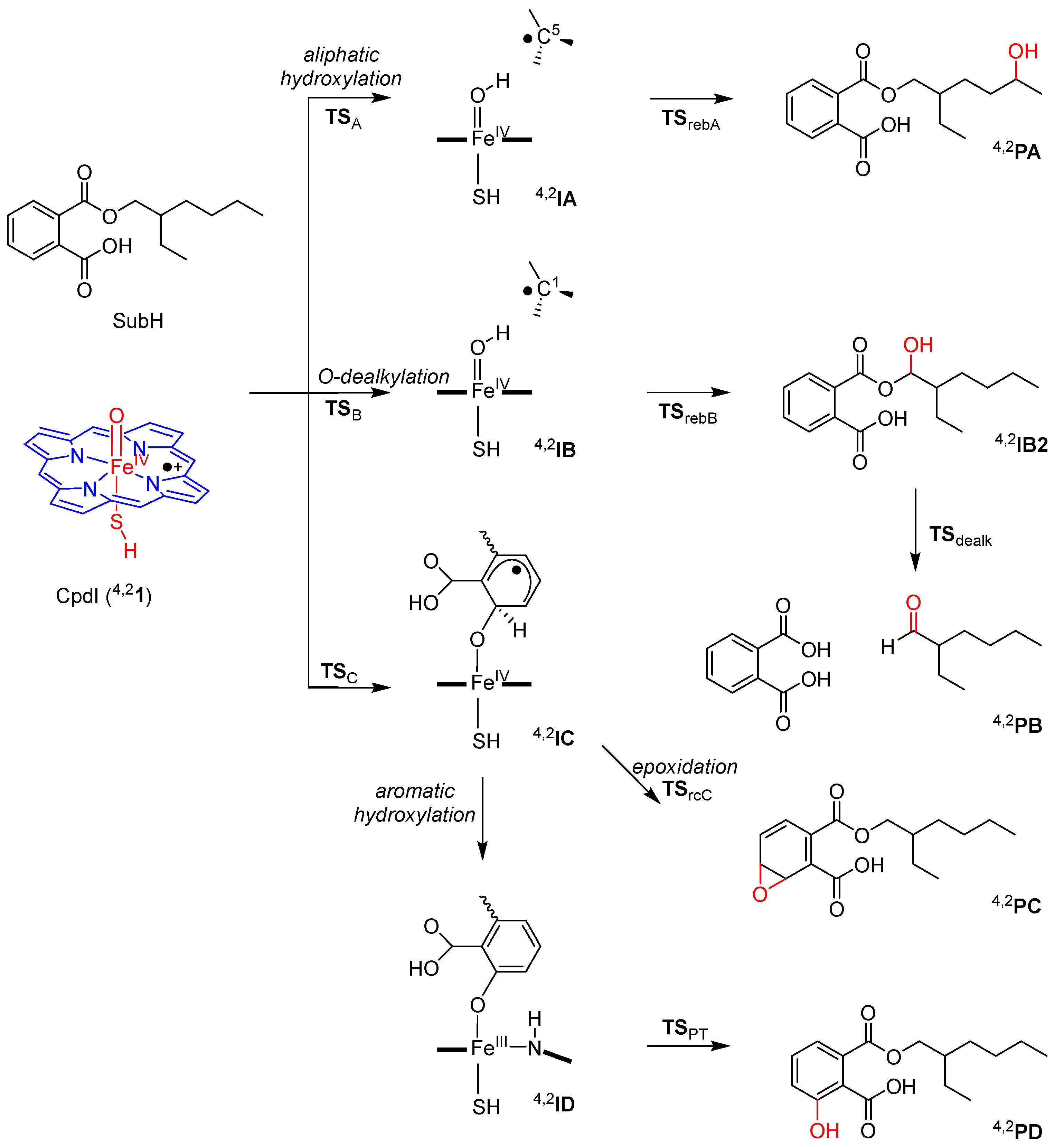
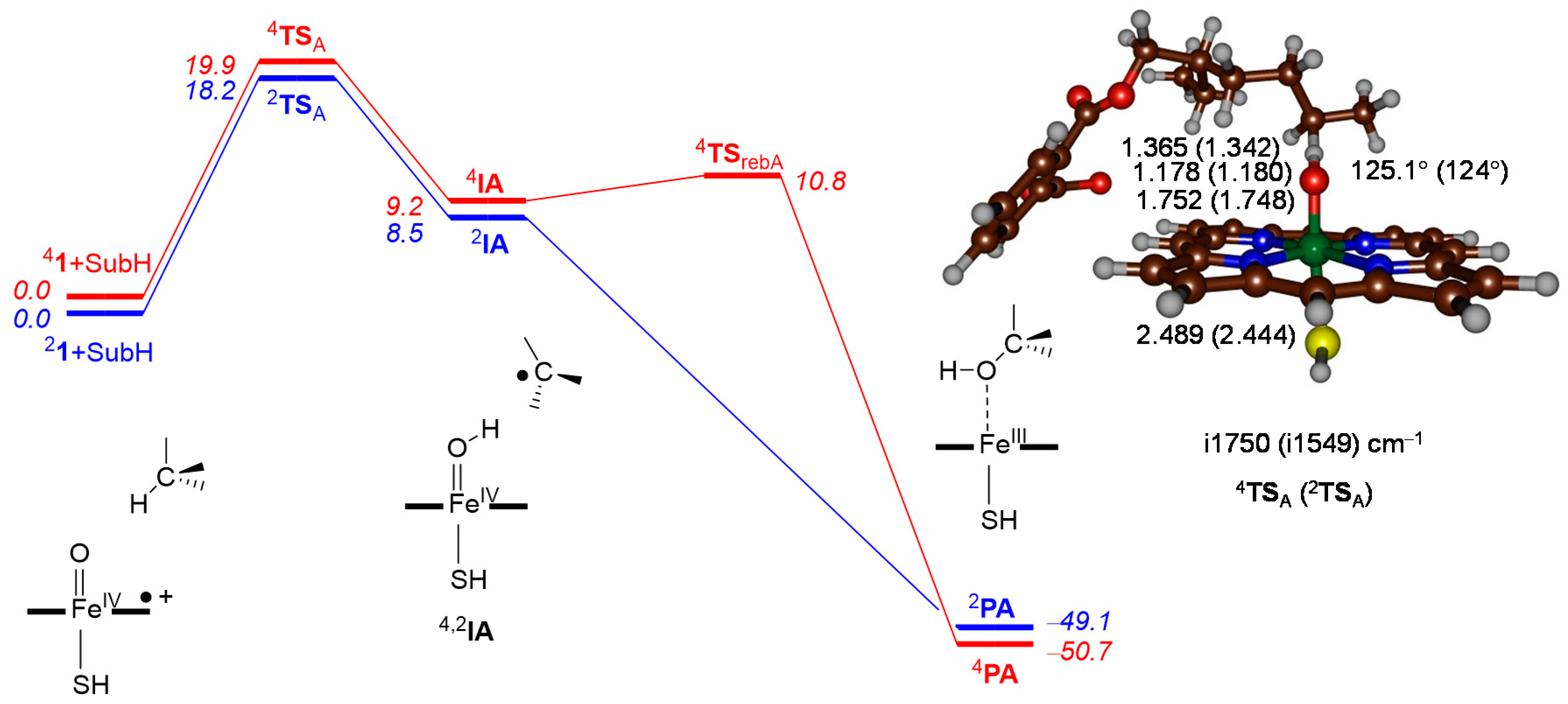
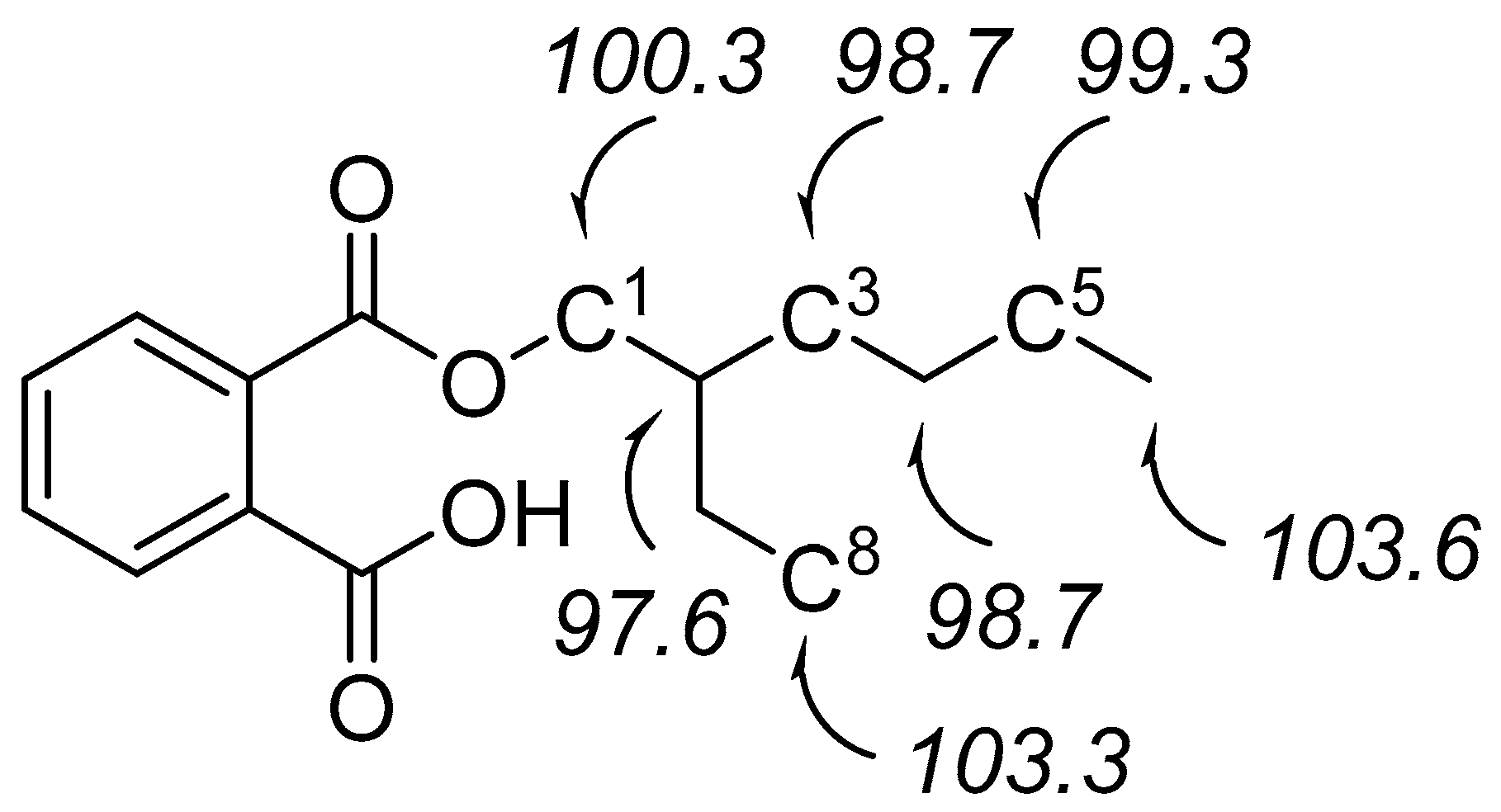
2.2. Phthalate Hydroxylation
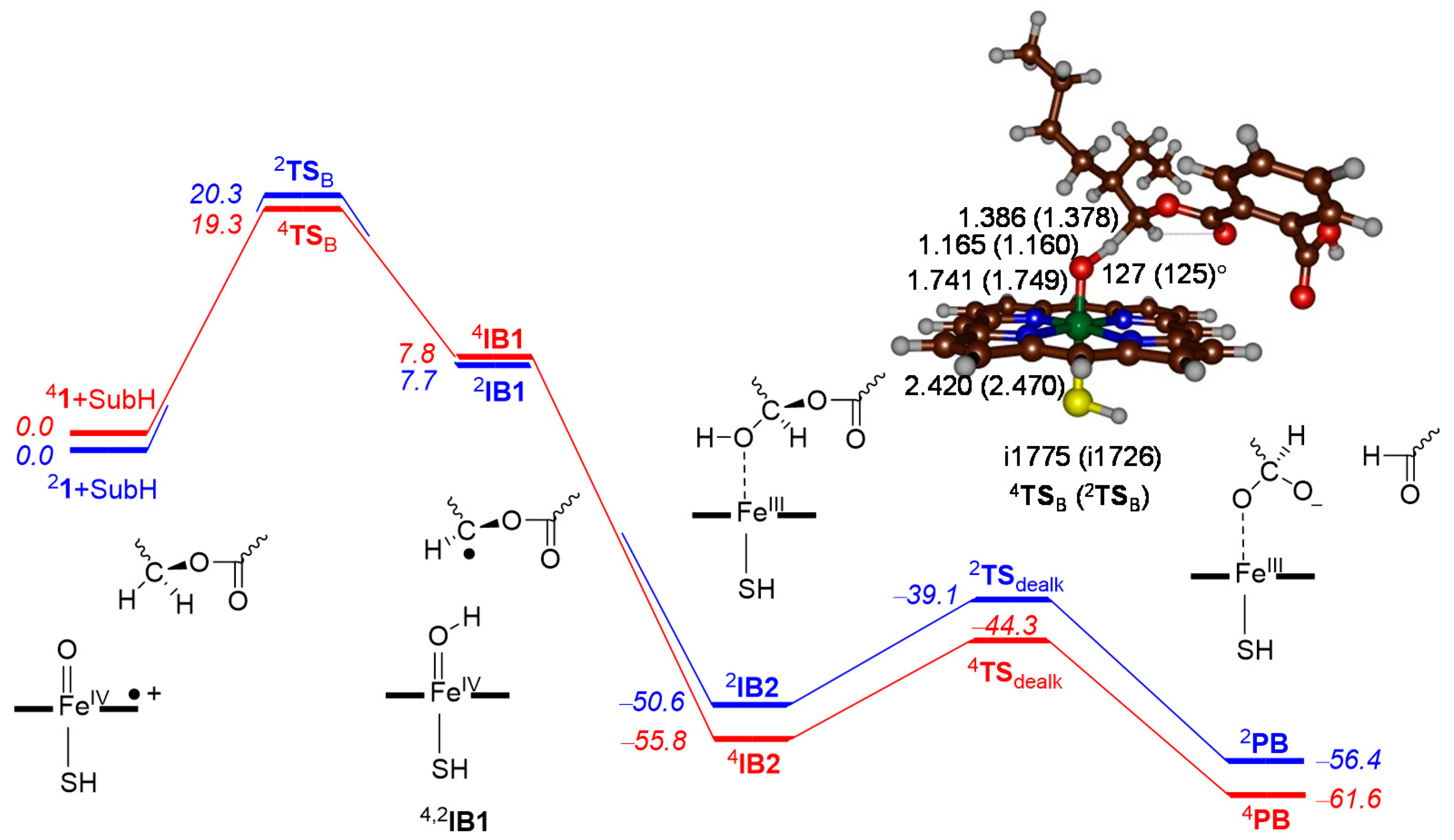
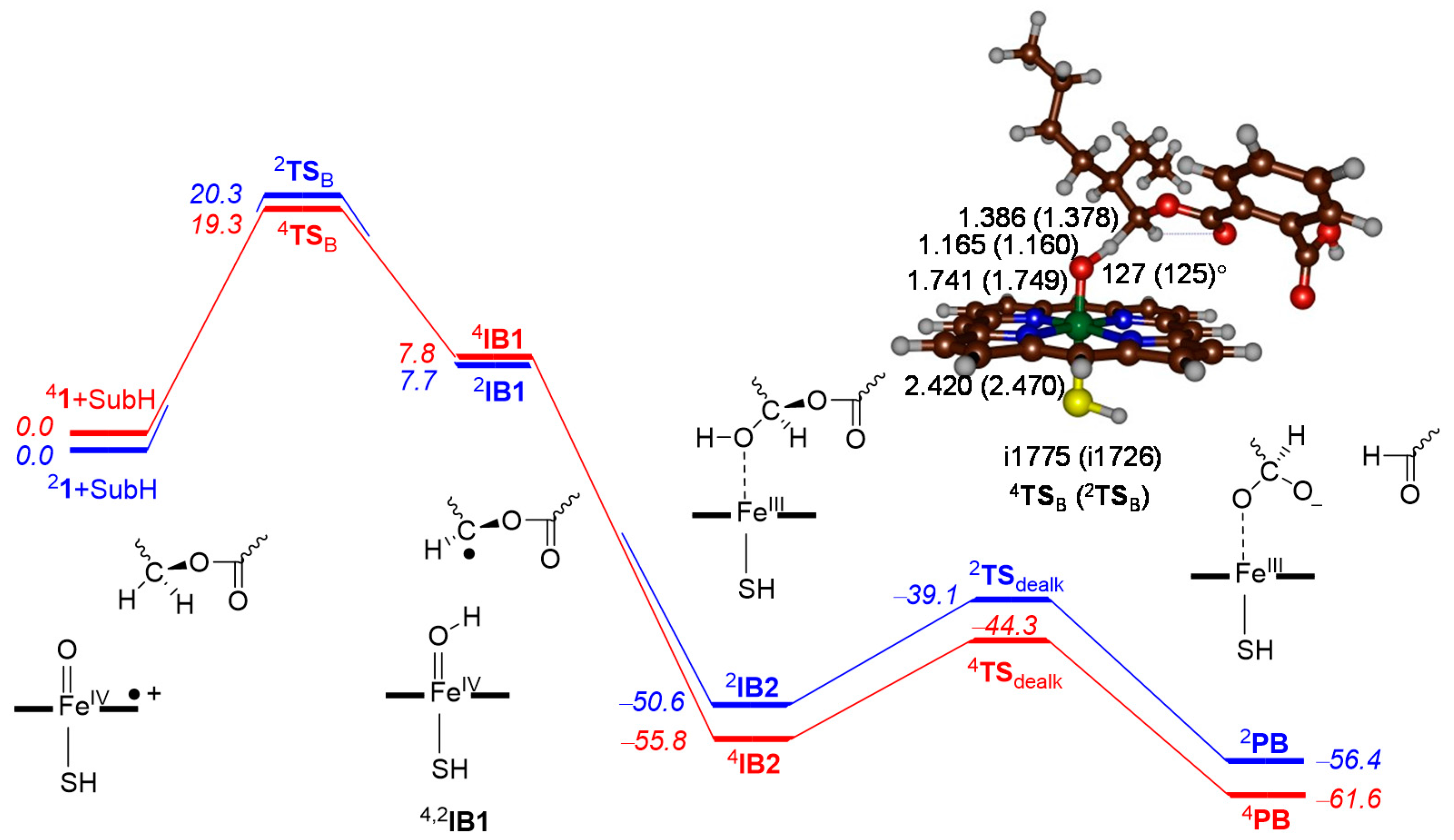
2.3. Phthalate O-Dealkylation
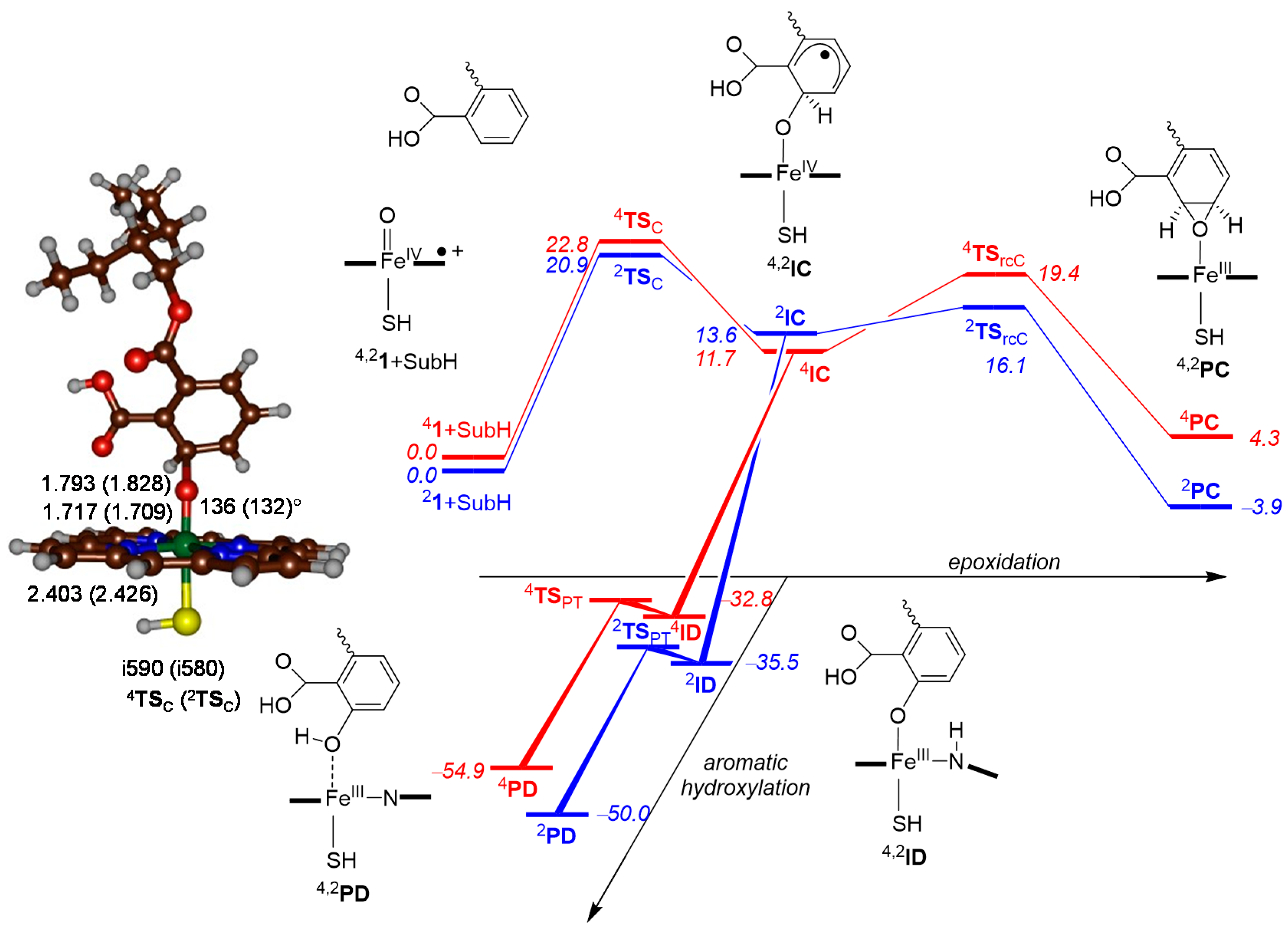
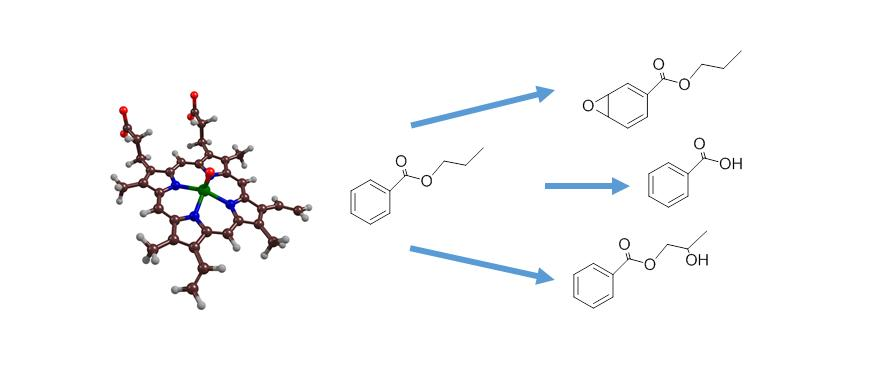
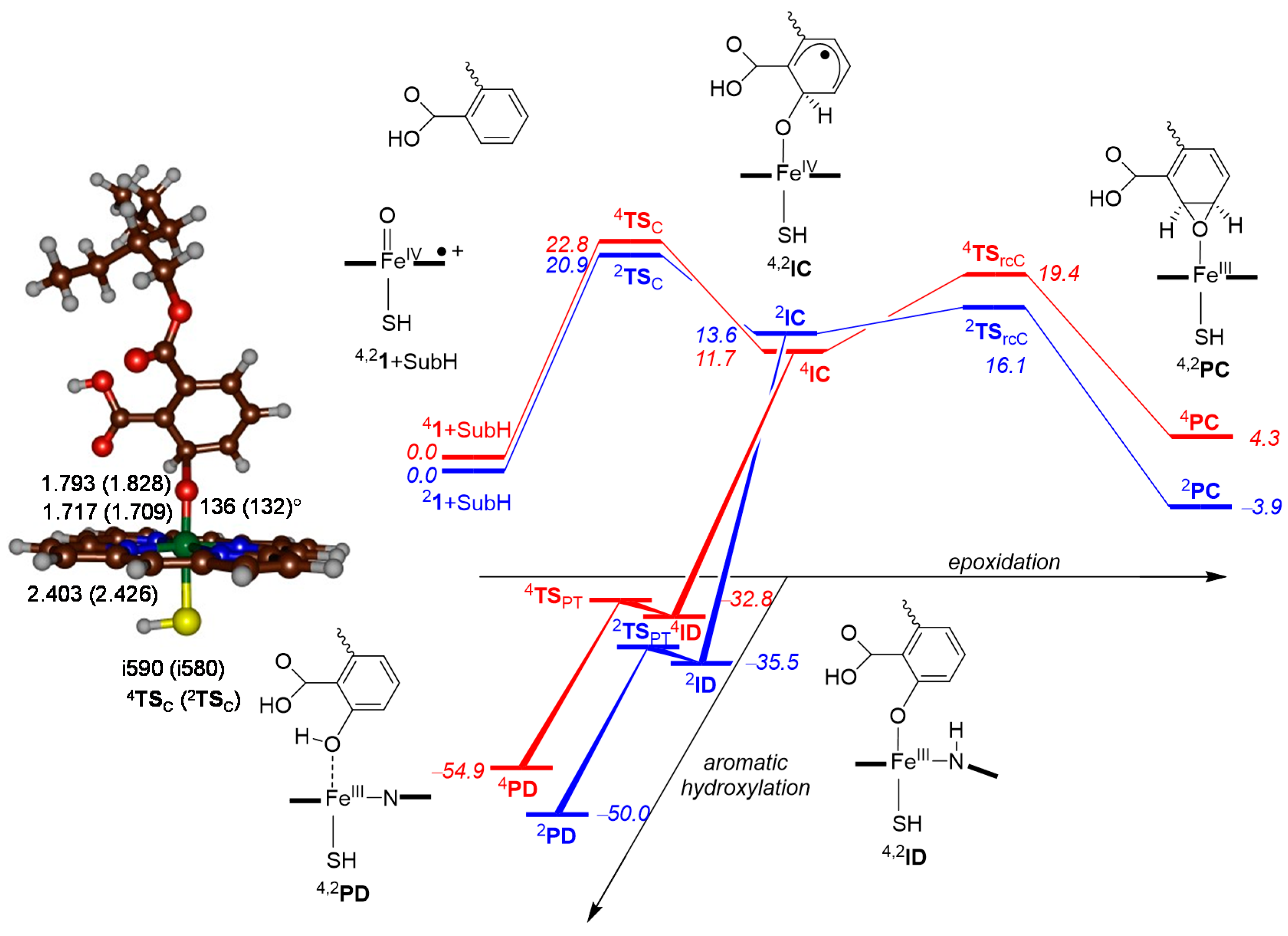
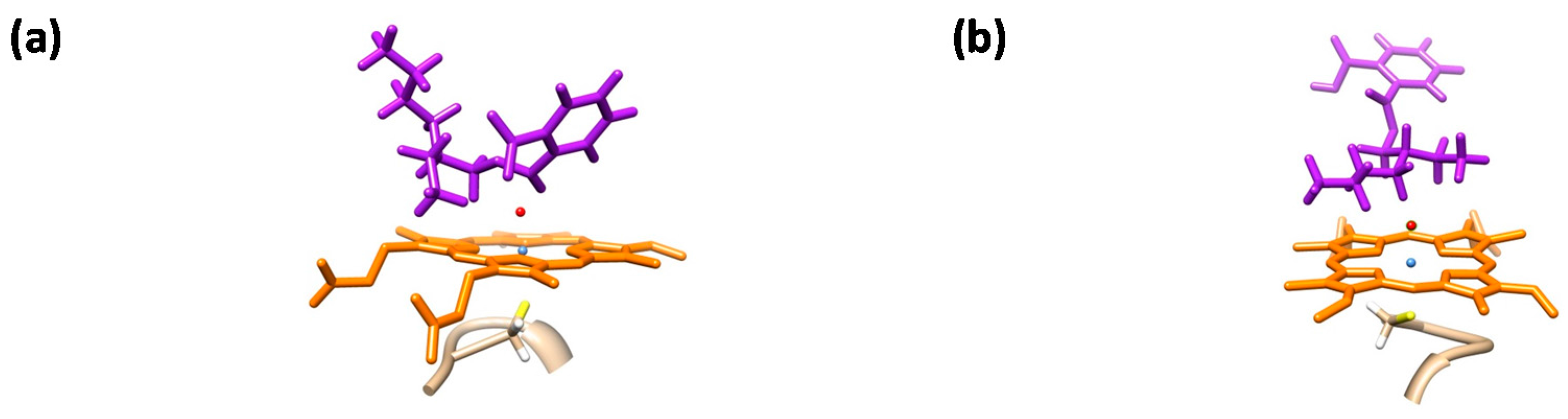
2.4. Phthalate Epoxidation and Aromatic Hydroxylation
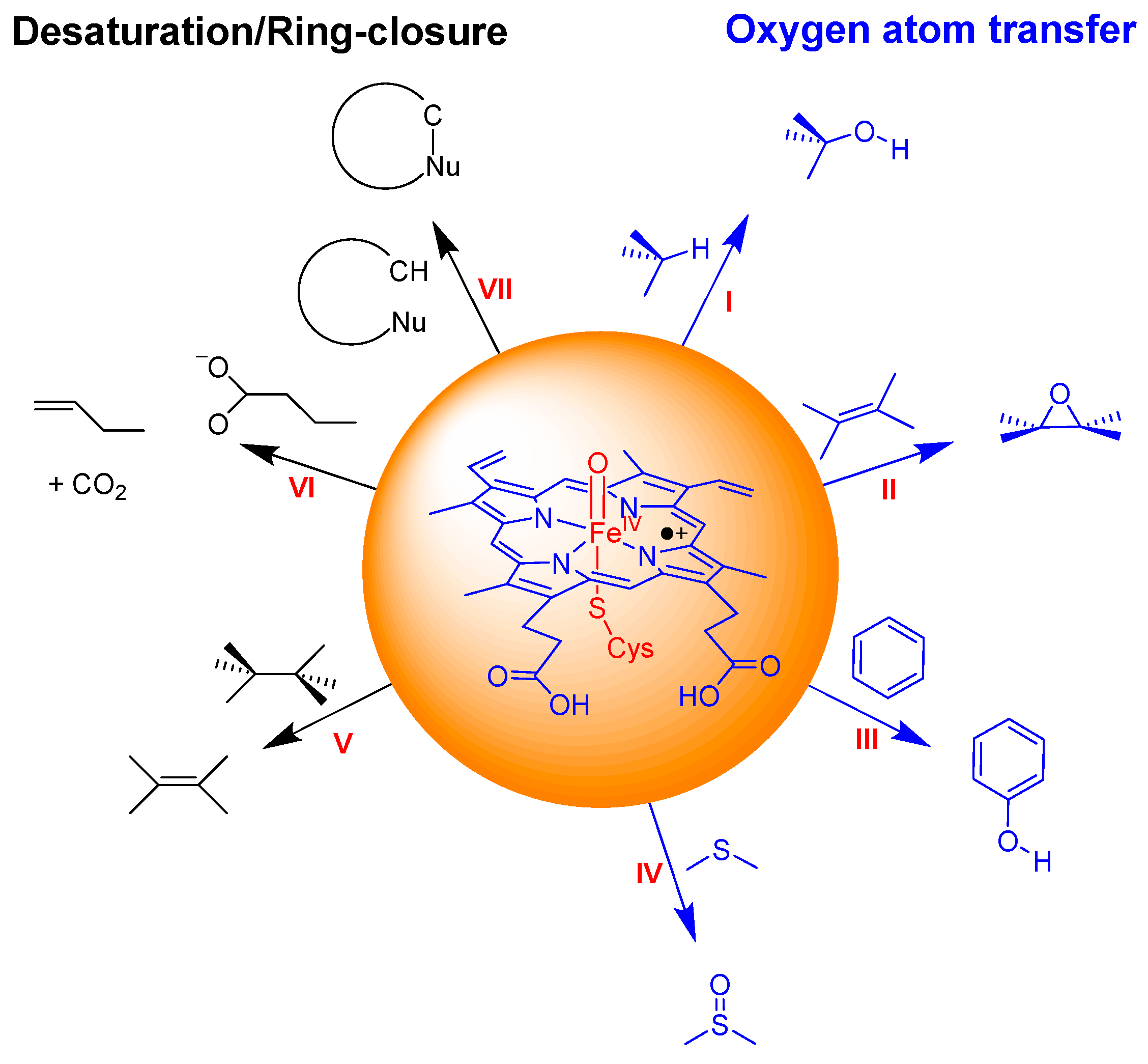
3. Discussion
3.1. Regioselectivity of Phthalate Oxidation by P450 CpdI
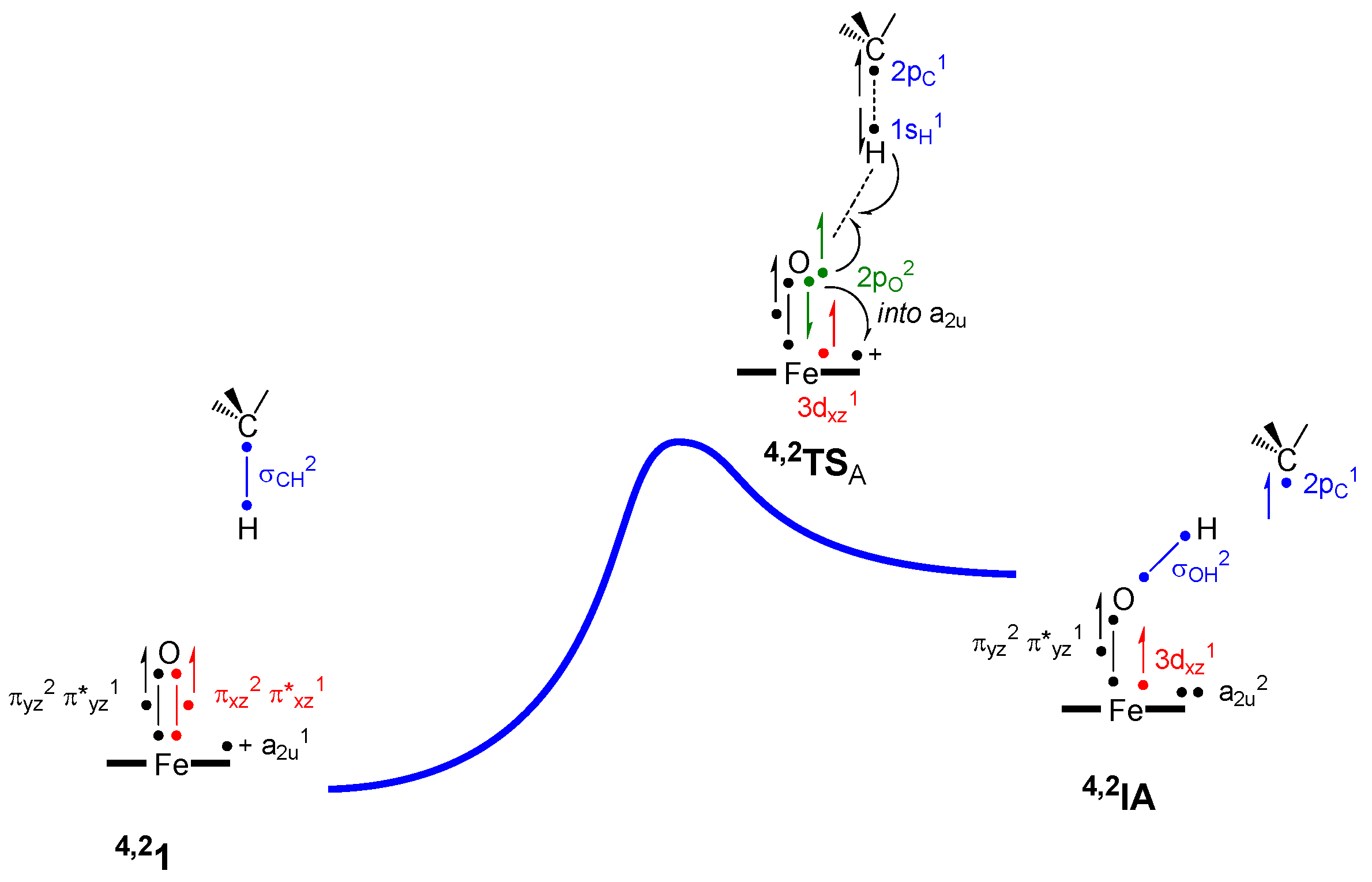
3.2. Features of the Hydrogen Atom Abstraction Step
4. Materials and Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2887. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; Girvan, H.M.; McLean, K.J. Variations on a (t)heme—Novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 2007, 24, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, X.; Guo, W. The cytochrome P450 superfamily: Key players in plant development and defense. J. Integr. Agricult. 2015, 14, 1673–1686. [Google Scholar] [CrossRef]
- Fujikura, K.; Ingelman-Sundberg, M.; Lauschke, V. Genetic variation in the human cytochrome P450 supergene family. Pharmacogenetics Genom. 2015, 12, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T. The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. Proc. Natl. Acad. Sci. USA 2003, 100, 3569–3574. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nakajima, H.; Ueno, T. Reactivities of oxo and peroxo intermediates studied by hemoprotein mutants. Acc. Chem. Res. 2007, 40, 554–562. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Nam, W. High-valent iron-oxo porphyrins in oxygenation reactions. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010; pp. 85–140, ISBN-13 978-981-4307-23-9. [Google Scholar]
- Belcher, J.; McLean, K.J.; Matthews, S.; Woodward, L.S.; Fischer, K.; Rigby, S.E.J.; Nelson, D.R.; Potts, D.; Baynham, M.T.; Parker, D.A.; et al. Structure and biochemical properties of the alkene producing cytochrome P450 OleTJE (CYP152L1) from the Jeotgalicoccus sp. 8456 bacterium. J. Biol. Chem. 2014, 289, 6535–6550. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Hirao, H.; de Visser, S.P.; Zheng, J.; Wang, D.; Thiel, W.; Shaik, S. New features in the catalytic cycle of cytochrome P450 during the formation of Compound I from Compound 0. J. Phys. Chem. B 2005, 109, 19946–19951. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Rittle, J.; Green, M.T. Cytochrome P450 Compound I: Capture, characterization, and C–H bond activation kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Davydov, R.; Perera, R.; Jin, S.; Yang, T.-C.; Bryson, T.A.; Sono, M.; Dawson, J.H.; Hoffman, B.M. Substrate modulation of the properties and reactivity of the oxy-ferrous and hydroperoxo-ferric intermediates of cytochrome P450cam as shown by cryoreduction-EPR/ENDOR spectroscopy. J. Am. Chem. Soc. 2005, 127, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Ryu, Y.O.; Song, R.; Nam, W. Oxoiron(IV) porphyrin π-cation radical complexes with a chameleon behavior in cytochrome P450 model reactions. J. Biol. Inorg. Chem. 2005, 10, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Sharma, P.K.; Shaik, S. Searching for the second oxidant in the catalytic cycle of cytochrome P450: A theoretical investigation of the iron(III)-hydroperoxo species and its epoxidation pathways. J. Am. Chem. Soc. 2002, 124, 2806–2817. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.K.; Kevorkiants, R.; de Visser, S.P.; Kumar, D.; Shaik, S. Porphyrin trap its own terminator! Concerted and stepwise porphyrin degradation mechanisms induced by heme-oxygenase vs. cytochrome P450. Angew. Chem. Int. Ed. 2004, 43, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Thibon, A.; Jollet, V.; Ribal, C.; Sénéchal-David, K.; Billon, L.; Sorokin, A.B.; Banse, F. Hydroxylation of aromatics with the help of a non-haem FeOOH: A mechanistic study under single-turnover and catalytic conditions. Chem. Eur. J. 2012, 18, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Mechanistic insight into halide oxidation by non-heme iron complexes. Haloperoxidase versus halogenase activity. Chem. Commun. 2013, 49, 10926–10928. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Quesne, M.G.; Sastri, C.V.; Banse, F.; de Visser, S.P. Differences and comparisons of the properties and reactivities of iron(III)-hydroperoxo complexes with saturated coordination sphere. Chem. Eur. J. 2015, 21, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Shaik, S. A Model “Rebound” Mechanism of Hydroxylation by Cytochrome P450: Stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- Kamachi, T.; Yoshizawa, K. A theoretical study on the mechanism of camphor hydroxylation by compound I of cytochrome P450. J. Am. Chem. Soc. 2003, 125, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Cohen, S.; de Visser, S.P.; Sharma, P.K.; Kumar, D.; Kozuch, S.; Ogliaro, F.; Danovich, D. The “rebound controversy”: An overview and theoretical modeling of the rebound step in C–H hydroxylation by cytochrome P450. Eur. J. Inorg. Chem. 2004, 207–226. [Google Scholar] [CrossRef]
- De Visser, S.P.; Ogliaro, F.; Harris, N.; Shaik, S. Multi-state epoxidation of ethene by cytochrome P450: A quantum chemical study. J. Am. Chem. Soc. 2001, 123, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. Hydrogen bonding modulates the selectivity of enzymatic oxidation by P450: A chameleon oxidant behavior of Compound I. Angew. Chem. Int. Ed. 2002, 41, 1947–1951. [Google Scholar] [CrossRef]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef] [PubMed]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef]
- Sharma, P.K.; de Visser, S.P.; Shaik, S. Can a single oxidant with two spin states masquerade as two different oxidants? A study of the sulfoxidation mechanism by cytochrome P450. J. Am. Chem. Soc. 2003, 125, 8698–8699. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Hirao, H.; Shaik, S. Sulfoxidation mechanisms catalyzed by cytochrome P450 and horseradish peroxidase models: Spin selection induced by the ligand. Biochemistry 2005, 44, 8148–8158. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Effect of the axial ligand on substrate sulfoxidation mediated by iron(IV)-oxo porphyrin cation radical oxidants. Chem. Eur. J. 2011, 17, 6196–6205. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Shaik, S. A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. J. Am. Chem. Soc. 2003, 125, 7413–7424. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Oh, K.; Han, A.