Ceria: Recent Results on Dopant-Induced Surface Phenomena †

 ,
, 
Abstract
1. Introduction
2. Sample Preparation
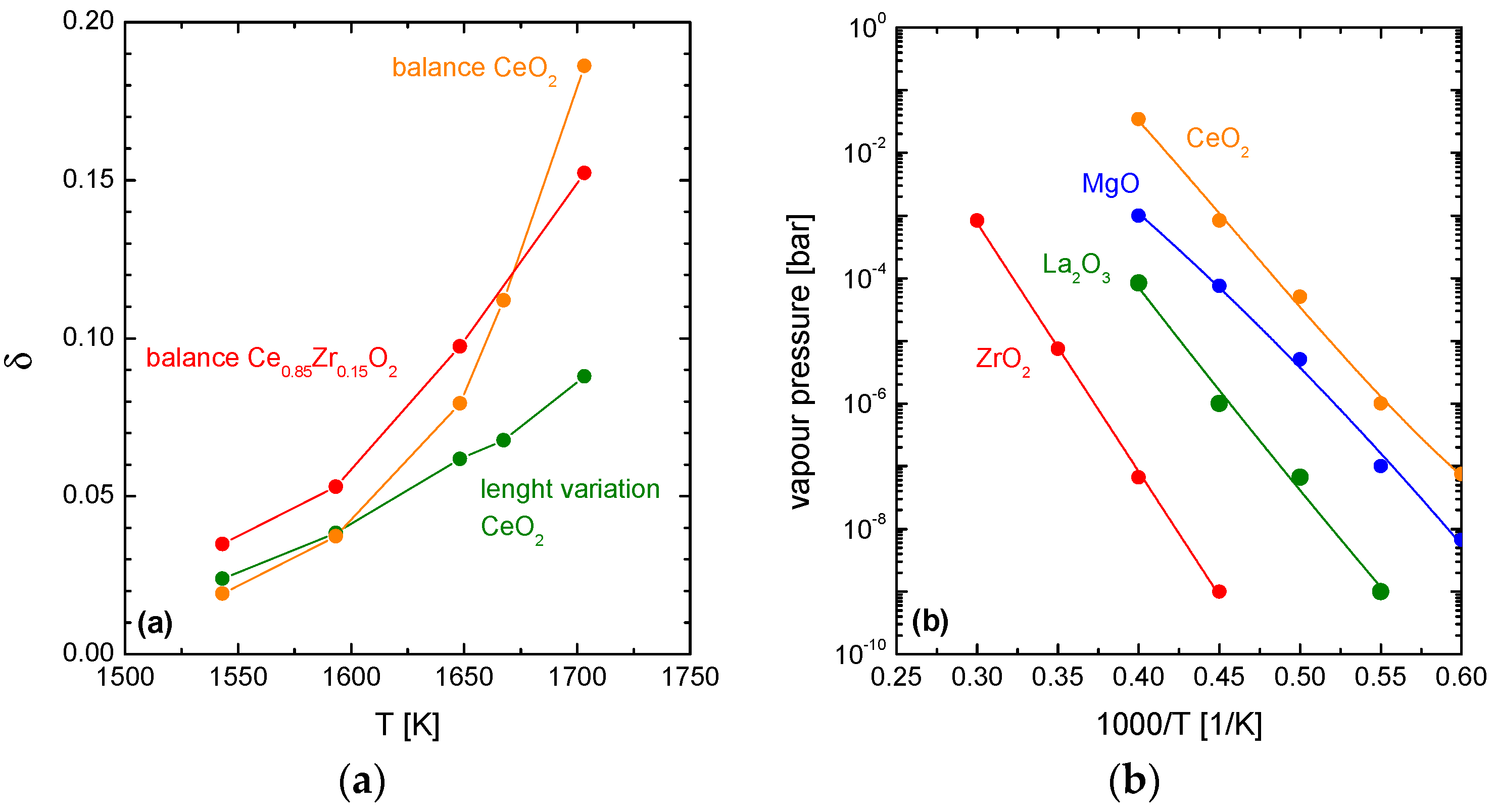
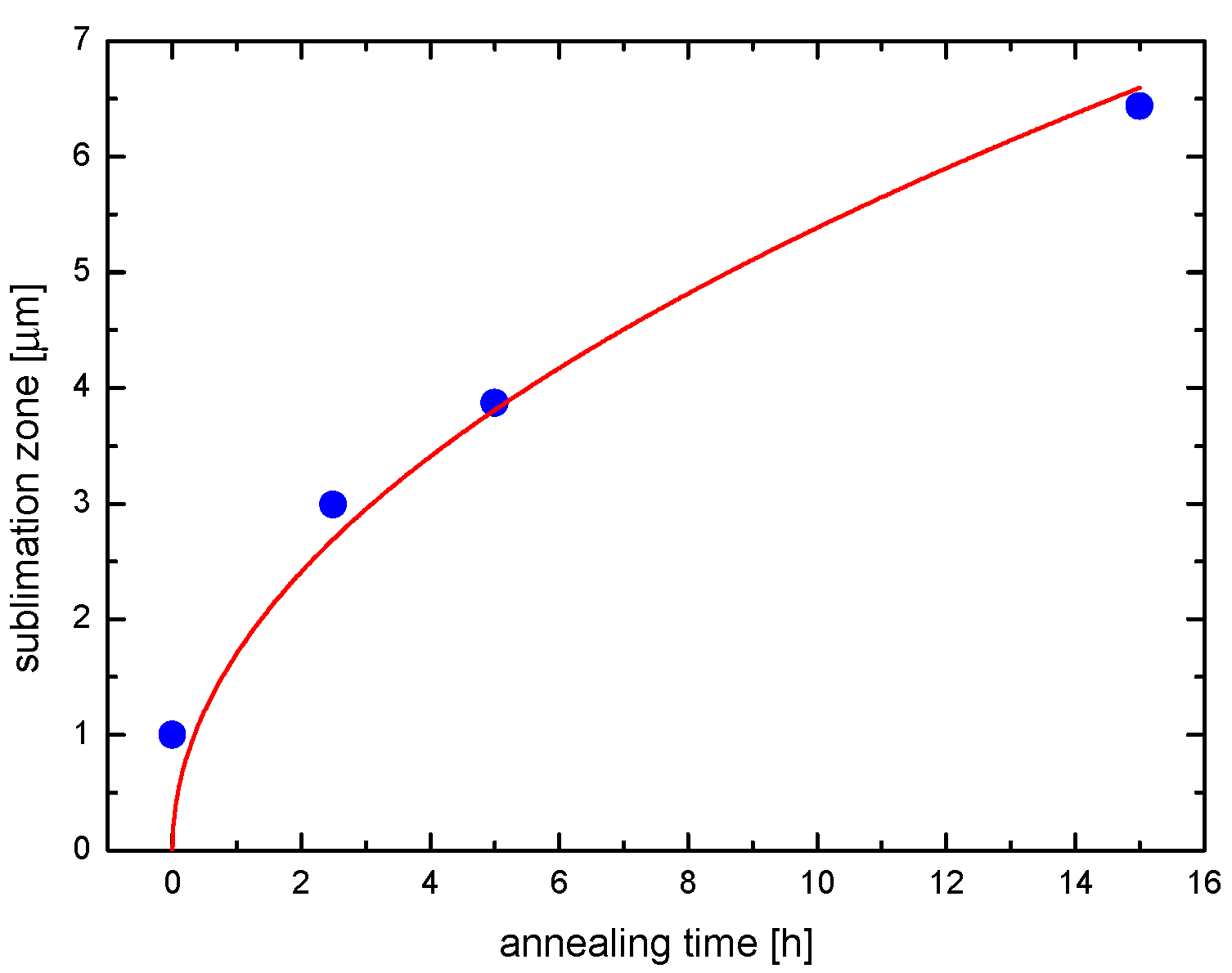
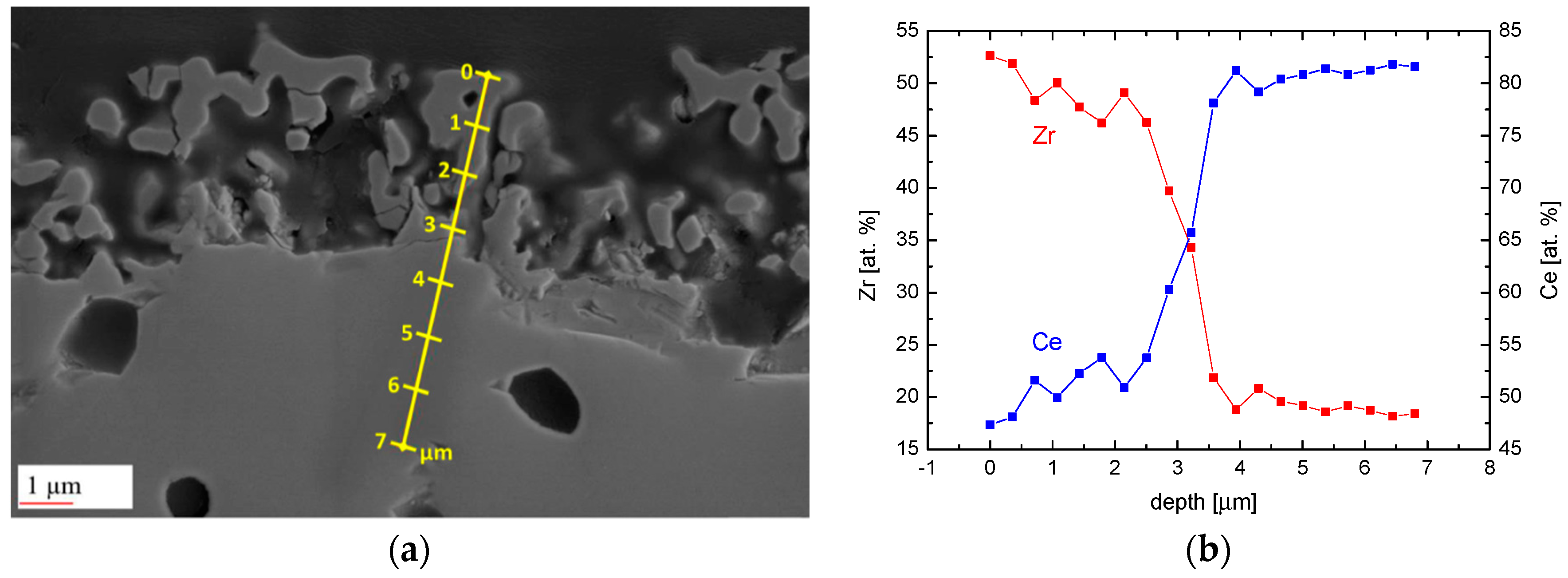
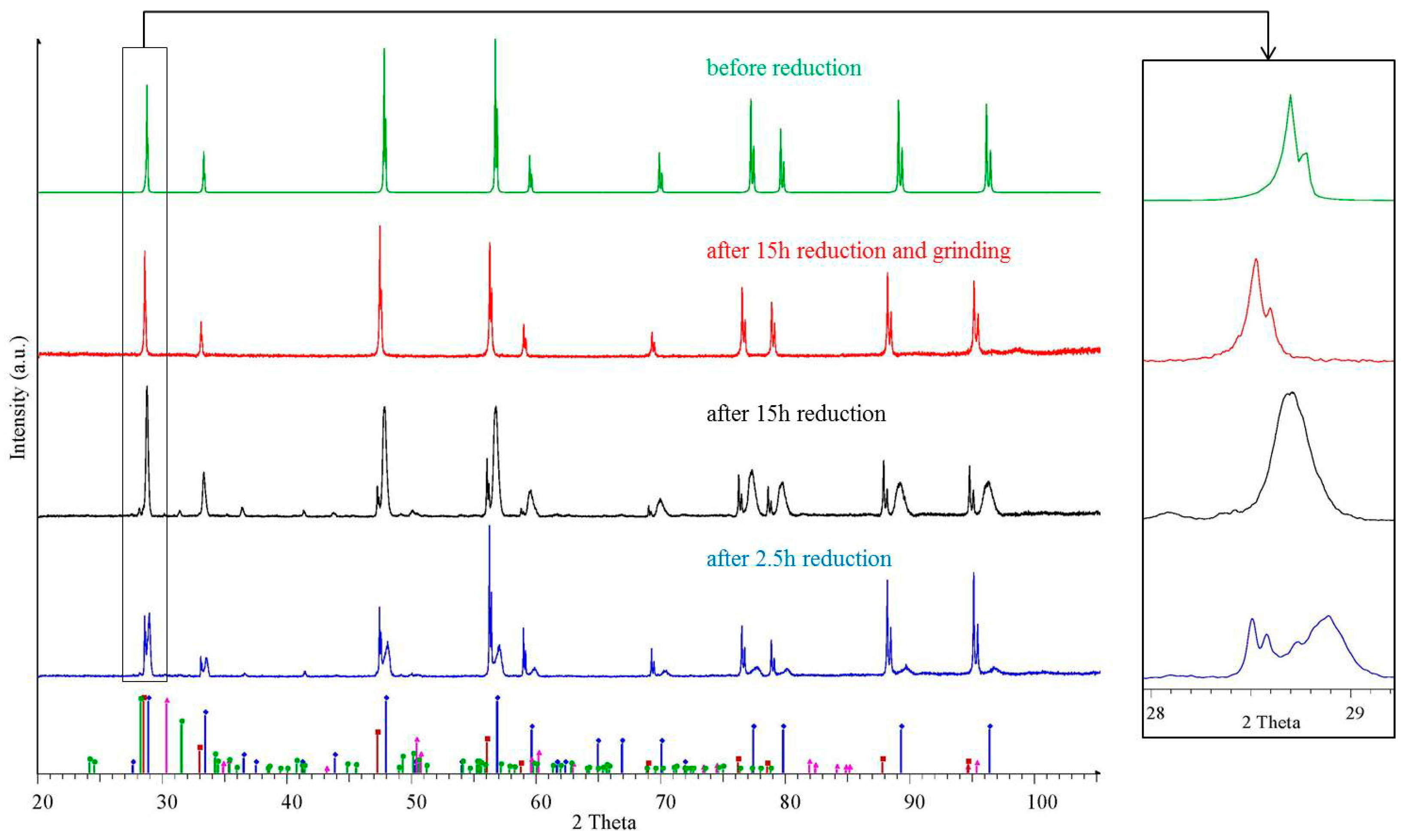
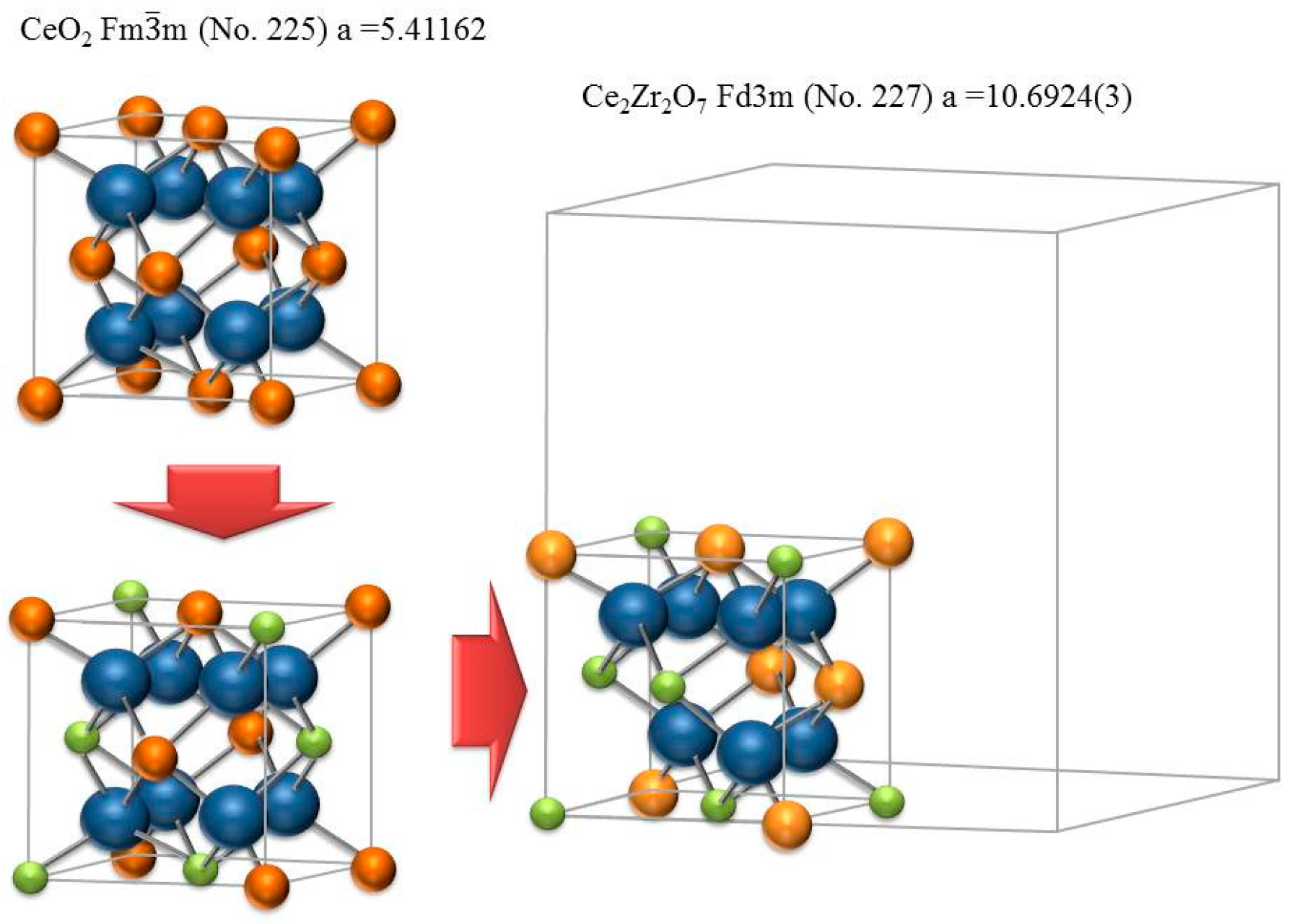