-R.; Nam, W. Combined experimental and theoretical study on aromatic hydroxylation by mononuclear nonheme iron(IV)-oxo complexes. Inorg. Chem. 2007, 46, 4632–4641. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Axial ligand effect on the rate constant of aromatic hydroxylation by iron(IV)-oxo complexes mimicking cytochrome P450 enzymes. J. Phys. Chem. B 2012, 116, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.-C.; Zou, Y.; Watanabe, K.; Walsh, C.T.; Tang, Y. Oxidative cyclization in natural product biosynthesis. Chem. Rev. 2017, 107, 5226–5333. [Google Scholar] [CrossRef] [PubMed]
- Rettie, A.E.; Rettenmeier, A.W.; Howald, W.N.; Baillie, T.A. Cytochrome P-450-catalyzed formation of delta4-VPA, a toxic metabolite of valproic acid. Science 1987, 235, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.P.; Parkinson, A.; Forkert, P.-G. Isozyme-selective metabolism of ethyl carbamate by cytochrome P450 (CYP2E1) and carboxylesterase (hydrolase A) enzymes in murine liver microsomes. Drug Metabol. Disp. 1998, 26, 60–65. [Google Scholar]
- Guengerich, F.P.; Kim, D.H. Enzymic oxidation of ethyl carbamate to vinyl carbamate and its role as an intermediate in the formation of 1,N6-ethenoadenosine. Chem. Res. Toxicol. 1991, 4, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Shaik, S. Oxygen economy of cytochrome P450: What is the origin of the mixed functionality as a dehydrogenase–oxidase enzyme compared with its normal function? J. Am. Chem. Soc. 2004, 126, 5072–5073. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef] [PubMed]
- Rude, M.A.; Baron, T.S.; Brubaker, S.; Alibhai, M.; Del Cardayre, S.B.; Schirmer, A. Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus species. Appl. Environm. Microbiol. 2011, 77, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.L.; Hsieh, C.H.; Makris, T.M. Decarboxylation of fatty acids to terminal alkenes by cytochrome P450 compound I. J. Am. Chem. Soc. 2015, 137, 4940–4943. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Quesne, M.G.; de Visser, S.P. Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: What drives the reaction to biofuel production? Chem. Eur. J. 2016, 22, 5478–5483. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Joo, H.; Campbell, J.L., Jr.; Clewell, R.A.; Andersen, M.E.; Clewell, H.J., III. In vitro metabolism of di(2-ethylhexyl) phthalate (DEHP) by various tissues and cytochrome P450s of human and rat. Toxicol. In Vitro 2012, 26, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Kaneti, J.; Shaik, S. The experimentally elusive oxidant of cytochrome P450: A theoretical “trapping” defining more closely the “real” species. ChemBioChem 2001, 2, 848–851. [Google Scholar] [CrossRef]
- De Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; de Visser, S.P. Oxygen atom transfer using an iron(IV)-oxo embedded in a tetracyclic N-heterocyclic carbene system: How does the reactivity compare to Cytochrome P450 Compound I? Chem. Eur. J. 2017, 23, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Shaik, S. How does product isotope effect prove the operation of a two-state “rebound” mechanism in C–H hydroxylation by cytochrome P450? J. Am. Chem. Soc. 2003, 125, 13024–13025. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Cohen, S.; Shaik, S. Radical clock substrates, their C–H hydroxylation mechanism by cytochrome P450 and other reactivity patterns: What does theory reveal about the clocks’ behavior? J. Am. Chem. Soc. 2004, 126, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Differences in and comparison of the catalytic properties of heme and non-heme enzymes with a central oxo-iron group. Angew. Chem. Int. Ed. 2006, 45, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Tan, L.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef] [PubMed]
- Green, M.T. Evidence for sulfur-based radicals in thiolate Compound I intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Schöneboom, J.C.; Lin, H.; Reuter, N.; Thiel, W.; Cohen, S.; Ogliaro, F.; Shaik, S. The elusive oxidant species of cytochrome P450 enzymes: Characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 2002, 124, 8142–8151. [Google Scholar] [CrossRef] [PubMed]
- Bathelt, C.M.; Mulholland, A.J.; Harvey, J.N. QM/MM modeling of benzene hydroxylation in human cytochrome P450 2C9. J. Phys. Chem. A 2008, 112, 13149–13156. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.S.; Sutcliffe, M.J.; de Visser, S.P. Quantum mechanics/molecular mechanics studies on the sulfoxidation of dimethyl sulfide by Compound I and Compound 0 of Cytochrome P450: Which is the better oxidant? J. Phys. Chem. A 2009, 113, 11635–11642. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; Cohen, S.; de Visser, S.P.; Shaik, S. Medium polarization and hydrogen bonding effects on Compound I of cytochrome P450: what kind of a radical is it really? J. Am. Chem. Soc. 2000, 122, 12892–12893. [Google Scholar] [CrossRef]
- De Visser, S.P. What external perturbations influence the electronic properties of catalase Compound I? Inorg. Chem. 2006, 45, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 2010, 132, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Barman, P.; Upadhyay, P.; Faponle, A.S.; Kumar, J.; Nag, S.S.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Deformylation reaction by a nonheme manganese(III)-peroxo complex via initial hydrogen atom abstraction. Angew. Chem. Int. Ed. 2016, 55, 11091–11095. [Google Scholar] [CrossRef] [PubMed]
- Timmins, A.; Saint-André, M.; de Visser, S.P. Understanding how prolyl-4-hydroxylase structure steers a ferryl oxidant toward scission of a strong C–H bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. Eur. J. 2006, 12, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; de Visser, S.P.; Ogliaro, F.; Schwarz, H.; Detlef Schröder, D. Two-state reactivity (TSR) mechanisms of hydroxylation and epoxidation by cytochrome P450 revealed by theory. Curr. Opin. Chem. Biol. 2002, 6, 556–567. [Google Scholar] [CrossRef]
- Sharma, P.K.; de Visser, S.P.; Ogliaro, F.; Shaik, S. Is the ruthenium analogue of Compound I of cytochrome P450 an efficient oxidant? A theoretical investigation of the methane hydroxylation reaction. J. Am. Chem. Soc. 2003, 125, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Kumar, D.; de Visser, S.P. Methane hydroxylation by axially ligated iron(IV)-oxo porphyrin cation radical models. Int. J. Sc. Technol. 2015, 1, 26–40. [Google Scholar] [CrossRef]
- Schyman, P.; Usharani, D.; Wang, Y.; Shaik, S. Brain Chemistry: How does P450 catalyze the O-demethylation reaction of 5-methoxytryptamine to yield serotonin? J. Phys. Chem. B. 2010, 114, 7078–7089. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Tahsini, L.; Nam, W. How does the axial ligand of cytochrome P450 biomimetics influence the regioselectivity of aliphatic versus aromatic hydroxylation? Chem. Eur. J. 2009, 15, 5577–5587. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate reactivity in styrene epoxidation by Compound I of cytochrome P450: Mechanisms of products and side products formation. Chem. Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Latifi, R.; Kumar, S.; Rybak-Akimova, E.V.; Sainna, M.A.; de Visser, S.P. Rationalization of the barrier height for para-Z-styrene epoxidation by iron(IV)-oxo porphyrins with variable axial ligands. Inorg. Chem. 2013, 52, 7968–7979. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Shaik, S. How does ethene inactivate cytochrome P450 en route to its epoxidation? A density functional study. Angew. Chem. Int. Ed. 2001, 40, 2871–2874. [Google Scholar] [CrossRef]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 124, 11809–11826. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05Å resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Propene activation by the oxo-iron active species of taurine/α-ketoglutarate dioxygenase (TauD) enzyme. How does the catalysis compare to heme-enzymes? J. Am. Chem. Soc. 2006, 128, 9813–9824. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Faponle, A.S.; Barman, P.; Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Long-range electron transfer triggers mechanistic differences between iron(IV)-oxo and iron(IV)-imido oxidants. J. Am. Chem. Soc. 2014, 136, 17102–17115. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the enhanced reactivity of μ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Karamzadeh, B.; Kumar, D.; Sastry, G.N.; de Visser, S.P. Steric factors override thermodynamic driving force in regioselectivity of proline hydroxylation by prolyl-4-hydroxylase enzymes. J. Phys. Chem. A 2010, 114, 13234–13243. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Groves, J.T.; Sason Shaik, S. Chameleon states: High-valent metal-oxo species of cytochrome P450 and its ruthenium analog. Angew. Chem. Int. Ed. 2001, 40, 2874–2878. [Google Scholar] [CrossRef]