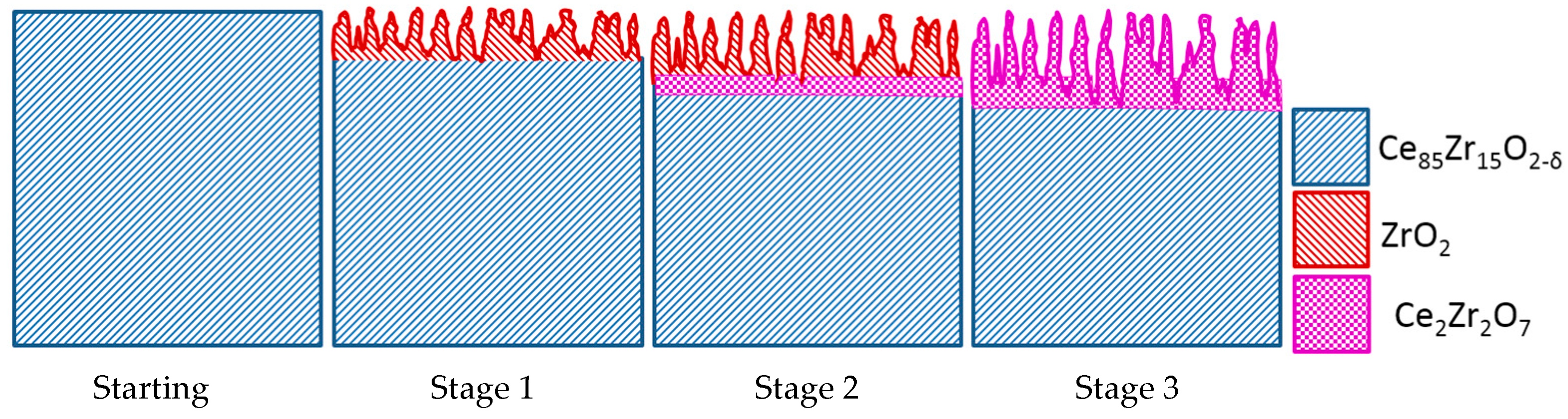
3. Degradation of (Ce,Zr)O2 Redox Ceramics by Selective Sublimation
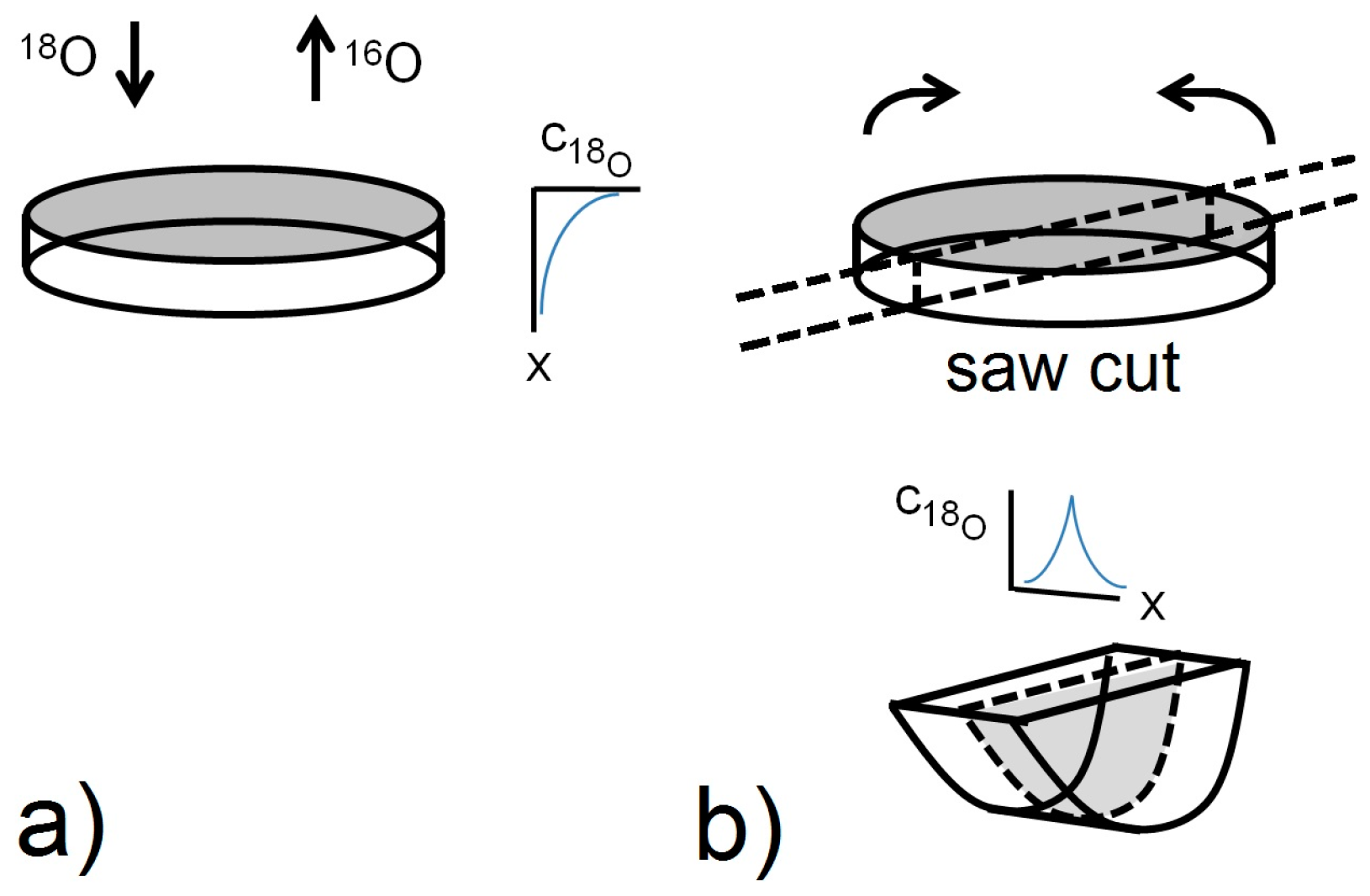
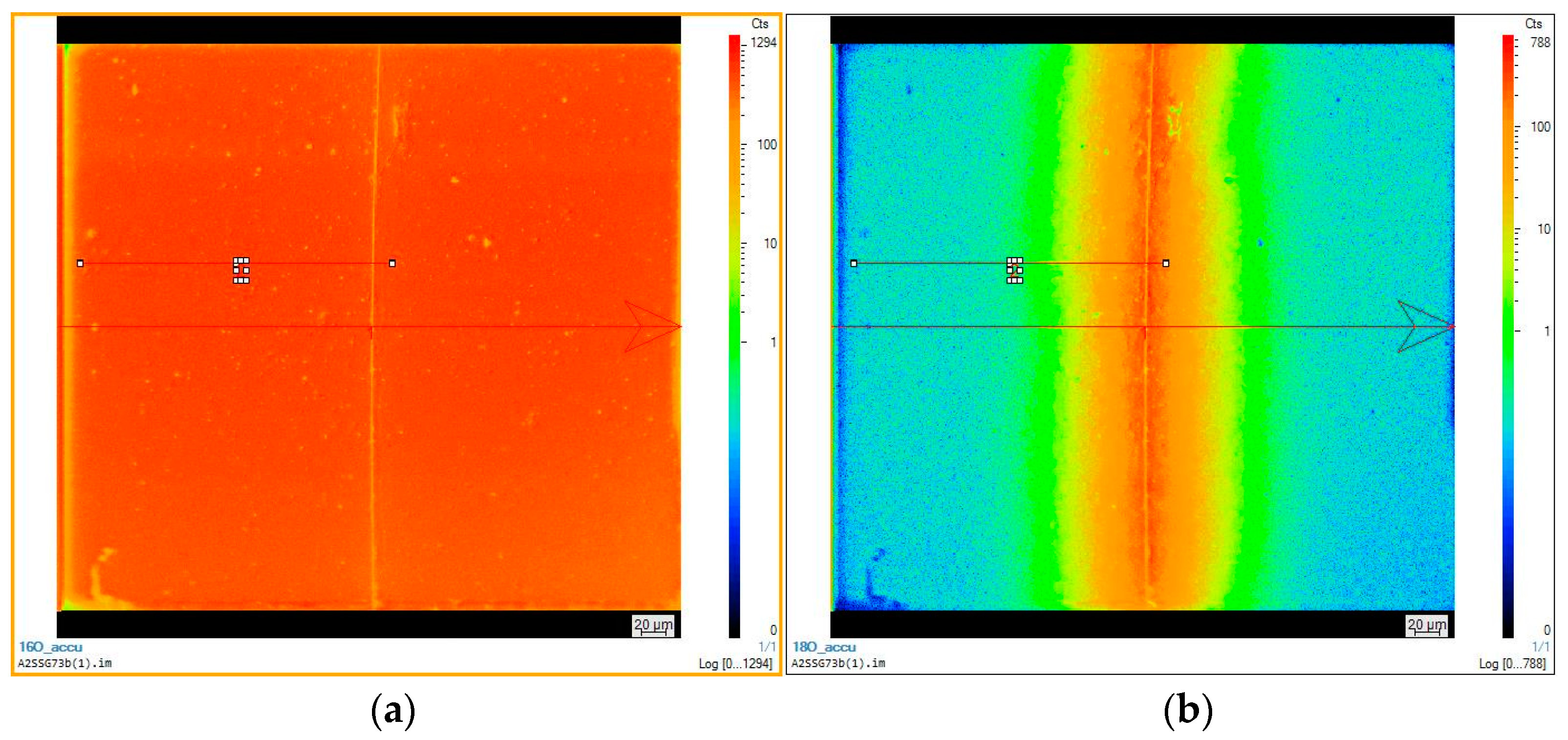
4. Surface Exchange and Bulk Diffusion of Oxygen from O2 and CO2 Atmospheres
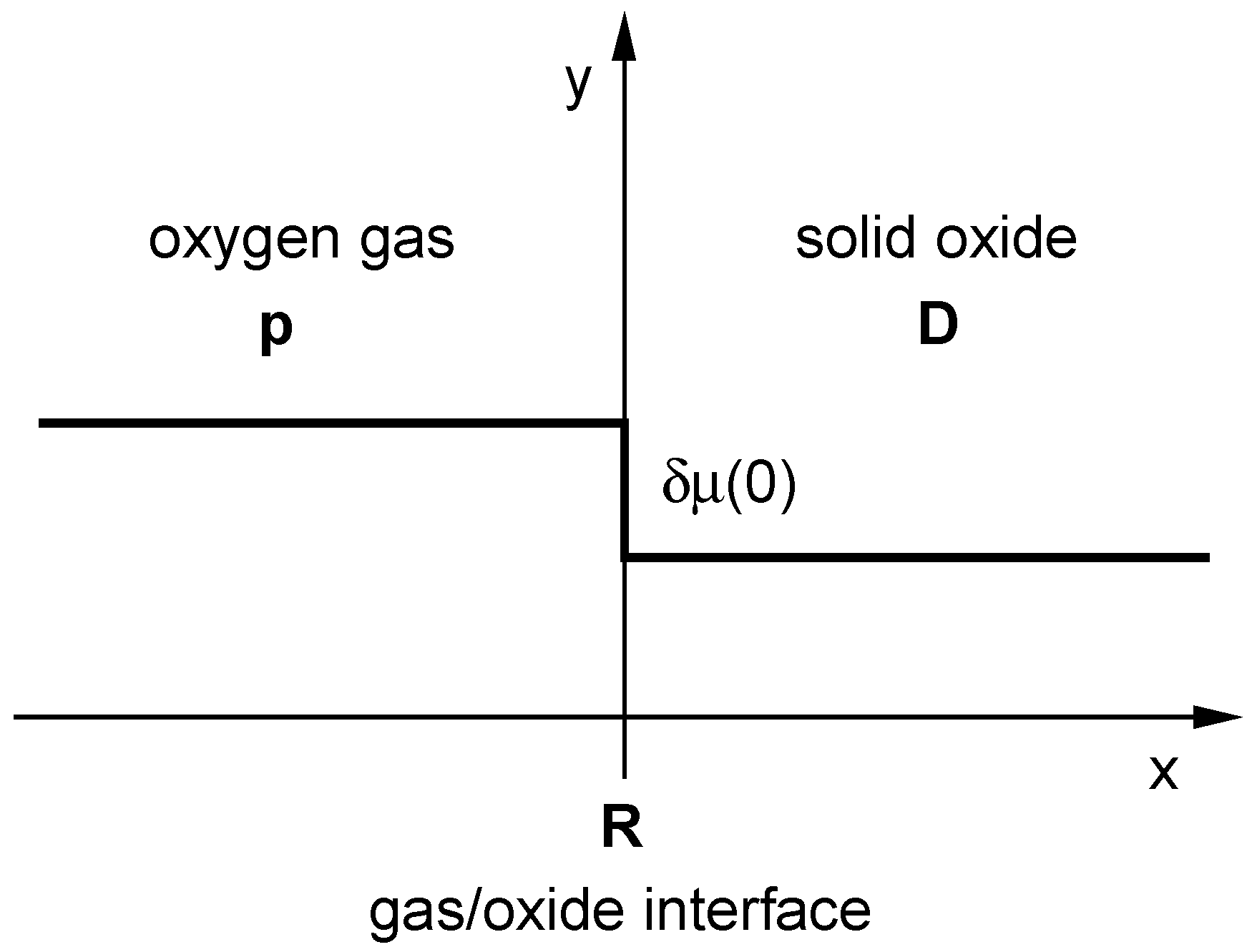
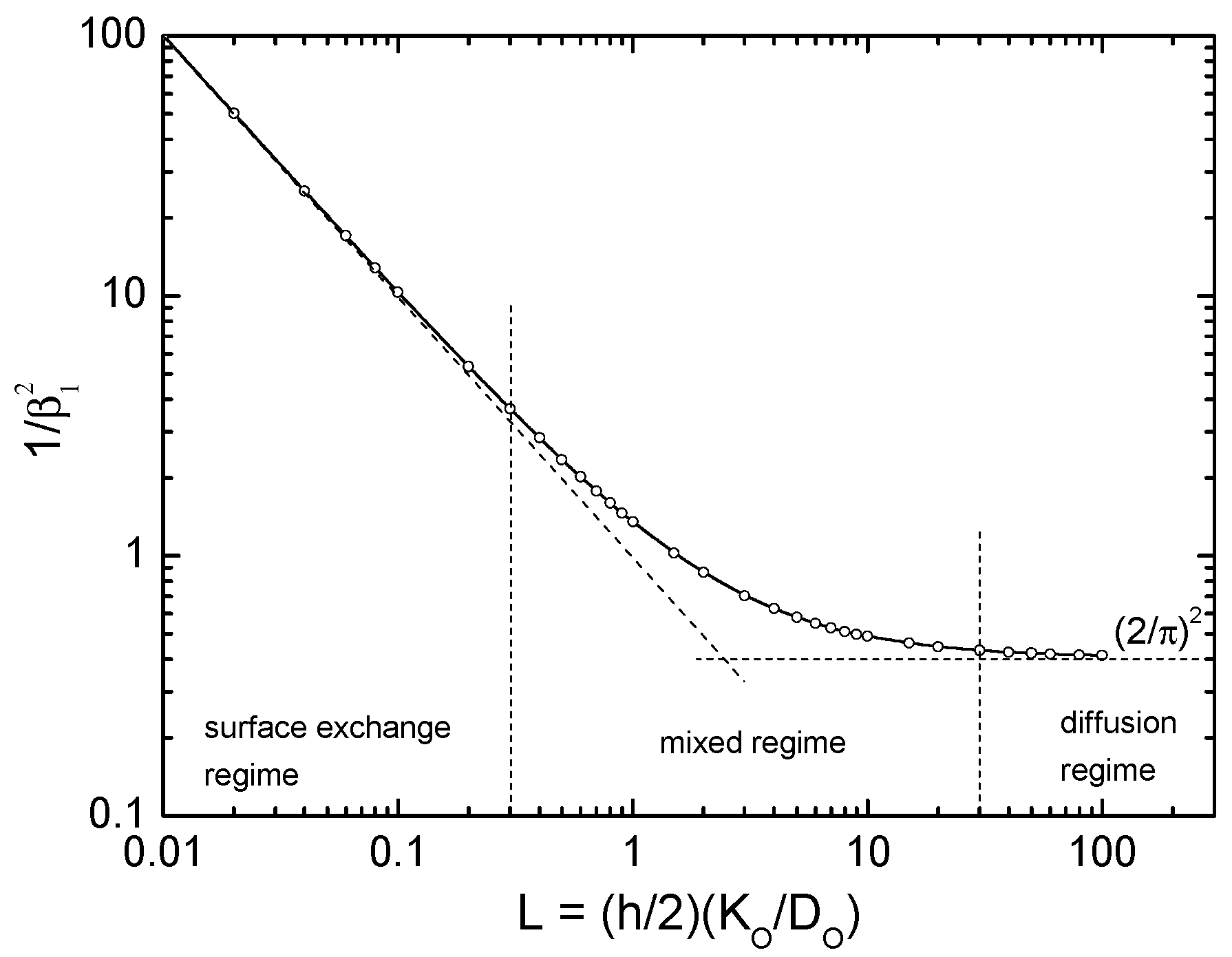
4.1. Revision of the Standard Picture of the Oxygen Exchange Kinetics
- (i)
- Check of on the basis of Table B1 in Appendix B ( for surface controlled kinetics and for diffusion controlled kinetics, respectively):
- (ii)