- De Visser, S.P.; Kumar, D.; Neumann, R.; Shaik, S. Computer-generated high-valent iron-oxo and manganese-oxo species with polyoxometalate ligands: How do they compare with the iron-oxo active species of heme enzymes? Angew. Chem. Int. Ed. 2004, 43, 5661–5665. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Nonheme ferric hydroperoxo intermediates are efficient oxidants of bromide oxidation. Chem. Commun. 2011, 47, 11044–11046. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Comparison of the reactivity of nonheme iron(IV)-oxo versus iron(IV)-imido complexes: Which is the better oxidant? Angew. Chem. Int. Ed. 2013, 52, 12288–12292. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, A.; Quesne, M.G.; Bui, S.; Heyes, D.J.; Steiner, R.A.; Scrutton, N.S.; de Visser, S.P. Catalytic mechanism of cofactor-free dioxygenases and how they circumvent spin-forbidden oxygenation of their substrates. J. Am. Chem. Soc. 2015, 137, 7474–7487. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Faponle, A.S.; de Visser, S.P. Substrate sulfoxidation by an iron(IV)-oxo complex: Benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 2016, 120, 9805–9814. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–272. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Barrier | Doublet 1 | Quartet 1 |
|---|---|---|
| 4,2TSA | 18.2 (32.4) | 19.9 (33.6) |
| 4,2TSB | 20.3 (34.2) | 19.3 (32.6) |
| 4,2TSC | 20.9 (34.8) | 22.8 (36.8) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantú Reinhard, F.G.; De Visser, S.P. Biodegradation of Cosmetics Products: A Computational Study of Cytochrome P450 Metabolism of Phthalates. Inorganics 2017, 5, 77. https://doi.org/10.3390/inorganics5040077
Cantú Reinhard FG, De Visser SP. Biodegradation of Cosmetics Products: A Computational Study of Cytochrome P450 Metabolism of Phthalates. Inorganics. 2017; 5(4):77. https://doi.org/10.3390/inorganics5040077
Chicago/Turabian StyleCantú Reinhard, Fabián G., and Sam P. De Visser. 2017. "Biodegradation of Cosmetics Products: A Computational Study of Cytochrome P450 Metabolism of Phthalates" Inorganics 5, no. 4: 77. https://doi.org/10.3390/inorganics5040077
APA StyleCantú Reinhard, F. G., & De Visser, S. P. (2017). Biodegradation of Cosmetics Products: A Computational Study of Cytochrome P450 Metabolism of Phthalates. Inorganics, 5(4), 77. https://doi.org/10.3390/inorganics5040077