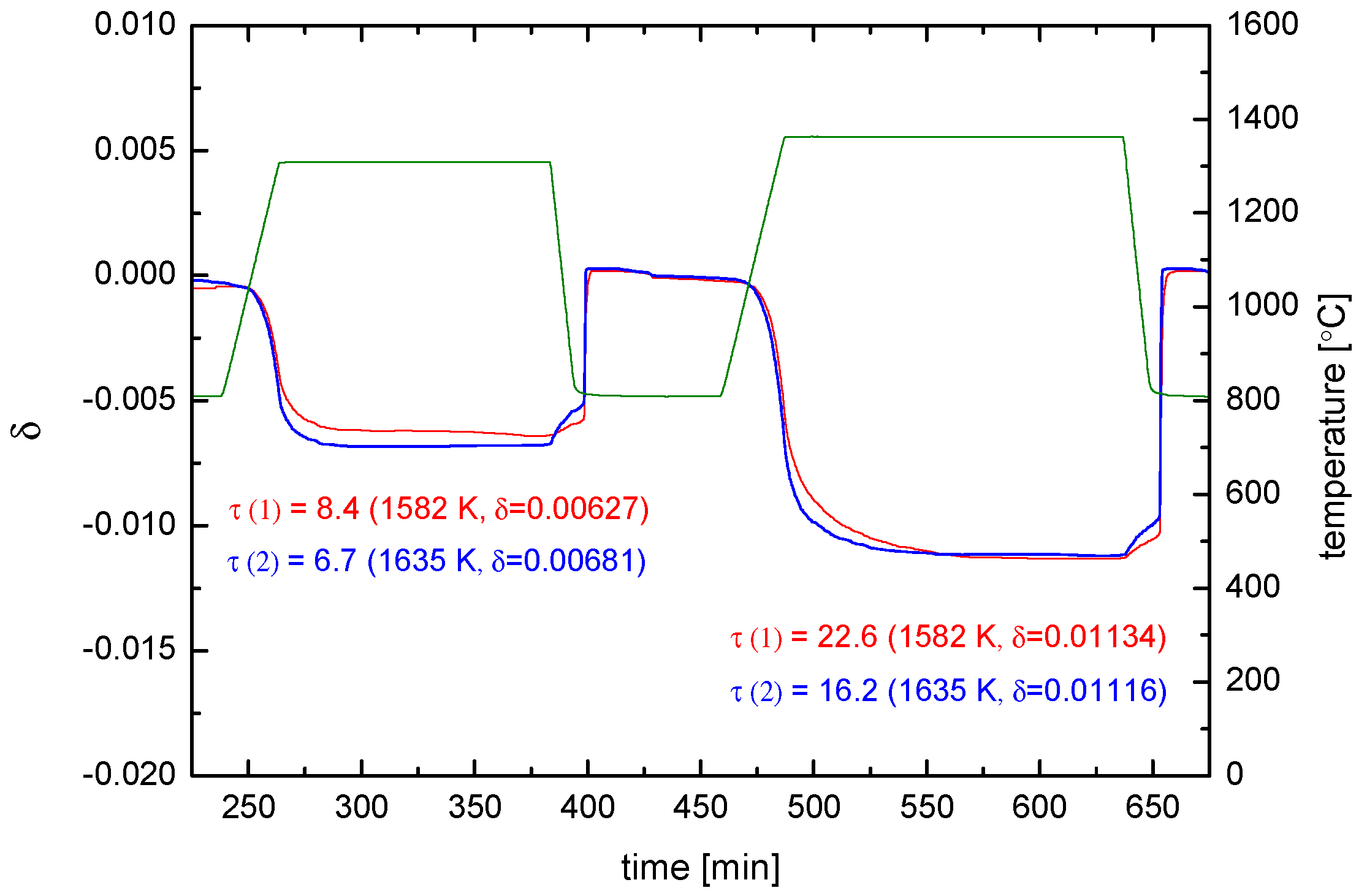
- (The experimental temperature interval (53 K) is too small to extract solid data for the activation enthalpy. The surprising behaviour of the values indicates that, however, the equilibrium exchange rate depends probably only very weakly on temperature for a given .
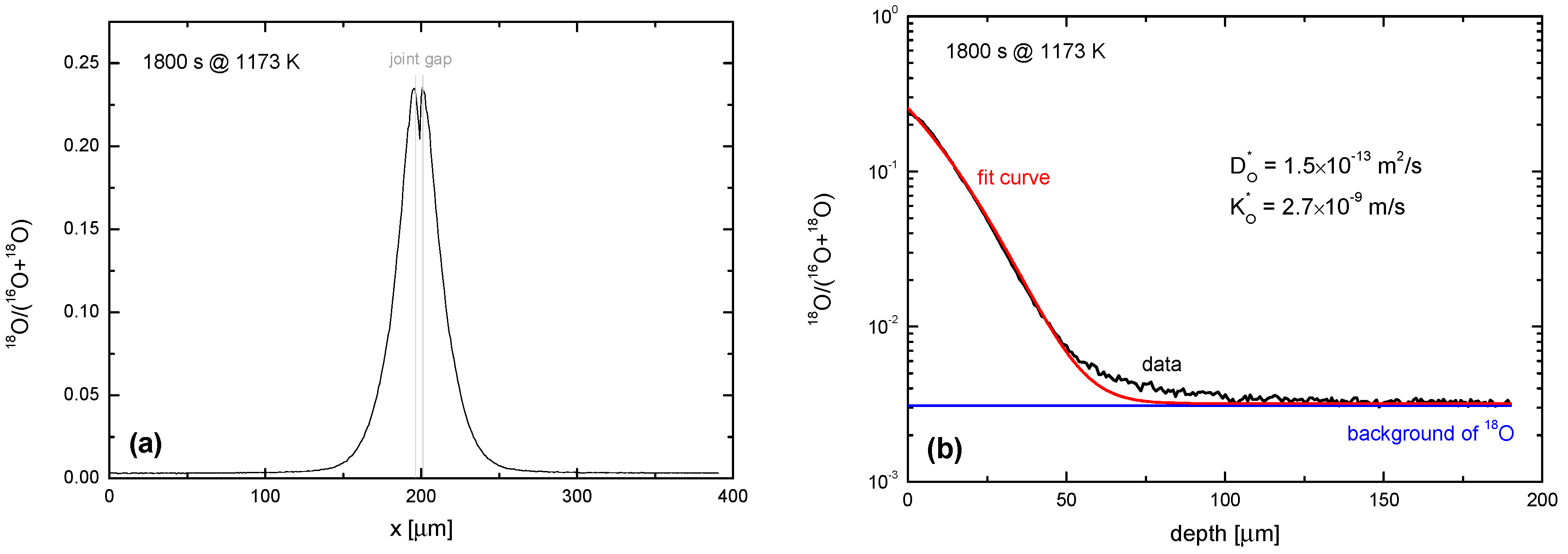
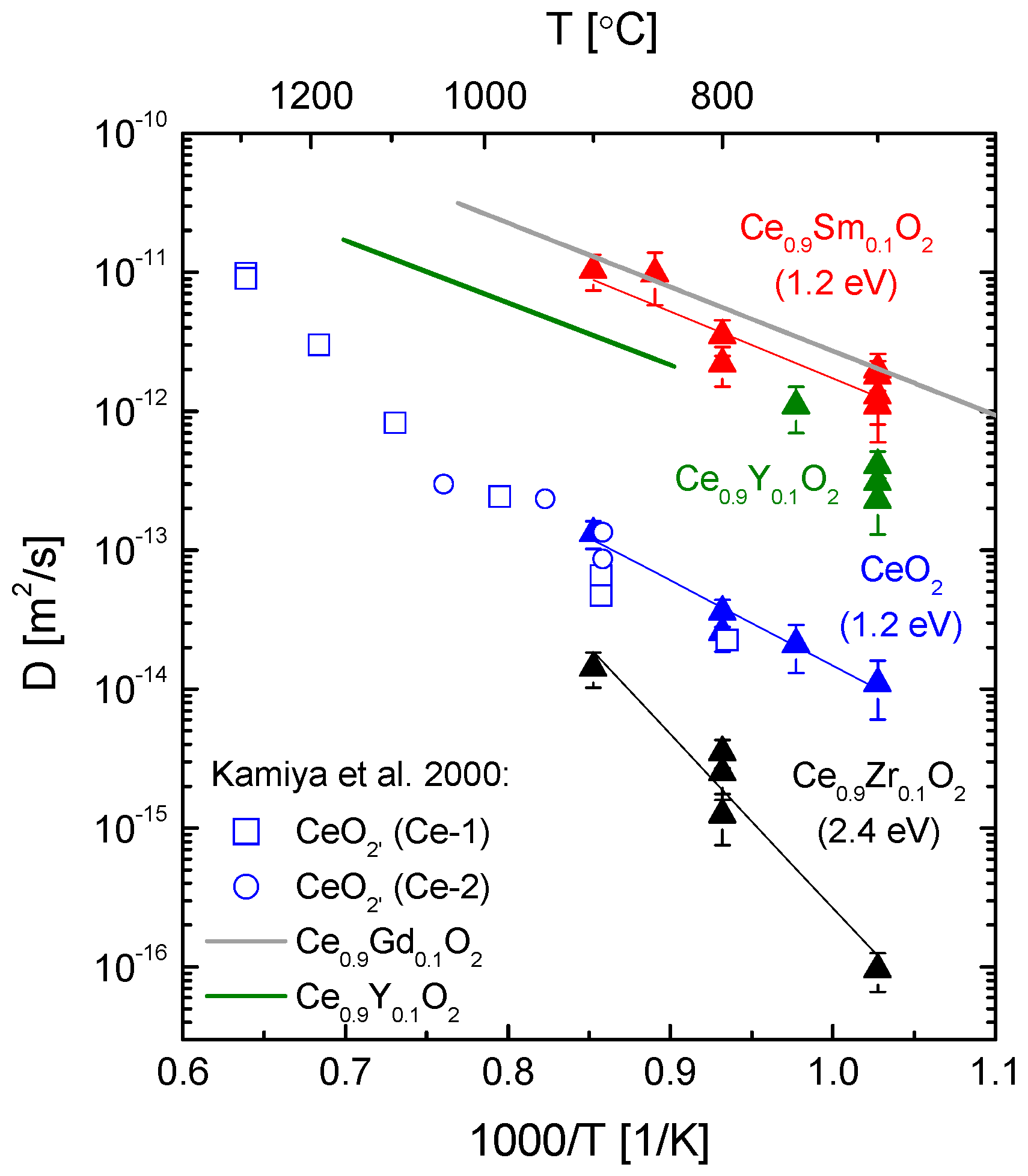
4.2. Oxygen Exchange from O2 Atmospheres
4.3. Oxygen Exchange from CO2 Atmospheres
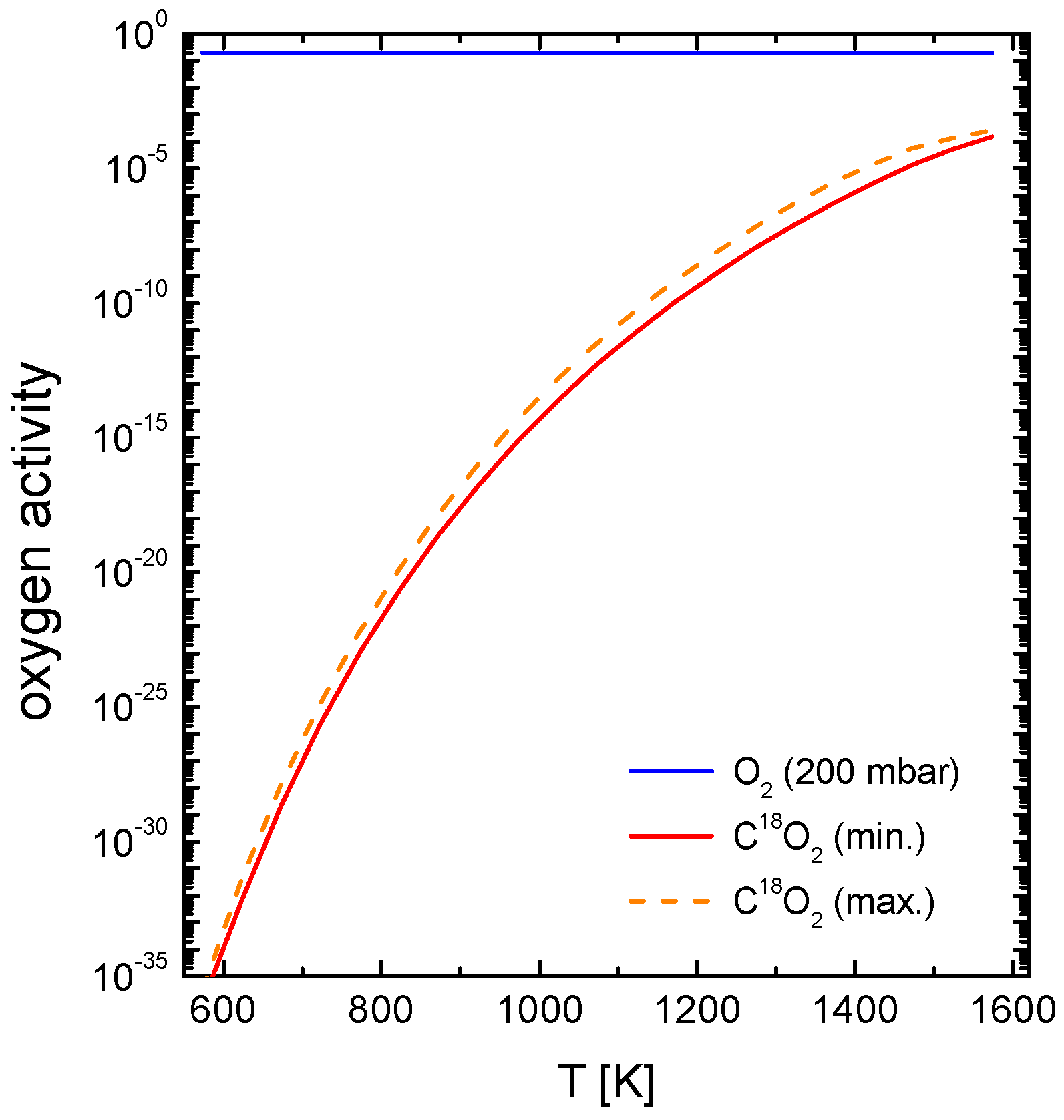
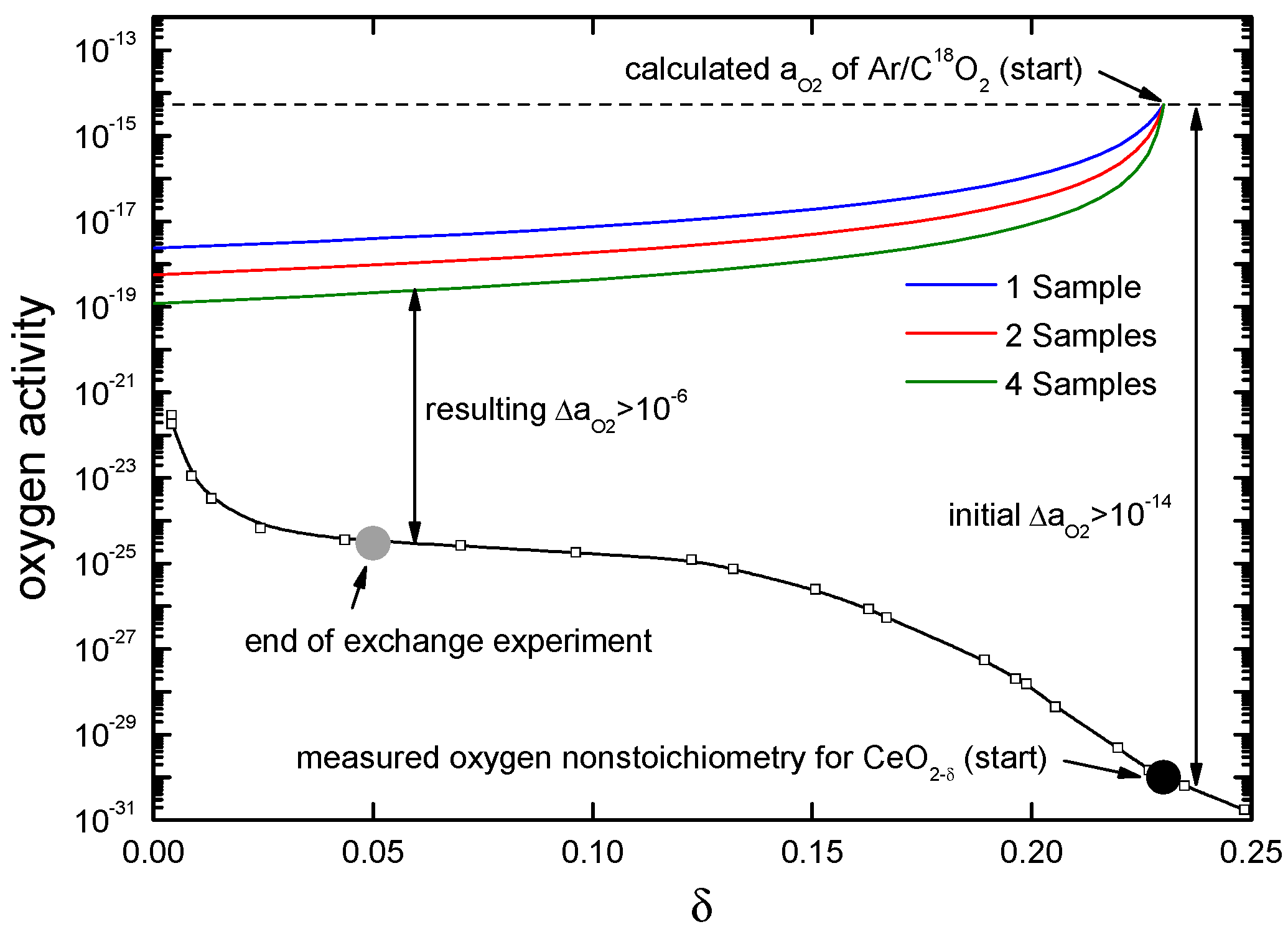
4.3.1. Measurements with a Chemical Potential Gradient at the Gas/Solid Interface
4.3.2. Measurements in Equilibrium
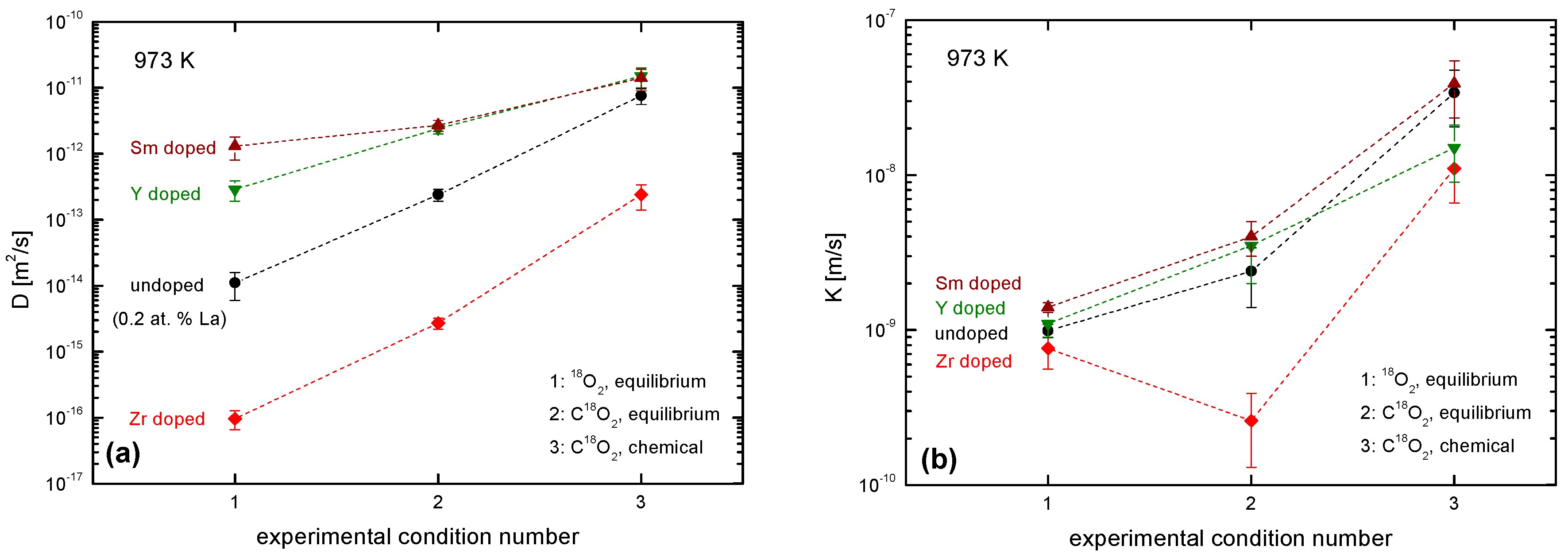
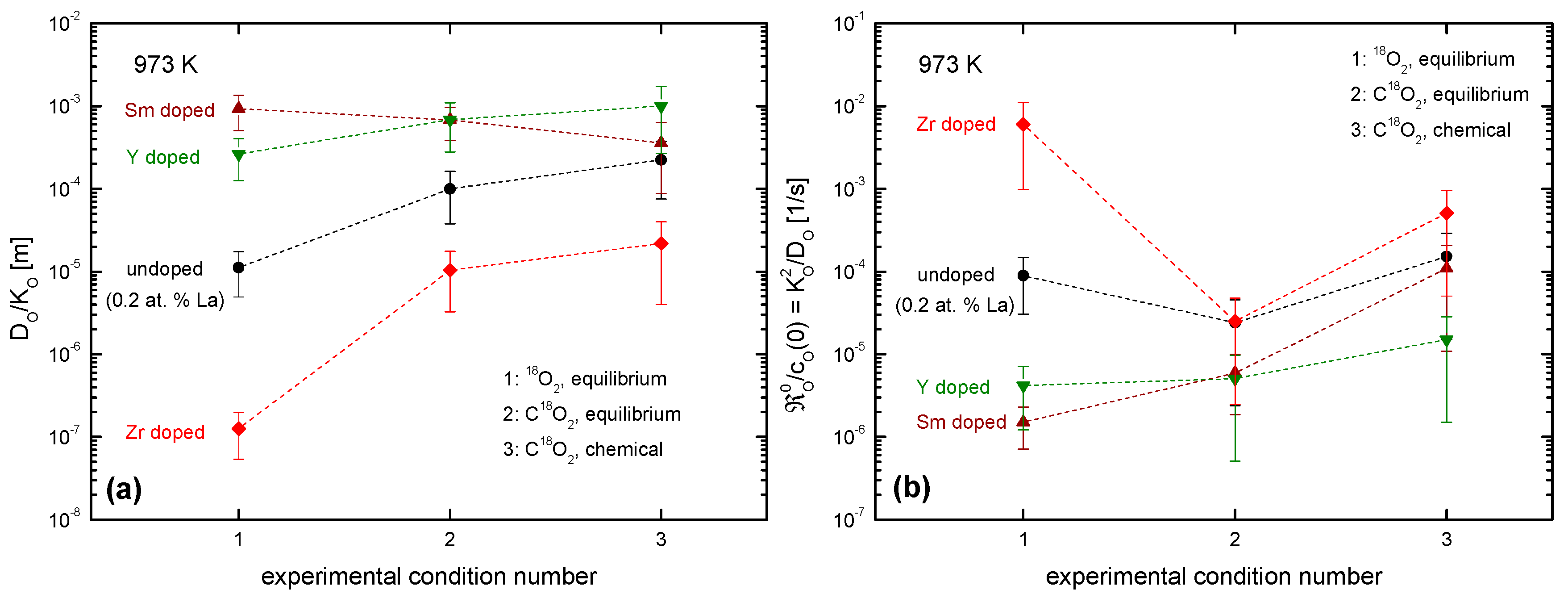
4.3.3. Comparison of the Oxygen Exchange Kinetics under Different Conditions
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A


Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Regime | |||||
|---|---|---|---|---|---|
| Surface Controlled | 1 | ||||
| Mixed Regime | |||||
| Diffusion Regime |
References
- Abanades, S.; Flamant, G. Thermochemical hydrogen production from a two-step solar-driven water-splitting cycle based on cerium oxides. Sol. Energy 2006, 80, 1611–1623. [Google Scholar] [CrossRef]
- Ackermann, S.; Scheffe, J.R.; Steinfeld, A. Diffusion of Oxygen in Ceria at Elevated Temperatures and Its Application to H2O/CO2 Splitting. J. Phys. Chem. C 2014, 118, 5216–5225. [Google Scholar] [CrossRef]
- Bulfin, B.; Lowe, A.J.; Keogh, K.A.; Murphy, B.E.; Lübben, O.; Krasnikov, S.A.; Shvets, I.V. Analytical Model of CeO2 Oxidation and Reduction. J. Phys. Chem. C 2013, 117, 24129–24137. [Google Scholar] [CrossRef]
- Chueh, W.C.; Haile, S.M. A thermochemical study of ceria: Exploiting an old material for new modes of energy conversion and CO2 mitigation. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2010, 368, 3269–3294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Shah, P.R.; Montini, T.; Fornasiero, P.; Gorte, R.J. Oxidation enthalpies for reduction of ceria surfaces. Surf. Sci. 2007, 601, 2512–2519. [Google Scholar] [CrossRef]
- Shah, P.R.; Kim, T.; Zhou, G.; Fornasiero, P.; Gorte, R.J. Evidence for Entropy Effects in the Reduction of Ceria–Zirconia Solutions. Chem. Mater. 2006, 18, 5363–5369. [Google Scholar] [CrossRef]
- Zhou, G.; Shah, P.R.; Kim, T.; Fornasiero, P.; Gorte, R.J. Oxidation entropies and enthalpies of ceria–zirconia solid solutions. Catal. Today 2007, 123, 86–93. [Google Scholar] [CrossRef]
- Kim, T.; Vohs, J.M.; Gorte, R.J. Thermodynamic Investigation of the Redox Properties of Ceria–Zirconia Solid Solutions. Ind. Eng. Chem. Res. 2006, 45, 5561–5565. [Google Scholar] [CrossRef]
- Bulfin, B.; Hoffmann, L.; Oliveira, L.; Knoblauch, N.; Call, F.; Roeb, M.; Sattler, C.; Schmücker, M. Statistical thermodynamics of non-stoichiometric ceria and ceria zirconia solid solutions. Phys. Chem. Chem. Phys. 2016, 18, 23147–23154. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Bishop, S.R.; Rupp, J.L.M.; Tuller, H.L. Structural characterization and oxygen nonstoichiometry of ceria–zirconia (Ce1−xZrxO2−δ) solid solutions. Acta Mater. 2013, 61, 4277–4288. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, C.-K.; Haile, S.M. Ceria–Zirconia Solid Solutions (Ce1–xZrxO2−δ, x ≤ 0.2) for Solar Thermochemical Water Splitting: A Thermodynamic Study. Chem. Mater. 2014, 26, 6073–6082. [Google Scholar] [CrossRef]
- Takacs, M.; Scheffe, J.R.; Steinfeld, A. Oxygen nonstoichiometry and thermodynamic characterization of Zr doped ceria in the 1573–1773 K temperature range. Phys. Chem. Chem. Phys. 2015, 17, 7813–7822. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, L.; Van der Biest, O.; Vleugels, J. Influence of the oxygen partial pressure on the reduction of CeO2 and CeO2–ZrO2 ceramics. Solid State Sci. 2005, 7, 539–544. [Google Scholar] [CrossRef]
- Huang, S.; Li, L.; Vleugels, J.; Wang, P.; Van der Biest, O. Thermodynamic prediction of the nonstoichiometric phase Zr1–zCezO2–x in the ZrO2–CeO1.5–CeO2 system. J. Eur. Ceram. Soc. 2003, 23, 99–106. [Google Scholar] [CrossRef]
- Sanjuan, M.L.; Oliete, P.B.; Varez, A.; Sanz, J. The role of Ce reduction in the segregation of metastable phases in the ZrO2–CeO2 system. J. Eur. Ceram. Soc. 2012, 32, 689–696. [Google Scholar] [CrossRef]
- Montini, T.; Banares, M.A.; Hickey, N.; Di Monte, R.; Fornasiero, P.; Kaspar, J.; Graziani, M. Promotion of reduction in Ce0.5Zr0.5O2: The pyrochlore structure as effect rather than cause? Phys. Chem. Chem. Phys. 2004, 6, 1–3. [Google Scholar] [CrossRef]
- Achary, N.S.; Sali, S.K.; Kulkarni, N.K.; Krishna, P.S.R.; Shinde, A.B.; Tyagi, A.K. Intercalation/Deintercalation of Oxygen: A Sequential Evolution of Phases in Ce2O3/CeO2−ZrO2 Pyrochlores. Chem. Mater. 2009, 21, 5848–5859. [Google Scholar] [CrossRef]
- Conesa, J.C. Computer Modeling of Local Level Structures in (Ce, Zr) Mixed Oxide. J. Phys. Chem. B 2003, 107, 8840–8853. [Google Scholar] [CrossRef]
- Wang, H.-F.; Guo, Y.-L.; Lu, G.-Z.; Hu, P. Maximizing the Localized Relaxation: The Origin of the Outstanding Oxygen Storage Capacity of κ-Ce2Zr2O8. Angew. Chem. Int. Ed. 2009, 48, 8289–8292. [Google Scholar] [CrossRef] [PubMed]
- Montini, T.; Hickey, N.; Fornasiero, P.; Graziani, M.; Bañares, M.A.; Martinez-Huerta, M.; Alessandri, I.; Depero, L.E. Variations in the Extent of Pyrochlore-Type Cation Ordering in Ce2Zr2O8: A t′−κ Pathway to Low-Temperature Reduction. Chem. Mater. 2005, 17, 1157–1166. [Google Scholar] [CrossRef]
- Pérez-Omil, J.A.; Bernal, S.; Calvino, J.J.; Hernández, J.C.; Mira, C.; Rodríguez-Luque, M.P.; Erni, R.; Browning, N.D. Combined HREM and HAADF Scanning Transmission Electron Microscopy: A Powerful Tool for Investigating Structural Changes in Thermally Aged Ceria−Zirconia Mixed Oxides. Chem. Mater. 2005, 17, 4282–4285. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. CO oxidation for the characterization of reducibility in oxygen storage components of three-way automotive catalysts. Appl. Catal. B 1997, 14, 105–115. [Google Scholar] [CrossRef]
- Gorte, R.J. Ceria in catalysis: From automotive applications to the water–gas shift reaction. AIChE J. 2010, 56, 1126–1135. [Google Scholar] [CrossRef]
- Scheffe, J.R.; Steinfeld, A. Thermodynamic Analysis of Cerium-Based Oxides for Solar Thermochemical Fuel Production. Energy Fuels 2012, 26, 1928–1936. [Google Scholar] [CrossRef]
- Manning, P.S.; Sirman, J.D.; Kilner, J.A. Oxygen self-diffusion and surface exchange studies electrolytes having the fluorite structure of oxide. Solid State Ion. 1997, 93, 125–132. [Google Scholar] [CrossRef]
- Kamiya, M.; Shimada, E.; Ikuma, Y.; Komatsu, M.; Haneda, H. Intrinsic and Extrinsic Oxygen Diffusion and Surface Exchange Reaction in Cerium Oxide. J. Electrochem. Soc. 2000, 147, 1222–1227. [Google Scholar] [CrossRef]
- Katsuki, M.; Wang, S.; Yasumoto, K.; Dokiya, M. The oxygen transport in Gd-doped ceria. Solid State Ion. 2002, 154–155, 589–595. [Google Scholar] [CrossRef]
- Stan, M.; Zhu, Y.T.; Jiang, H. Kinetics of oxygen removal from ceria. J. Appl. Phys. 2004, 95, 3358–3361. [Google Scholar] [CrossRef]
- Armstrong, E.N.; Duncan, K.L.; Oh, D.J.; Weaver, J.F.; Wachsman, E.D. Determination of Surface Exchange Coefficients of LSM, LSCF, YSZ, GDC Constituent Materials in Composite SOFC Cathodes. J. Electrochem. Soc. 2011, 158, B492–B499. [Google Scholar] [CrossRef]
- Gopal, C.B.; Haile, S.M. An electrical conductivity relaxation study of oxygen transport in samarium doped ceria. J. Mater. Chem. A 2014, 2, 2405–2417. [Google Scholar] [CrossRef]
- Rutman, J.; Kilo, M.; Weber, S.; Riess, I. Tracer surface exchange and diffusion of oxygen in nano crystals of Gd doped CeO2. Solid State Ion. 2014, 265, 29–37. [Google Scholar] [CrossRef]
- Maier, J. On the correlation of macroscopic and microscopic rate constants in solid state chemistry. Solid State Ion. 1998, 112, 197–228. [Google Scholar] [CrossRef]
- Knoblauch, N.; Dörrer, L.; Fielitz, P.; Schmücker, M.; Borchardt, G. Surface controlled reduction kinetics of nominally un-doped polycrystalline CeO2. Phys. Chem. Chem. Phys. 2015, 17, 5849–5860. [Google Scholar] [CrossRef] [PubMed]
- Fielitz, P.; Borchardt, G. Oxygen exchange at gas/oxide interfaces: How the apparent activation energy of the surface exchange coefficient depends on the kinetic regime. Phys. Chem. Chem. Phys. 2016, 18, 22031–22038. [Google Scholar] [CrossRef] [PubMed]
- Rührup, V.; Wiemhöfer, H. Ionic Conductivity of Gd- and Y-Doped Ceria–Zirconia Solid Solutions. Z. Naturforsch. 2006, 61b, 916–922. [Google Scholar] [CrossRef]
- Ji, H.-I.; Davenport, T.C.; Gopal, C.B.; Haile, S.M. Extreme high temperature redox kinetics in ceria: Exploration of the transition from gas-phase to material-kinetic limitations. Phys. Chem. Chem. Phys. 2016, 18, 21554–21561. [Google Scholar] [CrossRef] [PubMed]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to form a Capacitor. U.S. Patent 3.330.697, 11 July 1967. [Google Scholar]
- Knoblauch, N.; Simon, H.; Schmücker, M. Chemically induced volume change of CeO2−δ and nonstoichiometric phases. Solid State Ion. 2017, 301, 43–52. [Google Scholar] [CrossRef]
- Ahrens, L.H. The use of ionization potentials Part 1. Ionic radii of the elements. Geochim. Cosmochim. Acta 1952, 2, 155–169. [Google Scholar] [CrossRef]
- Bishop, S.R. Chemical expansion of solid oxide fuel cell materials: A brief overview. Acta Mech. Sin. 2013, 29, 312–317. [Google Scholar] [CrossRef]
- Marrocchelli, D.; Bishop, S.R.; Tuller, H.L.; Yildiz, B. Understanding Chemical Expansion in Non-Stoichiometric Oxides: Ceria and Zirconia Case Studies. Adv. Funct. Mater. 2012, 22, 1958–1965. [Google Scholar] [CrossRef]
- Hull, S.; Norberg, S.T.; Ahmed, I.; Eriksson, S.G.; Marrocchelli, D.; Madden, P.A. Oxygen vacancy ordering within anion-deficient Ceria. J. Solid State Chem. 2009, 182, 2815–2821. [Google Scholar] [CrossRef]
- Schulz, U.; Saruhan, B.; Fritscher, K.; Leyens, C. Review on Advanced EB-PVD Ceramic Topcoats for TBC Applications. Int. J. Appl. Ceram. Technol. 2004, 1, 302–315. [Google Scholar] [CrossRef]
- Chen, P.L.; Chen, I.W. Role of Defect Interaction in Boundary Mobility and Cation Diffusivity of CeO2. J. Am. Ceram. Soc. 1994, 77, 2289–2297. [Google Scholar] [CrossRef]
- Beschnitt, S.; Zacherle, T.; De Souza, R.A. Computational Study of Cation Diffusion in Ceria. J. Phys. Chem. C 2015, 119, 27307–27315. [Google Scholar] [CrossRef]
- Kilner, J.A.; De Souza, R.A.; Fullarton, I.C. Surface exchange of oxygen in mixed conducting perovskite oxides. Solid State Ion. 1996, 86–88, 703–709. [Google Scholar] [CrossRef]
- Kilner, J.A.; Skinner, S.J.; Brongersma, H.H. The isotope exchange depth profiling (IEDP) technique using SIMS and LEIS. J. Solid State Electrochem. 2011, 15, 861–876. [Google Scholar] [CrossRef]
- Blair, J.; Mebane, D.S. A Bayesian approach to electrical conductivity relaxation and isotope exchange/secondary ion mass spectrometry. Solid State Ion. 2015, 270, 47–53. [Google Scholar] [CrossRef]
- Ananyev, M.V.; Tropin, E.S.; Eremin, V.A.; Farlenkov, A.S.; Smirnov, A.S.; Kolchugin, A.A.; Porotnikova, N.M.; Khodimchuk, A.V.; Berenov, A.V.; Kurumchin, E.K. Oxygen isotope exchange in La2NiO4±δ. Phys. Chem. Chem. Phys. 2016, 18, 9102–9111. [Google Scholar] [CrossRef] [PubMed]
- Panlener, R.J.; Blumenthal, R.N.; Garnier, J.E. A Thermodynamic Study of Nonstoichiometric Cerium Dioxide. J. Phys. Chem. Solids 1975, 36, 1213–1222. [Google Scholar] [CrossRef]
- Kilner, J.A.; Steele, B.C.H. Oxygen self-diffusion studies using negative-ion secondary ion mass spectrometry (SIMS). Solid State Ion. 1984, 12, 89–97. [Google Scholar] [CrossRef]
- Chater, R.J.; Carter, S.; Kilner, J.A.; Steele, B.C.H. Development of a novel SIMS technique for oxygen self-diffusion and surface exchange coefficient measurements in oxides of high diffusivity. Solid State Ion. 1992, 53–56, 859–867. [Google Scholar] [CrossRef]
- Fielitz, P.; Borchardt, G. On the accurate measurement of oxygen self-diffusivities and surface exchange coefficients in oxides via SIMS depth profiling. Solid State Ion. 2001, 144, 71–80. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Oxford University Press: Oxford, UK, 1975. [Google Scholar]
- De Souza, R.A.; Zehnpfenning, J.; Martin, M.; Maier, J. Determining oxygen isotope profiles in oxides with Time-of-Flight SIMS. Solid State Ion. 2005, 176, 1465–1471. [Google Scholar] [CrossRef]
- De Souza, R.A. A universal empirical expression for the isotope surface exchange coefficients (k*) of acceptor-doped perovskite and fluorite oxides. Phys. Chem. Chem. Phys. 2006, 8, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, B.A.; van Hassel, B.A.; Vinke, I.C.; de Vries, K.J.; Burggraaf, A.J. The Oxygen transfer process on oxide/noble metal electrodes, studied with impedance spectroscopy, DC polarization and isotope exchange. Electrochem. Acta 1993, 38, 1817–1825. [Google Scholar] [CrossRef]
- Chueh, W.C.; McDaniel, A.H.; Grass, M.E.; Hao, Y.; Jabeen, N.; Liu, Z.; Haile, S.M.; McCarty, K.F.; Bluhm, H.; El Gabaly, F. Highly Enhanced Concentration and Stability of Reactive Ce3+ on Doped CeO2 Surface Revealed In Operando. Chem. Mater. 2012, 24, 1876–1882. [Google Scholar] [CrossRef]
- Murugan, B.; Ramaswamy, A.V. Defect-Site Promoted Surface Reorganization in Nanocrystalline Ceria for the Low-Temperature Activation of Ethylbenzene. J. Am. Chem. Soc. 2007, 129, 3062–3063. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Parker, S.C.; Watson, G.W. CeO2 catalysed conversion of CO, NO2 and NO from first principles energetics. Phys. Chem. Chem. Phys. 2006, 8, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Sayle, T.X.T.; Parker, S.C.; Catlow, R.A. Surface Oxygen Vacancy Formation on CeO2 and Its Role in the Oxidation of Carbon Monoxide. J. Chem. Soc. Chem. Commun. 1992, 977–978. [Google Scholar] [CrossRef]
- Fleig, J. On the current–voltage characteristics of charge transfer reactions at mixed conducting electrodes on solid electrolytes. Phys. Chem. Chem. Phys. 2005, 7, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Bale, C.W.; Bélisle, E. Available online: http://www.crct.polymtl.ca/equiweb.php (accessed on 7 August 2016).
- Bevan, D.J.M.; Kordis, J. Mixed Oxides Of The Type MO2(Fluorite)–M2O3—I Oxygen Dissociation Pressures And Phase Relationships In The System CeO2–Ce2O3 At High Temperatures. J. Inorg. Nucl. Chem. 1964, 26, 1509–1523. [Google Scholar] [CrossRef]
- Bueno-López, A.; Krishna, K.; Makkee, M. Oxygen exchange mechanism between isotopic CO2 and Pt/CeO2. Appl. Catal. A Gen. 2008, 342, 144–149. [Google Scholar] [CrossRef]
- Jones, J.; Xiong, H.; DeLaRiva, A.T.; Peterson, E.J.; Pham, H.; Challa, S.R.; Qi, G.; Oh, S.; Wiebenga, M.H.; Hernández, X.I.P.; et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Merkle, R.; Maier, J. How Is Oxygen Incorporated into Oxides? A Comprehensive Kinetic Study of a Simple Solid-State Reaction with SrTiO3 as a Model Material. Angew. Chem. Int. Ed. 2008, 47, 3874–3894. [Google Scholar] [CrossRef] [PubMed]
- Maier, J. Interaction of oxygen with oxides: How to interpret measured effective rate constants? Solid State Ion. 2000, 135, 575–588. [Google Scholar] [CrossRef]
- Armstrong, E.N.; Duncan, K.L.; Wachsman, E.D. Effect of A and B-site cations on surface exchange coefficient for ABO3 perovskite materials. Phys. Chem. Chem. Phys. 2013, 15, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Carslaw, H.S.; Jaeger, J.C. Conduction of Heat in Solids, 2nd ed.; Clarendon Press: Wotton-under-Edge, UK, 1959. [Google Scholar]


















| Temperature | Sample #1 () | Sample #2 () |
|---|---|---|
| 1582 K | ||
| 1635 K |
| Condition Number | Gas Atmosphere | in the Gas Phase | Experimental Condition/ in the Sample Bulk |
|---|---|---|---|
| 1 | 18O2 | 0.2 | equilibrium ()/0.2 |
| 2 | ≈10−15 | equilibrium ()/≈10−15 | |
| 3 | ≈10−15 → 10−19 | chem. pot. grad. ()/≈10−30 → 10−25 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knoblauch, N.; Simon, H.; Dörrer, L.; Uxa, D.; Beschnitt, S.; Fielitz, P.; Wendelstorf, J.; Spitzer, K.-H.; Schmücker, M.; Borchardt, G. Ceria: Recent Results on Dopant-Induced Surface Phenomena. Inorganics 2017, 5, 76. https://doi.org/10.3390/inorganics5040076
Knoblauch N, Simon H, Dörrer L, Uxa D, Beschnitt S, Fielitz P, Wendelstorf J, Spitzer K-H, Schmücker M, Borchardt G. Ceria: Recent Results on Dopant-Induced Surface Phenomena. Inorganics. 2017; 5(4):76. https://doi.org/10.3390/inorganics5040076
Chicago/Turabian StyleKnoblauch, Nicole, Heike Simon, Lars Dörrer, Daniel Uxa, Stefan Beschnitt, Peter Fielitz, Jens Wendelstorf, Karl-Heinz Spitzer, Martin Schmücker, and Günter Borchardt. 2017. "Ceria: Recent Results on Dopant-Induced Surface Phenomena" Inorganics 5, no. 4: 76. https://doi.org/10.3390/inorganics5040076
APA StyleKnoblauch, N., Simon, H., Dörrer, L., Uxa, D., Beschnitt, S., Fielitz, P., Wendelstorf, J., Spitzer, K.-H., Schmücker, M., & Borchardt, G. (2017). Ceria: Recent Results on Dopant-Induced Surface Phenomena. Inorganics, 5(4), 76. https://doi.org/10.3390/inorganics5040076